

Abstract
Many gene expression analysis techniques rely on material isolated from heterogeneous populations of cells from tissue homogenates or cells in culture.1,2,3 In the case of the brain, regions such as the hippocampus contain a complex arrangement of different cell types, each with distinct mRNA profiles. The ability to harvest single cells allows for a more in depth investigation into the molecular differences between and within cell populations. We describe a simple and rapid method for harvesting cells for further processing. Pipettes often used in electrophysiology are utilized to isolate (using aspiration) a cell of interest and conveniently deposit it into an Eppendorf tube for further processing with any number of molecular biology techniques. Our protocol can be modified for the harvest of dendrites from cell culture or even individual cells from acute slices.
We also describe the aRNA amplification method as a major downstream application of single cell isolations. This method was developed previously by our lab as an alternative to other gene expression analysis techniques such as reverse-transcription or real-time polymerase chain reaction (PCR).4,5,6,7,8 This technique provides for linear amplification of the polyadenylated RNA beginning with only femtograms of material and resulting in microgram amounts of antisense RNA. The linearly amplified material provides a more accurate estimation than PCR exponential amplification of the relative abundance of components of the transcriptome of the isolated cell. The basic procedure consists of two rounds of amplification. Briefly, a T7 RNA polymerase promoter site is incorporated into double stranded cDNA created from the mRNA transcripts. An overnight in vitro transcription (IVT) reaction is then performed in which T7 RNA polymerase produces many antisense transcripts from the double stranded cDNA. The second round repeats this process but with some technical differences since the starting material is antisense RNA. It is standard to repeat the second round, resulting in three rounds of amplification. Often, the third round in vitro transcription reaction is performed using biotinylated nucleoside triphosphates so that the antisense RNA produced can be hybridized and detected on a microarray.7,8
Keywords: Neuroscience, Issue 50, single-cell, transcriptome, aRNA amplification, RT-PCR, molecular biology, gene expression
Protocol
1. Cell Culture
Our lab uses primary neuronal rat hippocampal cultures for the experiments below. The following describes the modified Banker protocol for how these cell cultures are created and maintained.9 There are, of course, an exhaustive number of permutations of these cell culturing techniques and any established method tailored to the specific needs of a particular laboratory that provides a consistently healthy supply of cells would be a suitable substitute.
Five autoclaved, round, 12 mm coverslips are placed in a 35 mm dish and coated with 80 μg/mL Poly-D-Lysine (MW 70-150,000) in borate buffer O/N, rinsed with water (cell culture grade), then coated with 1 μg/mL Laminin in borate buffer O/N then rinsed again with water. NG media (MEM with Earle's salts and GlutaMAX supplemented with glucose (0.6% w/v), penicillin (100 U/mL), streptomycin (100 μg/mL), horse serum (10% v/v)) is added and dishes are stored in an incubator (5% CO2 at 37°C). The PDL150/laminin serves as a substrate for the isolated neurons to grow on as a monolayer. The coverslips can be coated up to two weeks before plating.
On the day of plating, primary neurons from embryonic day 18 rat hippocampi are plated in NG media by adding 1.5 mL of a 100,000 cells/mL suspension. Four to six hours after plating, each 35 mm dish is treated with 0.75 μL of 5 mM cytosine beta-D-arabinofuranoside (ara-C) to prevent growth of any contaminating glial cells. Twenty-four hours later, the plating media is changed to MEM with Earle's salts and L-glutamine supplemented with 0.6% w/v glucose, 1 mM sodium pyruvate, and B27. Cells are permitted to grow for at least two weeks before use in any experiments to allow complete development of processes.
2. Preparing Pipettes
Note on RNases: The skin, saliva, and even breath are major sources of RNases, enzymes that degrade RNA. It is imperative that RNase-free technique be observed from this point on in the protocol so that the sample does not become contaminated with RNases and subsequently degrade. This includes always wearing gloves when handling the samples and reagents, never talking over samples or reagents, and using new boxes of sterilized pipette tips and tubes. Often, pipettes that are not designated exclusively for RNA work are decontaminated by wiping them down with an RNase treatment solution such as RNase AWAY (Molecular Bioproducts). However, these solutions will inhibit any downstream enzymatic reactions so it is also important to prevent contamination of your samples with these treatments.
Autoclave 1.5 mm outer diameter (O.D.) glass pipettes.
