

Abstract
Haloenol pyran-2-ones and morpholin-2-ones were synthesized and evaluated as inhibitors of cell growth in two different prostate human cancer cell lines (PC-3 and LNCaP). Analogs derived from L- and D-phenylglycine were found to be the most effective antagonists of LNCaP and PC-3 cell growth. Additional studies reveal that the inhibitors induced G2/M arrest and the (S)-enantiomer of the phenylglycine-based derivatives was a more potent inhibitor of cytosolic iPLA2β.
Keywords: Haloenol, Pyranones, Morpholinones, Phospholipase A2, Prostate cancer
Haloenol pyranones1 are mechanism-based inhibitors of serine proteases due to their ability to alkylate enzyme active sites following ring hydrolysis and unmasking of a reactive α-haloketone functionality (Figure 1). To date, the most evaluated of these inhibitors is bromoenol lactone, (E)-6- (bromomethylene)tetrahydro-3-(1-naphthalenyl)-2H-pyran-2-one (4) or BEL. Interestingly, the popularity of BEL stems not as a deactivator of serine proteases, but rather for its ability to inhibit Ca2+-independent phospholipases A2 (iPLA2), which are responsible for the catabolism of membrane glycerophospholipids. Over the last 20 years, BEL has enabled researchers to probe the role of iPLA2 in pathologies involving oxidative stress and inflammation including cardiovascular2, Alzheimer’s3 and Parkinson’s diseases4, diabetes mellitus5, and more recently, carcinogenesis6,7.
Figure 1.

Mechanism of serinase inhibition by haloenol pyran-2-ones.
Mammalian cells possess multiple isoforms of iPLA2 8. The most studied are cytosolic iPLA2β, (Group VIA-1 and A-2 PLA2) and the membrane localized iPLA2γ(Group VIB PLA2), which together govern the release of fatty acids arachidonic acid and 2- lysophospholipids from membrane phospholipids. For many years, phospholipid remodeling8,9 was thought to be the only function of these enzymes; however, beginning in the 1990’s researchers began finding evidence that iPLA2 participates in cell signaling10, proliferation11, and death4,12. It was established that the products arising from the breakdown of phospholipids functioned as signaling molecules for promoting cell growth and that the enzymes responsible for generating the lipids (i.e. PLA2) are in greater abundance in carcinoma cells13.
The effects of iPLA2γ and iPLA2β on cell signaling and proliferation have recently been studied by enantiomer-based inhibition6,14 strategies using (R)- and (S)-BEL, respectively (Figure 2). The mechanisms involved in their selectivity are currently under study although it was demonstrated that LNCaP and PC-3 prostate cancer cells display moderate increases in chemosensitivities to racemic BEL compared to the individual enantiomers6. These results suggest that the (R)- and (S)-conformers could be acting in a synergistic manner as cell growth inhibitors. The studies further established that enantiomers of haloenol pyranones may be used to selectively and pharmacologically inhibit iPLA2γ, iPLA2β, and possibly other enzymes involved in critical cell processes. In this Letter, we report on the antineoplastic activities of haloenol pyran-2-one analogs of BEL against prostate cancer. In addition, the evaluation of novel haloenol morpholin-2-ones constructed from L- and D-amino acids and their inhibitory effects on the cell cycle and iPLA2 activity are described.
Figure 2.

Chemical structures of BEL and haloenol morpholinones.
To evaluate whether analogs of BEL could have similar inhibitory effects on iPLA2 and prostate cancer growth, we set forth to synthesize various haloenol pyran-2-ones from α-substituted and unsubstituted acetylenic acids. Standard E-specific haloenol lactonization procedures1,15 with N-halosuccinimides (X = Br, I) were used to generate the pyranone analogs (Scheme 1). In the case of the phenyl analog 4, the acid precursor 3 required preparation from phenylacetic acid and 4-bromobut-1-yne using classical enolate chemistry1g. Subsequent attempts to separate the (R)- and (S)-enantiomers of lactone 4 by chiral HPLC were unsuccessful, which led to us to consider the use of chiral pool amino acids to construct novel iPLA2 inhibitors containing a E-haloenol morpholin-2-one framework (Figure 2).
Scheme 1.

Synthesis of unsubstituted and monophenyl BEL analogs.
