

Abstract
The efficient generation of genetic diversity represents an invaluable molecular tool that can be used to label DNA synthesis, to create unique molecular signatures, or to evolve proteins in the laboratory. Here, we present a protocol that allows the generation of large (>1011) mutant libraries for a given target sequence. This method is based on replication of a ColE1 plasmid encoding the desired sequence by a low-fidelity variant of DNA polymerase I (LF-Pol I). The target plasmid is transformed into a mutator strain of E. coli and plated on solid media, yielding between 0.2 and 1 mutations/kb, depending on the location of the target gene. Higher mutation frequencies are achieved by iterating this process of mutagenesis. Compared to alternative methods of mutagenesis, our protocol stands out for its simplicity, as no cloning or PCR are involved. Thus, our method is ideal for mutational labeling of plasmids or other Pol I templates or to explore large sections of sequence space for the evolution of activities not present in the original target. The tight spatial control that PCR or randomized oligonucleotide-based methods offer can also be achieved through subsequent cloning of specific sections of the library. Here we provide protocols showing how to create a random mutant library and how to establish drug-based selections in E. coli to identify mutants exhibiting new biochemical activities.
Keywords: Genetics, Issue 49, random mutagenesis, directed evolution, LB agar drug gradient, bacterial complementation, ColE1 plasmid, DNA polymerase I, replication fidelity, genetic adaptation, antimicrobials, methylating agents
Protocol
I. Generation of a Random Mutant Library
Pol I mediates initiation of ColE1 plasmid replication (reviewed in (1-3). Our method of mutagenesis is based on placing a target sequence in a ColE1 plasmid adjacent to the origin or replication and propagating it in cells expressing low-fidelity DNA polymerase I (LF-Pol I). LF-Pol I is a mutant DNA polymerase I encoding three mutations that decrease the fidelity of replication, namely I709N (in motif A), A759R (in motif B) and D424A (inactivating proofreading) 4,5. LF-Pol I is expressed in an E. coli strain, JS200, that has a temperature-sensitive allele of Pol I (polA12) (6) so that LF-Pol I becomes the predominant activity at 37 °C. Replication of the target sequence in polA12 cells under restrictive conditions results in the generation of a random mutant library. Mutagenesis is more efficient in saturated cultures 4. For reasons that are still unclear, mutagenesis is not continuous, i.e. the mutation frequency does not increase linearly with the number of generations once the culture reaches saturation, even if cells are allowed to expand further in fresh media. Therefore, further increasing the mutation load of the library requires iterative rounds of mutagenesis and plasmid recovery. Here we provide protocols for LF-Pol I mutagenesis. Note that the protocols presented here have been considerably simplified relative to our original description 4 in order to facilitate iteration of the process for achieving the desired mutation load (Fig 1).
Materials
- Cells: JS200 recA718 polA12 (ts) uvrA155 trpE65 lon-11 sulA
- JS200 WT-Pol I: JS200 cells expressing wild type (WT) Pol I
- JS200 LF-Pol I: JS200 expressing low fidelity (LF) Pol I
- Readout strain: JS200 WT-Pol I or (for complementation) a strain lacking a specific activity
- Target Plasmid
- Plasmid containing a ColE1 origin of replication with the target gene cloned in
1. Before Mutagenesis: Preparation of electro-competent JS200 LF-Pol I cells
Pick a single JS200 LF-Pol I colony from a LB plate grown at 30°C (permissive conditions) overnight containing the appropriate antibiotic selection for the plasmid bearing LF-Pol I (0.03mg/ mL chloramphenicol) and place the colony into a test tube containing 8 mL of LB with chloramphenicol. Grow the culture at 30°C and shaking at 250rpm overnight.
In the morning, expand the culture by pouring the 8 mL of JS200 LF-Pol I into a flask containing 400 mL LB with chloramphenicol. Let the culture grow at 30°C while shaking at 250rpm until it reaches an A600 of 0.4-0.7 (typically 3-4h).
Once at an A600 of 0.4-0.7, chill the culture on wet ice for 15 minutes.