Using a micropipette puller, pull pipettes to a diameter appropriate for electrophysiological recordings, which may vary based on your puller, filament, and pipettes.10 The bore size is not too critical since the tip will be broken prior to cell collection.
Carefully store the pipettes in a dust-free environment where the tips will remain intact (ideally use a micropipette storage jar).
To break the tip in a controlled fashion, hold a kimwipe taut between your fingers in one hand and very gently brush the tip of the pipette across the paper 1 to 2 times. The opening should be approximately 75-100% of the target cell size. Adjust this step until you achieve the desired result.
3. Preparing Culture, Tubes, and Microscope
Prepare desired number of 1.7 mL Eppendorf tubes. Add 2 μL of PBS, first strand buffer if material is to be used for single cell amplifications, or any other solution for storage of the harvested material.
For harvesting, you will need a light microscope with a 40x objective fitted with a micromanipulator. Use a pipette holder with flexible tubing leading away from the holder. Affix the tubing to a stable surface to prevent unwanted movement of the pipette during collection. At the end of the tubing, insert a needle so that a 1 mL syringe with a Luer-Lock connection can be attached and changed in between users.
If using coverslips, work with one coverslip at a time. If necessary, transfer a single coverslip to a new 35 mm dish* containing PBS or media of choice. The longer the cells are out of the incubator, the more unhealthy they will become and the more their mRNA profiles will deviate from that of a healthy cell. It is critical to preserve the health of the cells on the other coverslips by keeping them in the incubator when not working with them. Work as quickly as possible with cells outside of the incubator.
*With our setup it is necessary to use the lid of a 35 mm dish since the walls of the actual dish are too high to allow proper advancement of the micropipette towards the coverslip.
4. Harvesting Cells
Secure the micropipette in the micropipette holder. Affix micropipette holder to the insert on the micromanipulators. Set the 40x objective.
Place the dish on the microscope stage and find a region of cells suitable for harvest. In the case of primary neuronal cultures, the cell should be relatively isolated from its neighbors to prevent harvest of surrounding cells and/or processes. The population of mRNA transcripts between the soma and processes vary, so if interested in somatic mRNAs solely, care should be taken to prevent contamination of the soma with processes. Place the cell you wish to harvest in the center of the field of view.
Use the micromanipulator to advance the micropipette toward the solution in the dish into the light path. Lower the pipette tip into the solution. Apply positive pressure from this point on by blowing lightly through the syringe. Look through the eyepiece. The pipette should appear as a shadow in the field of view. Well above the plane of the cells, adjust the position of the pipette tip until it is within the field of view and focus on the pipette tip using the coarse focusing knobs. At this point, it should be assessed if the bore size of the pipette is too large or too small. Practice harvesting will provide the ability to determine if the correct bore size has been achieved at this point.
Now, advance the pipette toward the plane of the cells. It is important that the tip is not advanced too hurriedly, causing it to break in the region from which you wish to collect. To prevent this, use the coarse focus to advance the focal plane BEFORE advancing the pipette tip towards the cells using the micromanipulators. When you are close to the cells, use the fine focus until both the cells and the pipette tip are in focus. Stop applying positive pressure before you reach the plane of the cells to prevent blowing them off of the coverslip.
Use the micromanipulators to position the pipette tip so that it is touching the cell soma you wish to harvest.
Using the syringe, apply gentle suction by mouth until the cell enters the pipette tip. If the cell is not entering the pipette, gently move the tip closer to the cell and downward until it lifts from the coverslip.
5. Saving the Cell
Use the micromanipulator to immediately move the pipette up and out of the solution.
Remove the pipette from the holder.
Hold the 1.7 mL Eppendorf tube in one hand and gently break the tip of the pipette on the side of the tube toward the bottom. (Aim for the 0.1 mL mark.)
With the broken tip centered inside the tube, insert a needle attached to a 1-3 mL syringe into the top opening of the pipette. Rapidly depress the plunger forcing the solution in the pipette to spray out into the tube. The cell should remain in the very tip of the pipette and this should be adequate to properly expel the cell. Be careful not to touch the tip to any liquid on the sides of the tube as capillary action will bring the liquid back into the pipette.
Quickly spin down the contents of the tube using a desktop microfuge and immediately freeze (store at -80°C) or place on ice for further processing (preferable).