L- and D-phenylglycine (Phg), L-phenylalanine (Pha), and glycine (Gly) were chosen as base materials to perform the asymmetrical synthesis of morpholinone analogs (Scheme 2). Protected tert-butyl esters forms of the amino acids were first prepared from tert-butyl acetate16 then converted to the the corresponding N,N-propargyl α-amino esters 11–1317. Following deprotection of the carboxylic acid, bromo- and iodoenol morpholin- 2-one analogs 14–17 were generated in 6–23% yield under the conditions described for pyranones 2 and 4.
Scheme 2.

Synthesis of N-propargyl bromoenol morpholin-2-ones 14–17.
The synthesis of additional L-Phg-based analogs was also attempted from the monopropargyl intermediate 18 (Scheme 3). Benzylation of the secondary amine followed by acid deprotection and cyclization gave the corresponding N-benzyl bromoenol morpholin-2-one 20. Efforts to prepare the unsubstituted analog 21 were unsuccessful however, which was attributed to chemical instability of the N-protio ring system.
Scheme 3.

Synthesis of N-benzyl bromoenol morpholin-2-one 20.
Minimum inhibitory concentrations (IC50s) were determined by MTT staining for the haloenol pyranones (2, 4) and morpholinones (14–17, 20) against LNCaP cells, and the more resistant PC-3 human prostate cancer cell line. With racemic BEL as a haloenol standard, IC50 measurements were taken at 24, 48, and 72 h (Table 1). BEL was found to inhibit growth in a time-dependent manner at 5–13 μM and 14–34 μM of LNCaP and PC-3, respectively, over 72 h which corroborated previous findings6. Activity comparison of BEL to pyranones 2 revealed that the unsubstituted analogs were equally efficacious inhibitors at 5–10 and 14–32 μM for the corresponding cell lines. For the α-substituted phenyl analog 4, slightly enhanced activities were observed with IC50s ranging from 6–27 μM against PC-3.
Table 1.
IC50s (μM) against human prostate cancers after 24, 48, and 72 h exposure to haloenol inhibitorsa
| compd | LNCaP | PC-3 | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| 24 | 48 | 72 | 24 | 48 | 72 | |
| rac-BEL | 13 | 5 | 9 | 34 | 26 | 14 |
| 2a | 10 | 5 | 5 | 19 | 23 | 14 |
| 2b | 9 | 5 | 7 | 32 | 15 | 16 |
| rac-4 | 31 | 5 | 4 | 27 | 10 | 6 |
| (S)-14a | 8 | 3 | 3 | 15 | 13 | 5 |
| (R)-14b | 6 | 6 | 3 | 8 | 6 | 3 |
| (S)-15 | 26 | 23 | 20 | 21 | 21 | 25 |
| (S)-16 | 41 | 26 | 32 | 33 | 57 | 39 |
| 17 | 25 | 29 | 28 | 13 | 10 | 7 |
| (S)-20 | 3 | 4 | 3 | 4 | 1 | 4 |
Data represent the calculated IC50 using data assessed 3–5 experiments ran in duplicate using separate passages of cells assessing alteration in MTT staining.
The morpholinones analogs similarly demonstrated antineoplastic activity with IC50s reaching 3 μM for the Phg-based derivatives 14 (Table 1). The inhibitors also appeared to be more rapid-acting antagonists of prostate cancer growth compared to BEL and its phenyl pyranone analog 4. Moreover, activity comparison of the enantiomers revealed that (R)-14b was a more effective inhibitor than (S)-14a particularly against PC-3 cells (IC50 3–8 μM). As a compound derived from the unnatural D-form of Phg, the augmented activity of (R)-14b was attributed in part to higher proteolytic susceptibility (e.g. chymotrypsin) that the L-Phg-based (S)-14a may have in the cell.
Other haloenol morpholinones were found to have weaker inhibitory activities including the L-Pha- and Gly-derived analogs 16 and 17, respectively. Surprisingly, chemosensitivity for the iodoenol derivative 15 was also considerably lower than its bromoenol counterparts 14. Conversely, the N-benzyl L-Phg-based analog 20 proved to be the most potent antagonist in the study (IC50 1–4 μM). The compound demonstrated rapid and sustained inhibitory effects on cell proliferation for both LNCaP and PC-3 cells over the 72 h evaluation period.