Transfer the chilled culture to an appropriate container for centrifugation. Pellet cells by centrifugation at 4°C. Pour off the supernatant, and then add 10 mL of sterile and chilled (on wet ice) 10% glycerol to the container and re-suspend the cells using a serological pipette.
Transfer the re-suspended cells into a 50 mL conical tube. Fill the conical tube up to the 45 mL mark with 10% glycerol and then centrifuge for 15 minutes at 4°C and 4000rpm.
Pour off the supernatant, add once more 10 mL of 10% glycerol to the conical tube, and re-suspend the cells using a serological or normal pipette. Again, fill up the conical tube to the 45 mL mark with 10% glycerol and centrifuge for 15 minutes at 4°C and 4000rpm. Repeat this process twice more to remove all traces of salts.
After the final spin, re-suspend the pellet of cells in equal part 10% glycerol (i.e. re-suspend 2 mL of cells with 2 mL of 10% glycerol).
Aliquot between 100 μL and 500 μL of the suspended cells into several storage tubes. Quick-freeze the cells on dry ice and then store them at -80°C.
Before using the cells for electro-competent transformations, thaw the cells slowly on ice.
2. Mutagenesis: Transforming the target plasmid
Pipette 40 μL of the electro-competent JS200 LF-Pol I cells and between 30-250ng of the target plasmid DNA into a 2mm gap electroporation cuvette. Note #1: A ColE1 plasmid containing GFP can be carried through mutagenesis in parallel with the target gene as a control. After completion of the readout step, GFP can be plated on LB agar plates and visualized for mutagenesis. Colonies that appear dark or dim contain inactivating mutations.
Pulse the mixture in the electroporator at 1800V. Check the time constant (TC) to ensure uniform electroporation conditions for each sample; ideally TC = 5-6 sec.
Recover the cell/DNA mixture in 1 mL of LB broth for 40 minutes at 37°C (restrictive conditions) shaking at 250 rpm.
Plate 50 μL of the recovery culture on a 100mm LB agar Petri dish pre-warmed to 37°C containing both chloramphenicol and an appropriate concentration of an antibiotic selecting for the target plasmid. Note: #2: Aim to plate the cells at a "near lawn" concentration. A "near lawn" concentration is defined as a distinct but uncountable number of colonies (>1000 colonies/ 100mm dish). The dilution plated, if any, will depend on how electro-competent the cells are. If the cells are not very electro-competent and a "near lawn" is not achievable by plating the recovery culture "neat", then centrifuge the recovery culture for 2 minutes at 4000rpm, pour off the supernatant, re-suspend the cells in 50 μL of LB broth and plate the culture.
Incubate the Petri dishe(s) overnight at 37°C.
3. Mutagenesis: Plasmid recovery
The next day, wash the Petri dishes by pipetting 2 mL of LB broth over the cells. Transfer the bacterial colonies from the LB agar Petri dishes to the LB broth by "scrubbing" them off the plate with a sterilized triangle-shaped glass rod. Add 1 mL of LB broth first, collect the wash and repeat the procedure with the second mL of LB broth.
Isolate the plasmid DNA from the plate wash. (This plasmid DNA constitutes the library). Note #3: The wash collected from the LB plate may be too dense to mini-prep in its entirety. If this is the case, mini-prep the maximum allotted amount by your mini-prep kit (typically, this involved diluting your wash to OD=1 and prepping ~3 mL of the diluted culture) or scale up to a maxi-prep
4. Iteration
Restrict 1μg of the isolated plasmid DNA with a restriction enzyme that linearizes the LF-Pol I plasmid, but does not cut your target plasmid (Supplementary Fig 1).
Clean up the restriction digest using a DNA purification kit. Note #4: This step removes all traces of the restriction enzyme and its buffer. This step is essential to keep the salt concentration low for subsequent electro-competent transformations.
Re-transform 30-250ng of the restricted target-plasmid library back into fresh JS200 LF-Pol I cells to put the library through subsequent rounds of mutagenesis. Note #5: Linearizing the Pol I plasmid using a restriction enzyme ensures only the target plasmid gets transformed.
Repeat sections 2 and 3.