6. aRNA Amplification
- Round 1 First strand cDNA synthesis: Create mRNA/cDNA hybrids.
- For every 5 μL of single cell collection volume, add 1x of the following to the collection tube (on ice). The reaction can be scaled up for larger collection volumes (2x for 10 μL collection volumes, etc.). Create a master mix by multiplying the following reagents by the number of collection tubes plus 10-20% to account for pipetting error. Do this for every subsequent reaction in the aRNA ampification procedure.
- ON ICE
- 1.2 μL dNTP's (2.5 mM each)
- 2.4 μL 5x First strand buffer
- 0.3 μL T7-oligo(dT) primer (100 ng/μL)
- 1.2 μL DTT (100 mM)
- Bring the volume up to 10.25 μL with nuclease free water. Pipette mix and spin briefly using a microfuge.
- Incubate for 5 minutes at 70°C to denature any secondary structure of the mRNA. Immediately place on ice for at least 5 minutes.
- Add (again using a master mix):
- 0.3 μL RNasin (40 U/μL)
- 0.45 μL Superscript III (200 U/μL)
- 1 μL nuclease free water
- Pipette mix and spin briefly. Incubate for 1 hour at 42°C.
- Incubate at 70°C for 15 minutes to inactivate the Superscript. Continue on to the next step.
- Round 1 Second strand cDNA synthesis Create RNA primers from the mRNA portion of the mRNA/DNA hybrid to aid in synthesis of double stranded cDNA.
- ON ICE
- To the 12 μL first strand reaction, add:
- 8 μL nuclease free water
- 5.6 μL 5x Second strand buffer
- 0.75 μL dNTP mix (2.5 mM each)
- 0.25 μL DNA ligase (10 U/μL)
- 1 μL DNA polymerase I (10 U/μL)
- 0.25 μL Rnase H (2 U/μL)
- Add: 1 μL T4 DNA polymerase (5 U/μL). Mix by pipetting and spin briefly. Incubate 10 more minutes at 16°C.
- Clean up the reaction using the Qiagen MinElute kit as per the manufacturer's instructions with slight modifications: 11 Wash 2 times with 500 μL wash buffer instead of 1 time with 750 μL. Elute in nuclease free water.
- Concentrate to 2-4 μL using a Speedvac or by ethanol precipitation. For the ethanol precipitation, the glycogen acts as a nucleic acid carrier and is used when precipitating small amounts of DNA or RNA. The sodium from the sodium acetate helps neutralize the negative charge of the DNA backbone, aiding in precipitation. It is not necessary to make a master mix for routine precipitations.
- Add 40 μL DEPC water
- 1 μL glycogen (3 mg/mL)
- 1/10 volume (3 μL) 3M Sodium acetate
- 2.5 volumes (~125 μL) cold 100% ethanol
- Centrifuge for 20 minutes at 4°C.
- Remove the supernatant and wash the pellet with 800 μL 70% ethanol. Be sure to fully dislodge the pellet either by pipetting or vortexing to aid in removal of excess salt.
- Centrifuge for another 20 minutes at 4°C. Remove the supernatant and air.
- Resuspend in 4 μL of nuclease free water. Store at -20 or -80 °C or continue on to the next step.
- Round 1 In vitro transcription (IVT): Synthesize antisense RNA from the T7 promoter incorporated into the double stranded cDNA.
- This reaction is performed using the Ambion MEGAscript T7 kit as per the manufacturer's instructions except scaled for a 10 μL instead of a 20 μL reaction.12 It is imperative to assemble this reaction at room temperature and to keep the buffer at room temperature during assembly. The supplied buffer will precipitate the DNA if it is ice cold. Keep the NTPs and enzyme mix on ice when not in use.
- AT ROOM TEMPERATURE
- Transfer the resuspended double stranded cDNA to a thin walled PCR tube.
- Add 4 μL NTP mix (18.75 mM each)
- 1 μL 10x reaction buffer
- 1 μL 10x enzyme mix
- Incubate 14 hours at 37°C in a thermocycler (preferable) or incubator.
- This reaction can be cleaned up using two different methods followed by concentration of the sample using a Speedvac or ethanol precipitation. For cleanup using a kit, proceed to Method A. For clean up with a standard phenol/chloroform extraction, proceed to Method B.