To determine if growth inhibition was due to cytostatic or cytotoxic effects by the antagonists, cell viability was assessed by phase-contrast microscopy18. Comparisons of morphology were made by visual inspection of LNCaP cells following 72 h treatment with rac-BEL, rac-4, (S)-14a, and (R)-14b (Figure 3). Exposure to 5 μM of BEL and its monophenyl analog 4 induced little to no morphological changes in cell shape, differentiation, and death compared to the vehicle (DMSO) control. For the morpholinone analogs 14, apoptosis and/or necrosis was evident at the same concentrations particularly for the (R)-enantiomer. It was concluded from these microscopic images that prostate cancer cells had greater chemosensitivity to haloenol morpholinones than to the analogous haloenol pyranones which corroborated the IC50 data.
Figure 3.

Changes in morphology (left-40X magnification) and cell cycle (right) of LNCaP cells following treatment with rac-BEL, rac-4, (S)-14a, and (R)-14b.
The inhibitory effects by rac-BEL, pyranone 4, and morpholinones 14 were additionally assessed my monitoring changes in the cell cycle by flow cytometry with propidium iodide6b (Figure 3). Moderate increases of LNCaP cell counts in the G1 phase were observed following 24 h treatment with 5 and 10 μM of the test compounds. It is believed that the elevated G1 levels led to the decrease in S and G2/M phase cell percentages and the effects were greatest for 14a and 14b which induced complete cell cycle arrest at 10 μM. Likewise, on comparison to cultures treated with the 5 μM of the inhibitors, the increase of cells residing in S phase may have been due to the lack of cells entering the G2/M phase. These results further suggest that the cytotoxic effects of morpholinone-based analogs may be the result of DNA hypoploidy, which is associated with DNA fragmentation and apoptosis. Examples of agents that block mitosis by inhibiting chromosome replication include DNA alkylating agents (e.g. nitrogen mustards) and antagonists of glutathione S-transferase (e.g. α-chloroacetamides19), which protect cells from oxidative DNA damage.
Lastly, rac-BEL, pyranone 4, and morpholinones 14 were evaluated for their ability to inhibit iPLA2β from rat kidney. Cytosolic fractions were treated for 0.5 h with 0–100 μM of the compounds prior to inoculation with the arachidonoyl thiophosphatidylcholine, a hydrolysable thioester-containing probe of PLA2 activity6b. Both rac-BEL and its phenyl-substituted analog 4 demonstrated nearly identical efficacy to inhibit the enzyme in a concentration-dependent manner (Figure 4). Inhibitory activity was also noted for (S)-14a but to a lesser degree compared to pyranone-based antagonists. Little to no effects on iPLA2β activity was observed for (R)-14b which correlates to earlier findings6,14 that the (S)-enantiomer of BEL selectively inhibits cytosolic iPLA2β while (R)-BEL possesses higher affinity for microsomal iPLA2γ.
Figure 4.
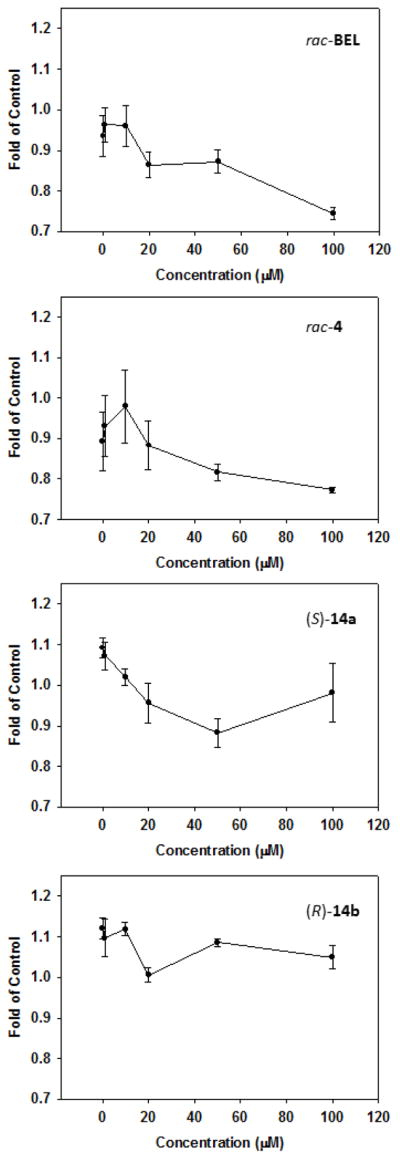
Inhibitory effects of rac-BEL, rac-4, (S)-14a, and (R)-14b on iPLA2β activity in rat kidney cytosol in the presence of 4 mM EGTA. Data are represented as the mean ± the S.E.M. of at least 3 separate experiments.