5. Readout
Restrict the isolated plasmid DNA with a restriction enzyme(s) that linearizes both the target plasmid and the Pol I plasmid. Run the digest on a 1% agarose gel to ensure quantity and quality of the plasmids. Restricting ~400ng of the isolated plasmid DNA is usually sufficient for analysis.
Restrict 1μg of the isolated plasmid DNA with a restriction enzyme that linearizes the LF-Pol I plasmid, but does not cut your target plasmid.
Clean up the restriction digest using a DNA purification kit.
Transform the restricted target plasmid library into a readout strain to characterize the mutations.
II. Mutant Screen and Trait Analysis Using Gradient Growth Plates.
In order to illustrate how the vast genetic diversity present in our libraries can be coupled to a functional selection, here we provide a protocol for drug-based functional selections in vivo in E. coli. This method is based upon growth along a drug gradient on solid agar. This allows the simultaneous characterization of multiple (up to 12) samples over a range of concentrations, providing a wider dynamic range than a single drug treatment. Another advantage is that the non-linear readout of this assay buffers moderate differences in viability, within a 2-fold range. Thus, this cytotoxicity-resistance assay provides a robust and rapid means to select mutant libraries and to determine the phenotypic profile of individual mutants. Fig. 2 shows an example of each of these uses: panel A shows selection of individual mutants from a human oxidative demethylase ABH2 library. Colonies growing above the WT threshold are selected for increased protection from cytotoxicity caused by the methylating agent methyl methane sulfonate (MMS) 7. Panel B shows an example of gradients used for individual clone characterization. The level of resistance to the third-generation cephalosporin antibiotic cefotaxime is shown on an agar gradient for WT β-lactamase and for two extended-spectrum mutants, R164H and E104K R164S G267R 4. Note that depending on the strength of the observed effects, more than a single plate may be necessary for adequate quantitation: while the 0.4mg/ml gradient allows for direct comparison to the control clones, the level of resistance of the most potent mutant can only be established using a higher concentration of cefotaxime (4mg/mL).
Materials
- Equipment
- 100x100x15mm square Petri dishes (Fisher Sci #0875711A)
- 100mm round Petri dish
- 25x75x1mm glass microscope slide (Fischer Sci #1255015)
- 50 mL tube graduated
- Media
- LB agar: melted and equilibrated in a water bath at 56°C Note #6: temperature of the media may affect the stability and thus activity of the drug or compound being examined.
- Soft Agar: melted and equilibrated in a water bath to 42°C
1. Construction of the gradient
Mark ten lanes, spaced evenly across one edge of the bottom of the square Petri dish.
Place the dish on an incline such that the bottom marked edge is elevated 7mm; a thick grass or other flat object can be used as support to elevate the dish. Pour 25 mL of warm (~56°C) LB agar mixed well with an appropriate concentration of the selecting agent into the inclined dish. This is the bottom layer of the gradient. Make sure the LB agar evenly coats the Petri dish such that the elevated, marked end of the dish contains ~1mm of LB agar and the lowered part contains ~8mm of LB agar. Then allow agar to set for 10-15 minutes. Note #7: For hydrophobic selecting agents, 0.1% surfactant (antifoam B emulsion) may be added to the LB agar to facilitate suspension and uniform distribution of the drug. Add surfactant to the warm sterile LB agar with vigorous shaking prior to the addition of the selecting agent. The surfactant should suspend as a fine haze of small droplets, large droplets indicate the media is too hot and may inhibit uniform distribution of the test drug.
After the first 25 mL of LB agar has hardened, the dish is moved to a flat surface. Next, 25 mL of LB agar without the selecting agent is poured to overlay the first LB agar surface. This is the top layer of the gradient. Make sure the LB agar covers the entire surface of the bottom layer. Cover with lid askew for ventilation, and allow agar to set for 10-15 minutes. Note #8: Be aware of aerosol hazards and potential volatilization of test compound; pour gradients in a chemical or biological safety hood if prescribed by chemical safety requirements. Note #9: Gradient dishes should be used within 4h to preserve the gradient of the drug or test compound concentration.