- Method A:
- Clean up the reaction using the AMBION Megaclear kit as per the manufacturer's instructions with some modifications13: Wash 2 times with 500 μL wash buffer instead of 1 time with 750 μL. For the elution step, add 50 μL of nuclease free water to the center of the column, incubate at 70 °C for 10 minutes. Spin at 10,000g for 1 min. Repeat in a fresh tube. Combine eluates. Concentrate sample to 2-4 μL using a Speedvac OR by ethanol precipitation.
- For ethanol precipitation: Ammonium acetate is used in place of sodium acetate in this case because it is efficient at preventing co-precipitation of free nucleotides with the nucleic acid. This is important because of the high concentration of NTP's used in the IVT reaction, which can inhibit downstream reactions.
- 2 μL glycogen (5 mg/mL)
- 0.1 volumes (10 μL) 5M Ammonium acetate
- 2.5 volumes (~275 μL) cold ethanol
- Precipitate at -80°C (30 min. to O/N).*
- Centrifuge for 20 minutes at 4°C.
- Remove the supernatant and wash the pellet with 800 μL 70% ethanol (in nuclease free water). Be sure to fully dislodge the pellet to remove all of the excess salt.
- Centrifuge for another 20 minutes at 4°C.
- Remove supernatant and air dry.
- Resuspend pellet in 4 μL nuclease free water.
- Method B:
- Alternatively, the aRNA can be cleaned up with a standard phenol/chloroform extraction.
- Add 90 μL DEPC water
- 2 μL glycogen (5 mg/mL)
- 0.1 volumes (10 μL) 5M Ammonium acetate
- 1 volume (100 μL) phenol:chloroform:isoamyl alcohol 25:24:1 (equilibrated to pH 7.8-8.0)
- Vortex for 15 seconds. Centrifuge for 1.5 minutes at half maximal speed in a table top microcentrifuge. Transfer the top aqueous layer to a new tube containing 2.5 volumes (245 μL) of cold ethanol.
- Precipitate at -80°C (30 min. to O/N).
- Centrifuge for 20 minutes at 4°C.
- Remove supernatant and wash pellet with 800 μL 70% ethanol (in nuclease free water). Be sure to fully dislodge the pellet to remove all of the excess salt.
- Centrifuge for another 20 minutes at 4°C.
- Remove supernatant and air dry.
- Resuspend pellet in 4 μL nuclease free water.*The sample may freeze at this temperature during overnight incubations. It has been shown that this can actually increase yield of nucleic acids.
- Round 2 First strand cDNA synthesis
- ON ICE
- Add 1 μL Random primers (0.05 mg/mL)*
- Heat at 70°C for 10 minutes. Immediately place on ice for at least 5 minutes.
- Add 2 μL 5x First strand buffer
- 1 μL DTT (100 mM)
- 0.5 μL dNTP's (2.5 mM each)
- 0.5 μL RNasin (40 U/μL)
- 1 μL Superscript III (200 U/μL)
- Mix thoroughly by pipetting and spin briefly. Allow to sit at room temperature for 10 minutes. This step is required to allow extension of the short random primers before the reverse transcription reaction is performed.
- Incubate for 30 minutes at 42°C.
- Heat 5 minutes at 95°C to denature the RNA in DNA/RNA hybrids. Place on ice for at least 5 minutes. Store at -20°C or -80°C or immediately continue on to the next step.*The concentration of random primers is important. If the concentration is too high, the product will be more truncated with each round of amplification.
- Round 2 Second strand cDNA synthesis
- ON ICE
- Add 2 μL T7-oligo(dT) primer (10 ng/μL)*
- Heat for 5 minutes at 70°C. Immediately place on ice for at least 5 minutes. Spin briefly.
- Add 43.5 μL nuclease free water
- 15 μL 5x Second strand buffer
- 1.5 μL dNTP mix (2.5 mM each)
- 2 μL DNA polymerase I (10 U/μL)
- Mix thoroughly by pipetting and spin briefly. Incubate for 2 hours at 16°C.
- Add 2 μL T4 DNA polymerase (5 U/μL)
- Mix thoroughly by pipetting and spin briefly. Incubate 10 more minutes at 16°C.
- Clean up the reaction using the Qiagen Minelute kit as in Section 6.2.3.