In summary, haloenol pyran-2-ones were found to be efficacious inhibitors of prostate carcinoma cell growth and iPLA2β activity however, as with BEL, a definitive correlation could not be made. Novel haloenol morpholin-2-ones constructed asymmetrically from chiral amino acids were also discovered to be antagonists of cell proliferation. Differences in the effects on the cell cycle and iPLA2β activity suggested that the morpholinone analogs 14 may have a greater capacity to directly or indirectly cause DNA damage. Glutathione S-transferase which has a role in protecting DNA from oxidative damage is known to be inhibited by haloenol lactones20 and could be a primary or secondary target for the Phg-based derivatives. Finally, during the course of these studies it became apparent that the chemical instability of the haloenol pyranones and morpholinones would likely preclude them from being viable drug candidates for prostate cancer. Their use as research tools in the study of tumorigenesis and validation of new therapeutic targets may be of great value though to the drug discovery community.
Acknowledgments
Financial support was generously provided by the College of Pharmacy at the University of Georgia and in part by a Georgia Cancer Coalition Distinguished Scholar Grants and a NIH NIBIB (EB08153) to B.S.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Rai R, Katzenellenbogen JA. J Med Chem. 1992;35:4150. doi: 10.1021/jm00100a021. [DOI] [PubMed] [Google Scholar]; (b) Reed PE, Katzenellenbogen JA. J Biol Chem. 1991;266:13. [PubMed] [Google Scholar]; (c) Sofia MJ, Katzenellenbogen JA. J Med Chem. 1986;29:230. doi: 10.1021/jm00152a011. [DOI] [PubMed] [Google Scholar]; (d) Daniels SB, Katzenellenbogen JA. Biochemistry. 1986;25:1436. doi: 10.1021/bi00354a037. [DOI] [PubMed] [Google Scholar]; (e) Daniels SB, Cooney E, Sofia MJ, Chakravarty PK, Katzenellenbogen JA. J Biol Chem. 1983;258:15046. [PubMed] [Google Scholar]; (f) Chakravarty PK, Krafft GA, Katzenellenbogen JA. J Biol Chem. 1982;257:610. [PubMed] [Google Scholar]; (g) Krafft GA, Katzenellenbogen JA. J Am Chem Soc. 1981;103:5459. [Google Scholar]; (h) Rando RR. Science. 1974;185:320. doi: 10.1126/science.185.4148.320. [DOI] [PubMed] [Google Scholar]
- 2.McHowat J, Creer MH. Curr Med Chem Cardiovasc Hematol Agents. 2004;2:209. doi: 10.2174/1568016043356282. [DOI] [PubMed] [Google Scholar]
- 3.(a) Sun GY, Xu J, Jensen MD, Yu S, Wood WG, González FA, Simonyi A, Sun AY, Weisman GA. Mol Neurobiol. 2005;31:27. doi: 10.1385/MN:31:1-3:027. [DOI] [PubMed] [Google Scholar]; (b) Hoozemans JJM, Veerhuis R, Janssen I, Rozemuller AJM, Eikelenboom P. Exp Gerontol. 2001;36:559. doi: 10.1016/s0531-5565(00)00226-6. [DOI] [PubMed] [Google Scholar]
- 4.(a) Farooqui AA, Horrocks LA. Neuroscientist. 2006;12:245. doi: 10.1177/1073858405285923. [DOI] [PubMed] [Google Scholar]; (b) Farooqui AA, Ong WY, Horrocks LA. Pharmacol Rev. 2006;58:591. doi: 10.1124/pr.58.3.7. [DOI] [PubMed] [Google Scholar]