2. Stamp transfer of bacteria
Soft agar should be equilibrated to 42°C. Transfer 2 mL of liquid soft agar into the lid or bottom of a 100mm round Petri dish. Pipette 40 μL of the bacterial culture into the soft agar and then mix by rocking the plate. Note #10: The growth stage of a bacterial culture may impact its response to a drug or test compound. Cultures in log phase or overnight cultures in stationary phase should be used with consistency for uniform results. Cell density also can skew relative results, thus all cultures should be diluted to have matched A600 density values. Overnight cultures should be diluted to have an A600 density less than 1.0
Coat the long edge of the glass microscope slide with the soft agar mixture. Then, aligning the coated edge of the slide with the bottom mark (from low to high concentration) on the gradient dish, touch the slide to the surface of the agar. A soft touch is sufficient to transfer a ribbon of the soft agar to the gradient surface. The slide is then set aside for cleaning and reuse.
Repeat this process, for the remainder of the bacterial samples. Reference samples should be included on each gradient dish if multiple gradients are being run.
Incubate the gradient dish top down overnight at 37°C. Times and temperatures of incubation may differ for different readout bacterial strains but overnight at 37°C is typically sufficient for visible growth.
3. Imaging and Analyzing Growth
Imaging: After overnight growth, gradients can be directly imaged or fixed and stained with a solution of 0.2mg/ mL Acridine Orange in 95% EtOH, to enhance contrast. The plate is incubated at room temperature for 5 min, in the staining solution, then washed with 95% EtOH and then imaged over a UV light box. Note #11: Take care not to flush colonies from the plate but rather rock the staining solution and washes over the plate and remove the solutions from corners.
For phenotype analysis of individual mutant plasmids, the distance of growth against the concentration gradient is normalized to a standard on each gradient. These relative values can then be compared across gradients. Note #12: Depending on the nature of the cytotoxic effect, a sharp edge or a more diffuse edge can be observed (compare for example Panels A and B in Fig 2). In the case of diffuse edges, it is advisable to measure the edge of continuous growth, as individual colonies tend to show increased variability.
For library selection, individual colonies that grow at concentrations higher than the parental wild type control are isolated and sequenced to identify mutations contributing to increased protection.
III. Representative Results:
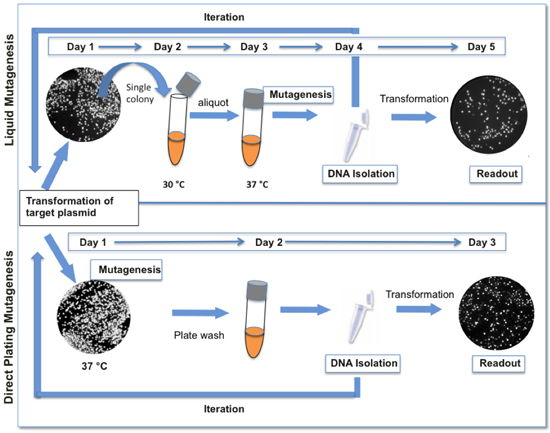 Figure 1. Comparison between liquid and direct plating mutagenesis protocols. The direct plating mutagenesis protocol presented here (bottom) is faster and requires fewer steps than our original liquid mutagenesis protocol (top). When GFP is used as a reporter, variations in fluorescence are indicative of the genetic diversity present in the library. Typically, one cycle of mutagenesis results in 12-18% colonies with appreciably decreased levels of fluorescence.
Figure 1. Comparison between liquid and direct plating mutagenesis protocols. The direct plating mutagenesis protocol presented here (bottom) is faster and requires fewer steps than our original liquid mutagenesis protocol (top). When GFP is used as a reporter, variations in fluorescence are indicative of the genetic diversity present in the library. Typically, one cycle of mutagenesis results in 12-18% colonies with appreciably decreased levels of fluorescence.