- Concentrate to 2-4 μL with a Speedvac or ethanol precipitation as in Sections 6.2.4 to 6.2.8. *Note the different stock concentration of T7-oligo(dT) primer compared to first round second strand cDNA synthesis.
- Round 2 In vitro transcriptionPerform as in Section 6.3.
- Repeat Sections 6.4 to 6.6 desired number of times. Note that each subsequent round of amplification results in shorter aRNA products. The number of rounds of amplification should be limited for this reason. Normally, two to three rounds of amplification are sufficient to amplify material harvested from a single cell to perform microarray analysis. In the case of microarray analysis, the third round IVT reaction is performed using the Illumina TotalPrep RNA Amplification Kit as per the manufacturer's instructions. This procedure incorporates biotin-labeled UTP into the aRNA that is then used for detection on a microarray chip.
- Measure the concentration of third round aRNA using a Nanodrop or Agilent Bioanalyzer with a Nano RNA kit.
7. Representative Results
Successful harvest of a single cell from primary neuronal cultures can be completed in less than 2 minutes, depending on aptitude (see Figure 1). However, time for harvest will vary between systems and with intervening experimental manipulation. Subjecting the single cell to the aRNA procedure (see Figures 4A and 4B) results in microgram amounts of total amplified aRNA and produces a characteristic broad peak when analyzed with a bioanalyzer (see Figure 3). Three rounds can be completed in a minimum of three days, allowing for quick analysis of single cell gene expression.
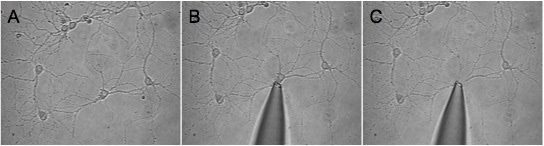 Figure 1. Shown is an example of a successful harvest of an isolated neuron. We selected a relatively low-density region (A) and advanced the pipette tip toward the desired cell (B). The third image (C) shows the field of view after the cell had been harvested. Note that the surrounding processes remain on the coverslip.
Figure 1. Shown is an example of a successful harvest of an isolated neuron. We selected a relatively low-density region (A) and advanced the pipette tip toward the desired cell (B). The third image (C) shows the field of view after the cell had been harvested. Note that the surrounding processes remain on the coverslip.
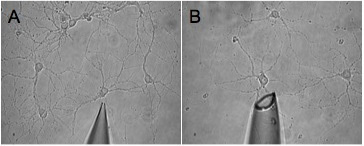 Figure 2. Shown are two images of pipette tips, which are an inappropriate size for effective harvesting. These tips will lead to incomplete harvest (A) and harvest of surrounding mileu (B) respectively.
Figure 2. Shown are two images of pipette tips, which are an inappropriate size for effective harvesting. These tips will lead to incomplete harvest (A) and harvest of surrounding mileu (B) respectively.
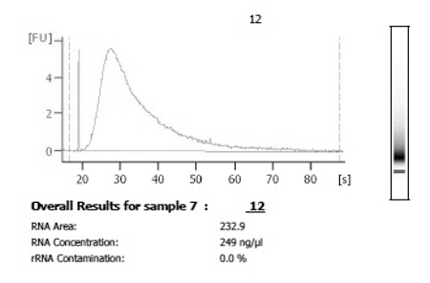 Figure 3. Following harvest and amplification, analysis using a bioanalyzer is recommended to examine the distribution and quantity of amplified RNA. A successful amplification from single cell material will yield total amounts in the low micrograms and will have a distribution that is smooth and wide.
Figure 3. Following harvest and amplification, analysis using a bioanalyzer is recommended to examine the distribution and quantity of amplified RNA. A successful amplification from single cell material will yield total amounts in the low micrograms and will have a distribution that is smooth and wide.
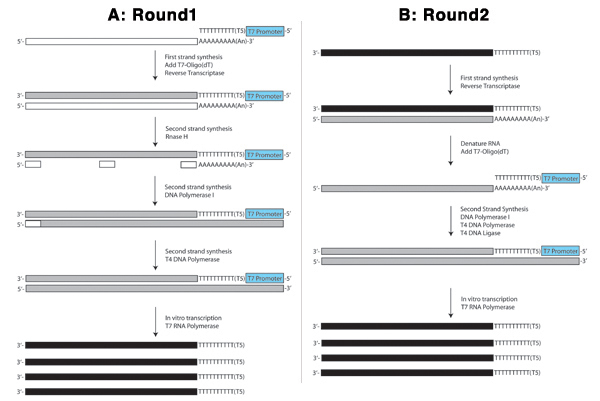 Figures 4. Schematics of the first round (A) and the second round (B) of the aRNA procedure are shown.