- 5.(a) Song K, Zhang X, Zhao CY, Ang NT, Ma ZMA. Mol Endocrinol. 2005;19:504. doi: 10.1210/me.2004-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ramanadham S, Song HW, Hsu FF, Zhang S, Crankshaw M, Grant GA, Newgard CB, Bao SZ, Ma ZM, Turk J. Biochemistry. 2003;42:13929. doi: 10.1021/bi034843p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk J. J Biol Chem. 1999;274:9607. doi: 10.1074/jbc.274.14.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ma Z, Ramanadham S, Hu Z, Turk J. Biochim Biophys Acta. 1998;1391:384. doi: 10.1016/s0005-2760(98)00027-7. [DOI] [PubMed] [Google Scholar]
- 6.(a) Sun B, Zhang XL, Yonz C, Cummings BS. Biochem Pharmacol. 2010;79:1727. doi: 10.1016/j.bcp.2010.02.005. [DOI] [PubMed] [Google Scholar]; (b) Sun B, Zhang X, Talathi S, Cummings BS. J Pharmacol Exp Ther. 2008;326:59. doi: 10.1124/jpet.108.138958. [DOI] [PubMed] [Google Scholar]
- 7.(a) Song Y, Wilkins P, Hu WH, Murthy KS, Chen J, Lee Z, Oyesanya R, Wu JH, Barbour SE, Fang XJ. Biochem J. 2007;406:427. doi: 10.1042/BJ20070631. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang XH, Zhao C, Seleznev K, Song K, Manfredi JJ, Ma ZA. J Cell Sci. 2006;119:1005. doi: 10.1242/jcs.02821. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang L, Peterson BL, Cummings BS. Biochem Pharmacol. 2005;70:1697. doi: 10.1016/j.bcp.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 8.(a) Cummings BC. Biochem Pharmacol. 2007;74:949. doi: 10.1016/j.bcp.2007.04.021. [DOI] [PubMed] [Google Scholar]; (b) Schaloske RH, Dennis EA. Biochim Biophys Acta. 2006;1761:1246. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]; (c) Balsinde J, Perez R, Balboa MA. Biochim Biophys Acta. 2006;1761:1344. doi: 10.1016/j.bbalip.2006.07.013. [DOI] [PubMed] [Google Scholar]; (d) Casas J, Gijon MA, Vigo AG, Crespo MS, Balsinde J, Balboa MA. J Biol Chem. 2006;281:6106. doi: 10.1074/jbc.M505230200. [DOI] [PubMed] [Google Scholar]; (e) Balboa MA, Balsinde J, Dennis EA. Biochem Biophys Res Commun. 2000;267:145. doi: 10.1006/bbrc.1999.1964. [DOI] [PubMed] [Google Scholar]
- 9.(a) Balsinde J, Balboa MA, Dennis EA. J Biol Chem. 1997;272:29317. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]; (b) Balsinde J, Dennis EA. J Biol Chem. 1997;272:16069. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]; (c) Balsinde J, Dennis EA. J Biol Chem. 1996;271:31937. doi: 10.1074/jbc.271.50.31937. [DOI] [PubMed] [Google Scholar]
- 10.(a) Hooks SB, Cummings BS. Biochem Pharmacol. 2008;76:1059. doi: 10.1016/j.bcp.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu J, Weng YI, Simonyi A, Krugh BW, Liao Z, Weisman GA, Sun GY. J Neurochem. 2002;83:259. doi: 10.1046/j.1471-4159.2002.01145.x. [DOI] [PubMed] [Google Scholar]; (c) Balsinde J, Balboa MA, Li WH, Llopis J, Dennis EA. J Immunol. 2000;164:5398. doi: 10.4049/jimmunol.164.10.5398. [DOI] [PubMed] [Google Scholar]; (d) Bonventre JV. J Lipid Mediat Cell Signal. 1997;17:71. doi: 10.1016/s0929-7855(97)00021-7. [DOI] [PubMed] [Google Scholar]; (e) Ramanadham S, Wolf MJ, Li B, Bohrer A, Turk J. Biochim Biophys Acta. 1997;1344:153. doi: 10.1016/s0005-2760(96)00139-7. [DOI] [PubMed] [Google Scholar]