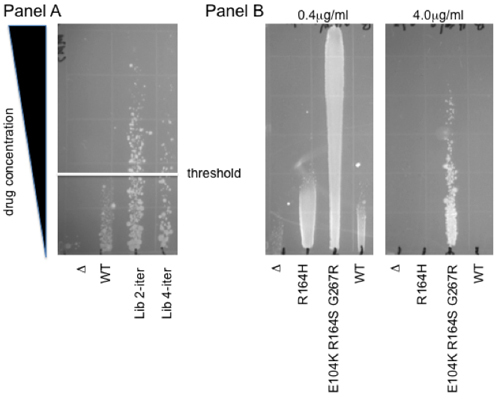 Figure 2. Gradient resistance assays. Panel A Selection for increased resistance to Methyl-Methanesulfonate (MMS). Plasmid libraries of the human oxidative demethylase ABH2 were selected for increased resistance to MMS. Two such libraries representing two and four iterations of the mutagenesis protocol are shown in comparison to the parental wild type (WT) and the empty vector (Δ). The white line denotes the threshold above which individual mutant colonies were isolated for further phenotypic analysis. Panel B Cefotaxime protection, indicative of extended-spectrum β-lactamase activity. R164H and E104K R164H G267R, two mutants of beta-lactamase previously identified following LF-Pol I mutagenesis coupled to aztreonam selection 4, are shown on a 0.4μg/mL and a 4μg/mL cefotaxime gradient. Note that the wild-type β-lactamase enzyme confers no protection relative to cells expressing an empty vector, Δ. Therefore, these mutants represent the evolution of a new biochemical activity 8,9.
Figure 2. Gradient resistance assays. Panel A Selection for increased resistance to Methyl-Methanesulfonate (MMS). Plasmid libraries of the human oxidative demethylase ABH2 were selected for increased resistance to MMS. Two such libraries representing two and four iterations of the mutagenesis protocol are shown in comparison to the parental wild type (WT) and the empty vector (Δ). The white line denotes the threshold above which individual mutant colonies were isolated for further phenotypic analysis. Panel B Cefotaxime protection, indicative of extended-spectrum β-lactamase activity. R164H and E104K R164H G267R, two mutants of beta-lactamase previously identified following LF-Pol I mutagenesis coupled to aztreonam selection 4, are shown on a 0.4μg/mL and a 4μg/mL cefotaxime gradient. Note that the wild-type β-lactamase enzyme confers no protection relative to cells expressing an empty vector, Δ. Therefore, these mutants represent the evolution of a new biochemical activity 8,9.
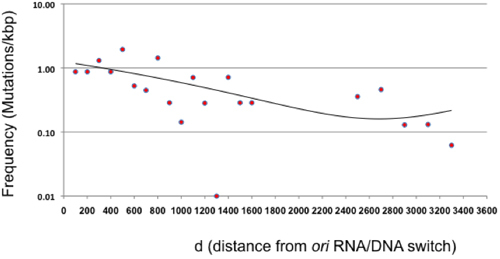 Figure 3. Mutation frequency as a function of distance from ori (d). The mutation frequency following a single cycle of direct plating mutagenesis is shown for 100 bp intervals relative to the RNA/DNA switch of the ColE1 origin of replication. The area between 1600 and 2400 is not represented because β-lactamase does not represent a neutral target. The points beyond β-lactamase represent 200 bp intervals given the overall low frequency of mutation in this area. The trend (binomial equation, with an R2=0.41) is shown as a line.
Figure 3. Mutation frequency as a function of distance from ori (d). The mutation frequency following a single cycle of direct plating mutagenesis is shown for 100 bp intervals relative to the RNA/DNA switch of the ColE1 origin of replication. The area between 1600 and 2400 is not represented because β-lactamase does not represent a neutral target. The points beyond β-lactamase represent 200 bp intervals given the overall low frequency of mutation in this area. The trend (binomial equation, with an R2=0.41) is shown as a line.
 Supplementary Figure 1. LF Pol I-containing Pol I plasmid. A Sequence (FastA format). Sequence information for determining the proper restriction enzyme(s) to use when linearizing the Pol I plasmid either for iteration (generation of a random mutation library step 4) or readout (step 5). B General features and restriction map of the plasmid. The location of the pSC101 origin of replication, the chloramphenicol resistance marker (CAT) and LF-Pol I gene are presented. The location of single restriction sites is indicated as well.