Figures 4. Schematics of the first round (A) and the second round (B) of the aRNA procedure are shown.
Discussion
Notes and Troubleshooting
You may want to take before and after images showing that the target cell was isolated and that the remaining penumbra was left untouched.
If too much solution is entering the pipette as you are harvesting, you can use 3 way Luer-Lock fittings and the plunger for the syringe to hold positive pressure in the tubing. The desired volume will vary with the type of processing but up to 5 μl should be fine for most applications.
General tips on molecular biological techniques
Vortex all stock tubes and spin them down before adding to a reaction. NEVER vortex enzymes.
Thoroughly mix reactions before adding the enzyme to ensure that the buffer is at the proper concentration.
Keep enzyme stocks at -20°C if possible by using a stratacooler. Otherwise, remove right before use, keep on ice, and return immediately to -20°C.
Always make master mixes when preparing more than one reaction to reduce pipetting errors. Calculate the volume required based on the number of samples and add 10-20%.
Store RNA at -80°C to retard degradation. RNA is most stable stored in buffer to retard apurination of RNA that occurs in acidic conditions.
General Outline of the aRNA Procedure
Figure 4A illustrates the first round of the aRNA procedure. In the first strand reaction, the poly-T portion of the T7-oligo(dT) primer selects for mRNA species (long white rectangle) by binding to the polyA tails. Some microRNAs are also polyadenylated and will be captured by this procedure. More importantly, however, the most abundant RNAs in the cell, ribosomal RNAs, will not. This oligo acts as a primer for Reverse Transcriptase to synthesize a complementary strand of cDNA (long grey rectangle) using the mRNA as a template. The T7 portion of the T7-oligo(dT) primer incorporates the T7 RNA polymerase promoter in frame with the sequence antisense to the starting mRNA. This is used later in the in vitro transcription reaction.
Next, the mRNA in the mRNA/DNA hybrid created in the preceding step is partially hydrolyzed by Rnase H creating RNA "primers" (small white rectangles) similar to the Okazaki fragments created in lagging strand DNA synthesis. DNA Polymerase I uses the RNA fragments to prime DNA synthesis using the DNA complementary to the mRNA as a template. When it reaches the next RNA fragment, its 5' to 3' nuclease activity removes the ribonucleotides and replaces them with deoxyribonucleotides. DNA ligase is added to ligate any strands where the replacement of the leading strand is not complete. T4 DNA Polymerase is added to fill in the areas where RNA fragments served as initial primers for DNA Polymerase I creating a blunt-ended double stranded cDNA that is then purified before performing the IVT reaction.
In the IVT reaction, T7 RNA polymerase binds to the T7 promoter incorporated into the double stranded cDNA and synthesizes antisense RNA molecules (long black rectangles) using the sense strand as a template. This serves as the amplification step in which thousands of antisense RNA molecules are produced from each double stranded cDNA molecule (Figure 4A).
The second round, as depicted in Figure 4B, begins with a reverse transcription reaction that is slightly different from that of the first round since the starting RNA is antisense (solid black rectangle) and lacks the polyadenylated tail that was targeted by the T7-oligo(dT) primer in the first round. Therefore, this reaction is primed with random primers (small grey rectangles) and the RNA subsequently denatured. The second strand reaction is then primed by the T7-oligo(dT) primer, which binds to the poly-A sequence at the 3' end of the sense RNA created in the preceding reverse transcription reaction. Another IVT reaction is performed in the same manner as in the first round. This second round is usually repeated at least once to achieve three rounds of amplification from a single cell.