- 11.(a) Hassan S, Carraway RE. Regul Pept. 2006;133:105. doi: 10.1016/j.regpep.2005.09.031. [DOI] [PubMed] [Google Scholar]; (b) Saavedra G, Zhang W, Peterson B, Cummings BS. J Pharmacol Exp Ther. 2006;318:1211. doi: 10.1124/jpet.106.105650. [DOI] [PubMed] [Google Scholar]; (c) Bao S, Bohrer A, Ramanadham S, Jin W, Zhang S, Turk J. J Biol Chem. 2006;281:187. doi: 10.1074/jbc.M509105200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Longo WE, Grossmann EM, Erickson B, Panesar N, Mazuski JE, Kaminski DL. J Surg Res. 1999;84:51. doi: 10.1006/jsre.1999.5603. [DOI] [PubMed] [Google Scholar]; (e) Teslenko V, Rogers M, Lefkowith JB. Biochim Biophys Acta. 1997;1344:189. doi: 10.1016/s0005-2760(96)00137-3. [DOI] [PubMed] [Google Scholar]
- 12.(a) Cummings BS, McHowat J, Schnellmann RG. J Pharmacol Exp Ther. 2000;294:793. [PubMed] [Google Scholar]; (b) Atsumi G, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I. J Biol Chem. 1998;273:13870. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- 13.(a) Dong Q, Patel M, Scott KF, Graham GG, Russell PJ, Sved P. Cancer Lett. 2006;240:9. doi: 10.1016/j.canlet.2005.08.012. [DOI] [PubMed] [Google Scholar]; (b) Jiang J, Neubauer BL, Graff JR, Chedid M, Thomas JE, Roehm NW, Zhang S, Eckert GJ, Koch MO, Eble JN, Cheng L. Am J Pathol. 2002;160:667. doi: 10.1016/S0002-9440(10)64886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Graff JR, Konicek BW, Deddens JA, Chedid M, Hurst BM, Colligan B, Neubauer BL, Carter HW, Carter JH. Clin Cancer Res. 2001;7:3857. [PubMed] [Google Scholar]; (d) Yamashita S, Yamashita J, Ogawa M. Br J Cancer. 1994;69:1166. doi: 10.1038/bjc.1994.229. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yamashita S, Ogawa M, Sakamoto K, Abe T, Arakawa H, Yamashita J. Clin Chim Acta. 1994;228:91. doi: 10.1016/0009-8981(94)90280-1. [DOI] [PubMed] [Google Scholar]; (f) Yamashita S, Yamashita J, Sakamoto K, Inada K, Nakashima Y, Murata K, Saishoji T, Nomura K, Ogawa M. Cancer. 1993;71:3058. doi: 10.1002/1097-0142(19930515)71:10<3058::aid-cncr2820711028>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 14.(a) Kinsey GR, Cummings BS, Beckett CS, Saavedra G, Zhang W, McHowat J, Schnellmann RG. Biochem Biophys Res Commun. 2005;327:287. doi: 10.1016/j.bbrc.2004.12.016. [DOI] [PubMed] [Google Scholar]; (b) Jenkins CM, Han X, Mancuso DJ, Gross RW. J Biol Chem. 2002;277:32807. doi: 10.1074/jbc.M202568200. [DOI] [PubMed] [Google Scholar]
- 15.Dai W, Katzenellenbogen JA. J Org Chem. 1991;56:6893. [Google Scholar]
- 16.Chen H, Feng Y, Xu Z, Ye T. Tetrahedron. 2005;61:11132. [Google Scholar]
- 17.Cho JH, Kim BM. Tetrahedron Lett. 2002;43:1273. doi: 10.1016/S0040-4039(02)01113-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cummings BS, Schnellmann RG. J Pharm Exp Therap. 2002;302:8. doi: 10.1124/jpet.302.1.8. [DOI] [PubMed] [Google Scholar]
- 19.Tsuboi K, Bachovchin DA, Speers AE, Spicer TP, Fernandez-Vega V, Hodder P, Rosen H, Cravatt BF. J Am Chem Soc. 2011;133:16605. doi: 10.1021/ja2066972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Z, Minhas GS, Wen D, Jiang H, Chen K, Zimniak P, Zheng J. J Med Chem. 2004;47:3282. doi: 10.1021/jm0499615. [DOI] [PubMed] [Google Scholar]