Supplementary Figure 1. LF Pol I-containing Pol I plasmid. A Sequence (FastA format). Sequence information for determining the proper restriction enzyme(s) to use when linearizing the Pol I plasmid either for iteration (generation of a random mutation library step 4) or readout (step 5). B General features and restriction map of the plasmid. The location of the pSC101 origin of replication, the chloramphenicol resistance marker (CAT) and LF-Pol I gene are presented. The location of single restriction sites is indicated as well.
| Library | Direct Plating | Liquid (1 day) | Liquid (3 days) | |
| Mutations (#) | 95 | 40 | 142 | |
| Clones sequenced | 288 | 96 | 190 | |
| Total Coverage (bp) | 182,000 | 102,000 | 213,000 | |
| Mutation freq (x103 bp) | 0.52 | 0.39 | 0.67 | |
| Freq d<1000 (x103 bp) | 0.92 | 0.41 | 0.70 |
Table 1. Mutation frequencies. Frequencies (expressed as # of mutations/bp) for d<1000, i.e. within the 1000 bp adjacent to the RNA/DNA switch, for three mutagenesis protocols: direct plating, liquid saturation (1 day), and liquid hypersaturation (3 days).
| Direct Plating | Liquid | ||
| Mutations (#) | 95 | 182 | |
| Spectrum (%) | |||
| A to G | A to G | 6.3 | 19.2 |
| T to C | 4.2 | 3.8 | |
| C to T | G to A | 27.4 | 13.7 |
| C to T | 35.8 | 35.2 | |
| A to T | A to T | 5.3 | 8.2 |
| T to A | 5.3 | 7.1 | |
| T to G | T to G | 1.1 | 1.1 |
| A to C | 0.0 | 2.2 | |
| G to T | G to T | 1.1 | 2.7 |
| C to A | 2.1 | 2.7 | |
| C to G | C to G | 2.1 | 1.1 |
| G to C | 5.3 | 2.7 | |
| Indels | Ins | 3.2 | 0.0 |
| Del | 1.1 | 0.0 | |
| A to N | 11.6 | 29.7 | |
| G to N | 33.7 | 19.2 | |
| T to N | 10.5 | 12.1 | |
| C to N | 40.0 | 39.0 | |
| Ts | 73.7 | 72.0 | |
| Tv | 22.1 | 28.0 | |
| Indels | 4.2 | 0 |
Table 2. Metrics of mutation for direct plating and liquid mutagenesis. The table presents the number of observed mutations (in number) and the mutation spectrum (in %) following a single cycle of mutagenesis. The spectrum is broken down by complementary pairs, by nucleotide changes, and by the type of mutation.
Discussion
This article presents a mutagenesis protocol that allows the generation of large random mutant libraries without the need for cloning or PCR. This method is based on error-prone replication of a plasmid encoding a sequence of interest. In theory, mutations should be largely restricted to the 100-300 bp located immediately downstream of the RNA/DNA switch, the size of leader strand intermediate produced by Pol I 2,10. We found that under the conditions presented in this protocol, Pol I mutations occur all over the plasmid, although diminishing in frequency as the distance from the plasmid ori increases (Figure 3). This finding implies that the transition or "switch" from Pol I to Pol III during ColE1 plasmid replication is much more gradual than previously reported, at least under our experimental conditions 2, and agrees with earlier studies suggesting a functional redundancy between Pol I and Pol III 11.
Our original protocol described mutagenesis in liquid cultures (4). This protocol yields 0.41 mutations/kb for d<1000, i.e. within the 1000 bp adjacent to the RNA/DNA switch (Table 1). Hypersaturation by leaving the liquid culture in the shaker for 3 days without the addition of any fresh media raises the mutation frequency to 0.70 mutations/kb but results in very poor plasmid yield (less than 1% compared to day 1; data not shown) (Table 1). Here we present a simplified protocol based on directly plating the transformation of the target plasmid on solid agar at 37 °C (Figure 1). This procedure produces the largest frequency of mutation/cycle of mutagenesis (0.92 mutations/kb for d<1000) and greatly facilitates iteration. Changing culture conditions to solid media also affects the mutation spectrum (Table 2). Direct plating mutagenesis reduces the marked asymmetry between complementary base pair substitutions seen in liquid culture (compare C→T vs. G→A and A→G vs. T→C). On the other hand, direct plating produced more indels (from less than 0.5% to 4%) and decreased the number of A→G mutations by 3-fold, leading to an overrepresentation of G/C mutations (74%). Overall, we believe that the greater simplicity and increased efficiency of the direct plating protocol presented here outweighs the benefits of the slightly more balanced mutation spectrum produced by liquid mutagenesis.