Applications
The techniques that we have presented in this article can be translated into a large number of applications. The single cell isolation protocol can be modified for use in acute slices.14 Although technically more challenging, the same principles apply in this alternate preparation. Additionally, if the size of the pipette is slightly adjusted, recordings of the physiology of the cells can be made before harvest allowing for a well-controlled investigation of molecular mechanisms behind physiological outputs. Another slight modification is to isolate processes from the cell soma.15 For this application, collect cell bodies with one pipette and then go back with a fresh pipette and collect 100-300 identified dendrites or axons per collection tube.16
Once cells have been harvested, comparisons of mRNA abundances and compositions can be made between different and even within the same cell populations. Incorporating biotinylated-UTP into the third round aRNA allows for microarray analysis to determine these relative mRNA abundances. The composition of the original mRNA population can also be determined after the aRNA procedure using next generation sequencing. The amplified aRNA can also be used to confirm cell phenotype conversion studies in which a full set of mRNAs from one cell type are transfected into a different cell type in order to induce transition of the phenotype of the latter cell type into that of the former, a procedure developed by the lab and known as TIPeR.17 These studies are particularly useful for studying disease states and cell phenotypes and such studies are currently ongoing in the lab. RT-PCR or qPCR can be performed on the amplified material to confirm the expression of cell-specific genes. Additionally, evaluations of the efficiency of transfection or transduction can be made at the single cell level.
Advantages and Limitations
As stated in the abstract, isolation of single cells for analysis eliminates the averaging effects seen with analysis of heterogeneous cell populations. These averaging effects misrepresent mRNA abundances within a single cell by over-representing abundant transcripts and averaging out and preventing detection of many low-abundance transcripts. Flow cytometry can be used to sort individual cells, but this method requires knowledge of cell specific markers and expensive equipment.18 Laser capture microdissection either with UV or IR laser capture microdissection systems allow for single cell and even subcellular capture but requires cells of interest to be located at the surface of very thin sections.19 Similar to flow cytometry, laser capture microdissection also requires expensive equipment.
One of the major benefits of the electrode based collection technique described above is that valuable electrophysiological data can be obtained from the cell of interest before harvest, allowing for functional and transcriptome analysis to be performed on the same cell.20 A downside of our technique is that it does require experience using micromanipulators. Investigators familiar with micromanipulators will find this technique very intuitive; however, individuals with no such experience will need to become comfortable with the fine movements required.
Inherent in any amplification technique is the preferential amplification of certain transcripts based on size and nucleotide composition.4,6,7 Polymerase chain reaction (PCR) based techniques such as reverse-transcription polymerase chain reaction (RT-PCR) and rapid amplification of cDNA ends (RACE) result in exponential amplification of transcripts, whereas the aRNA amplification procedure results in linear amplification. Thus, one of the major advantages of the aRNA procedure lies in its ability to better preserve the relative abundances of mRNA transcripts by only linearly amplifying any errors or biases that occur in the amplification process as opposed to exponentially amplifying said errors and biases.
The aRNA procedure coupled with microarray analysis allows for comparison of mRNA abundances between single cells of similar or different morphology, treated or untreated. Additionally, amplified material can be submitted for next generation sequencing.5,8 However, care should be taken in such analyses since, in contrast to the use of random primers for the procedure, the oligo-dT primed procedure described above biases amplification of the 3' ends of mRNA and generally results in slight shortening of subsequent amplified material with each round. Care should be taken in analyzing microarray results with standard methods as some false absent calls can arise from slightly shortened amplified material. In addition, while sequencing results will indeed provide the full 5' sequence of most of the original mRNA, the 5' sequences of some mRNA might be missed. For these reasons, the number of rounds of amplifications should be limited.
Disclosures
Dr. Eberwine is an inventor on the aRNA patent that has been licensed to LBS technologies in which Dr. Eberwine is a share-holder.
Acknowledgments
Thank you to Kevin Miyashiro for plating and maintaining cell cultures, to Dr. Terri Schochet for providing cell cultures for the pictures included in this document. In addition, thank you to Kevin Miyashiro, Dr. Peter Buckley, and Tiina Pertiz for input on the aRNA procedure. Funding for this work was from the National Institute on Aging, the National Institute on Mental Health and the Human Resources Fact Finder funds from the Commonwealth of Pennsylvania.
References
- Davis JE, Eberwine JH, Hinkle DA, Marciano PG, Meaney DF, McIntonsh TK. Methodological considerations regarding single-cell gene expression profiling for brain injury. Neurochemical Research. 2004;29:1113–1121. doi: 10.1023/b:nere.0000023598.04937.83. [DOI] [PubMed] [Google Scholar]
- Eberwine J. Single-cell molecular biology. Nature Neuroscience. 2001;4:1155–1156. doi: 10.1038/nn1101-1155. [DOI] [PubMed] [Google Scholar]
- Kamme F, Salunga R, Yu J, Tran D, Zhu J, Luo J, Bittner A, Guo H, Miller N, Wan J, Erlander M. Single-cell microarray analysis in hippocampus CA1: Demonstration and Validation of Cellular Heterogeneity. The Journal of Neuroscience. 2003;23:3607–3607. doi: 10.1523/JNEUROSCI.23-09-03607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle D, Glanzer J, Sarabi A, Pajunen T, Zielinski J, Belt B, Miyashiro K, McIntosh T, Eberwine J. Single neurons as experimental systems in molecular biology. Progress in Neurobiology. 2004;72:129–142. doi: 10.1016/j.pneurobio.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Elarova I, Ruben M, Tan F, Counts SE, Eberwine JH, Trojanowski JQ, Hemby SE, Mufson EJ, Che S. Single-cell gene expression analysis: Implications for Neurodegenerative and Neuropsychiatric Disorders. Neeurochemical Research. 2004;29:1053–1064. doi: 10.1023/b:nere.0000023593.77052.f7. [DOI] [PubMed] [Google Scholar]
- Eberwine J, Spencer K, Miyashirto K, Mackler S, Finnell R. Complementary DNA synthesis in situ: methods and applications. Methods Enxymol. 1992;216:80–100. doi: 10.1016/0076-6879(92)16011-8. [DOI] [PubMed] [Google Scholar]
- Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, Zettel M, Coleman P. Analysis of gene expression in single live neurons. Proc. Natl. Acad. Sci. USA. 1992;89:3010–3014. doi: 10.1073/pnas.89.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelz MB, Dent GW, Therianos S, Marciano PG, McIntosh TK, Coleman PD, Eberwine JH. Single-cell antisense RNA amplification and microarray analysis as a tool for studying neurological degeneration and restoration. Sci. Aging Knowledge Eviron. 2002;1:re1–re1. doi: 10.1126/sageke.2002.1.re1. [DOI] [PubMed] [Google Scholar]
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Research. 1977;126:397–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- P-1000 & P-97 Pipette Cookbook. rev. F. Sutter Instrument; 2010. [Google Scholar]
- MinElute Handbook For MinElute Reaction Cleanup Kit. Qiagen; 2004. pp. 28–29. [Google Scholar]
- Megascript kit. Ambion; pp. 7–8. [Google Scholar]
- MEGAclear kit. Applied Biosystems; pp. 4–5. [Google Scholar]
- Eberwine J, Kacharmina JE, Andrews C, Miyashiro K, McIntosh T, Becker K, Barrett T, Hinkle D, Dent G, Marciano P. mRNA expression analysis of tissue sections and single cells. The Journal of Neuroscience. 2001;21:8310–8314. doi: 10.1523/JNEUROSCI.21-21-08310.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl. Acad. Sci. USA. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashiro K, Dichter M, Eberwine J. On the nature and differential distribution of mRNAs in hippocampal neuritis: Implications for neuronal functioning. Proc. Natl. Acad. Sci. USA. 1994;91:10800–10804. doi: 10.1073/pnas.91.23.10800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sul J, Wo CK, Zeng F, Jochems J, Lee MT, Kim TK, Peritz T, Buckley P, Cappelleri DJ, Maronski M, Kim M, Kumar V, Meaney D, Kim J, Eberwine J. Transcriptional transfer produces a predictable cellular phenotype. Proc. Natl. Acad. Sci. USA. 2009;106:7624–7629. doi: 10.1073/pnas.0902161106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sow FB, Gallup JM, Sacco RE, Ackerman MR. Laser capture microdissection revisited as a tool for transcriptomic analysis: Application of an excel-based qPCR preparation software (PREXCEL-Q) Int. J. Biomed. Sci. 2010;5 [PMC free article] [PubMed] [Google Scholar]
- Vandewoestyne M, Deforce D. Laser capture microdissection in forensic research: a review. Int J Legal Med. 2010;124 doi: 10.1007/s00414-010-0499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberwine J, Bartfai T. Single cell transcriptomics of hypothalamic warm sensitive neurons that control core body temperature and fever response Signaling asymmetry and an extension of chemical neuroanatomy. Pharmacol. Ther. 2010. [DOI] [PMC free article] [PubMed]