Within the target plasmid, the mutations generated by our method are not restricted to the desired target sequence. However, the evolution of novel biochemical activities should be dependent on mutations within the target gene, as it represents a qualitative rather than a quantitative change. Thus, combining our plasmid mutagenesis with screening of mutants on gradient plates capitalizes on the strengths of our system (large libraries and availability of selection) for the evolution of new biochemical properties. Other applications of random mutagenesis such as optimizing existing enzymatic activities or randomizing specific areas of a gene do require cloning following random mutagenesis. In this case, having the library in a plasmid as opposed to a PCR amplification product improves the efficiency of cloning by facilitating amplification and restriction.
In sum, we demonstrate a simple protocol to create a random mutant library for a given target gene that stands out for its simplicity and for the diversity of the libraries generated. We show how this method can be coupled with functional selections for the efficient evolution of new biochemical activities. In addition, our in vivo-generated libraries can be easily cloned, allowing site-specific mutagenesis or optimization of existing activities.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work has been supported by K08 award CA116429-04 to MC and by a grant from the Conquer Cancer Now (CONCERN) foundation # 8501
References
- Camps M. Modulation of ColE1-like plasmid replication for recombinant gene expression. Recent Pat DNA Gene Seq. 2010;4:58–73. doi: 10.2174/187221510790410822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Tomizawa J. Initiation of replication of plasmid ColE1 DNA by RNA polymerase, ribonuclease H, and DNA polymerase I. Cold Spring Harb Symp Quant Biol. 1979;43 Pt 1:409–417. doi: 10.1101/sqb.1979.043.01.047. [DOI] [PubMed] [Google Scholar]
- Polisky B. ColE1 replication control circuitry: sense from antisense. Cell. 1988;55:929–932. doi: 10.1016/0092-8674(88)90235-8. [DOI] [PubMed] [Google Scholar]
- Camps M, Naukkarinen J, Johnson BP, Loeb LA. Targeted gene evolution in Escherichia coli using a highly error-prone DNA polymerase I. Proc Natl Acad Sci U S A. 2003;100:9727–9732. doi: 10.1073/pnas.1333928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai A, Loeb LA. In vivo mutagenesis by Escherichia coli DNA polymerase I. Ile(709) in motif A functions in base selection. J Biol Chem. 2001;276:46759–46764. doi: 10.1074/jbc.M104780200. [DOI] [PubMed] [Google Scholar]
- Uyemura D, Lehman IR. Biochemical characterization of mutant forms of DNA polymerase I from Escherichia coli. I. The polA12 mutation. J Biol Chem. 1976;251:4078–4084. [PubMed] [Google Scholar]
- Sedgwick B, Robins P, Lindahl T. Direct removal of alkylation damage from DNA by AlkB and related DNA dioxygenases. Methods Enzymol. 2006;408:108–120. doi: 10.1016/S0076-6879(06)08008-6. [DOI] [PubMed] [Google Scholar]
- Aharoni A, Gaidukov L, Khersonsky O, Mc QGS, Roodveldt C, Tawfik DS. The 'evolvability' of promiscuous protein functions. Nat Genet. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- Elena SF, Lenski RE. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet. 2003;4:457–469. doi: 10.1038/nrg1088. [DOI] [PubMed] [Google Scholar]
- Itoh T, Tomizawa J. FoFormation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proc Natl Acad Sci U S A. 1980;77:2450–2454. doi: 10.1073/pnas.77.5.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan SK, Moses RE. Sufficiency of the Klenow fragment for survival of polC(Ts) pcbA1 Escherichia coli at 43 degrees. C. J Bacteriol. 1988;170:456–458. doi: 10.1128/jb.170.1.456-458.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]