

Abstract
Store operated Ca2+ entry (SOCE), earlier termed capacitative Ca2+ entry, is a tightly regulated mechanism for influx of extracellular Ca2+ into cells to replenish depleted endoplasmic reticulum (ER) or sarcoplasmic reticulum (SR) Ca2+ stores1,2. Since Ca2+ is a ubiquitous second messenger, it is not surprising to see that SOCE plays important roles in a variety of cellular processes, including proliferation, apoptosis, gene transcription and motility. Due to its wide occurrence in nearly all cell types, including epithelial cells and skeletal muscles, this pathway has received great interest3,4. However, the heterogeneity of SOCE characteristics in different cell types and the physiological function are still not clear5-7.
The functional channel properties of SOCE can be revealed by patch-clamp studies, whereas a large body of knowledge about this pathway has been gained by fluorescence-based intracellular Ca2+ measurements because of its convenience and feasibility for high-throughput screening. The objective of this report is to summarize a few fluorescence-based methods to measure the activation of SOCE in monolayer cells, suspended cells and muscle fibers5,8-10. The most commonly used of these fluorescence methods is to directly monitor the dynamics of intracellular Ca2+ using the ratio of F340nm and F380nm (510 nm for emission wavelength) of the ratiometric Ca2+ indicator Fura-2. To isolate the activity of unidirectional SOCE from intracellular Ca2+ release and Ca2+ extrusion, a Mn2+ quenching assay is frequently used. Mn2+ is known to be able to permeate into cells via SOCE while it is impervious to the surface membrane extrusion processes or to ER uptake by Ca2+ pumps due to its very high affinity with Fura-2. As a result, the quenching of Fura-2 fluorescence induced by the entry of extracellular Mn2+ into the cells represents a measurement of activity of SOCE9. Ratiometric measurement and the Mn+2 quenching assays can be performed on a cuvette-based spectrofluorometer in a cell population mode or in a microscope-based system to visualize single cells. The advantage of single cell measurements is that individual cells subjected to gene manipulations can be selected using GFP or RFP reporters, allowing studies in genetically modified or mutated cells. The spatiotemporal characteristics of SOCE in structurally specialized skeletal muscle can be achieved in skinned muscle fibers by simultaneously monitoring the fluorescence of two low affinity Ca2+ indicators targeted to specific compartments of the muscle fiber, such as Fluo-5N in the SR and Rhod-5N in the transverse tubules9,11,12.
Keywords: Cellular Biology, Issue 60, Mn quenching, 2-APB, Fura-2, Orai1, esophageal squamous cell carcinoma, skinned muscle fiber
Protocol
1. Intracellular Ca2+ measurement for individual cells
KYSE-150, a human esophageal squamous cell carcinoma (ECSS) cell line is cultured in 5% CO2 atmosphere at 37°C in mixed RPMI 1640/Ham's F-12 medium (1:1) containing 5% fetal bovine serum.
KYSE-150 cells are transfected with the plasmids either containing shRNA specifically against Orai1 or a scrambled sequence. The plasmids also included a gene encoding red fluorescent protein (RFP) as a reporter, which is driven by a separate promoter.
The cells are grown in glass-bottom dishes (No. 1.5 cover glass, MatTek, MA) for 48 hours.
Remove the medium and rinse the cells with a balanced salt solution (BSS) (140 mM NaCl, 2.8 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, pH 7.2).
Add 1 ml BSS solution containing 2 μM Fura-2 acetoxymethyl ester (Molecular Probes) into the dish and wrap the whole dish with foil to protect from light.
Incubate the cells for 40 min at 37°C and then leave them for an additional 15 min period at room temperature to allow ester hydrolysis to be completed. Wash the cells with BSS twice.
Mount the dish on the stage of Nikon TE200 inverted microscope, which is connected to PTI spectrofluorometer, with excitation wavelengths at 350 and 390 nm and emission at 510 nm. The exact excitation wavelengths to be used can slightly vary depending on the optics of each individual system. The two wavelengths with best ratio dynamic are determined by excitation spectrum scanning of Fura-2 salt in solutions containing Ca2+ ranging from 0 to 39.8 μM (or higher concentration at which Fura-2 fluorescence is saturated).
Set up a gravity perfusion system with 4 different solutions in BSS: Ca2+ (2 mM), EGTA (0.5 mM), EGTA-TG (thapsigargin, 5 μM), Ca2+-2-APB (a SOCE inhibitor, 75 μM). Computer-controlled automated perfusion systems can also be used.
Select the cells expressing RFP in visualization mode.
Position the opening tip of the perfusion system at 10x magnification until the tip is right above the region of interest at a 45 degree angle.
Simultaneously record the fluorescence signals F350nm and F390nm. Ratio (F350nm/F390nm) is displayed in the third window. Change the perfusion solutions in the following order: Ca2+; EGTA; Ca2+; EGTA-TG; Ca2+; Ca2+-2-APB.
The above protocol can be modified for measurement of intracellular Ca2+ in cell suspension system.
Culture KYSE-150 cells in a T25 flask with the same culture condition as above.
When the cells reach 90% confluence, remove the medium and rinse the cells with a BSS solution.
Add 2.5 ml BSS solution containing 2 μM Fura-2 AM and incubate the cells for 40 min at 37°C followed by deesterification.
Remove the BSS, add 2.5 ml trypsin into the flask and incubate at 37°C until the cells detach. Then add 2.5 ml culture medium to stop the trypsinization and harvest the cells by centrifugation at 800 rpm for 5 min.
Wash the cells with culture medium one more time by centrifugation and re-suspend the cells' pellet into BSS solution (without addition of 2 mM CaCl2, containing μM range Ca2+) and count the cells number using a hematometer.
Add ~106 cells into a quartz cuvette (10 mm, Starna Cells) and fill the volume up to 2 ml with BSS.
Place the cuvette (stirring with a magnetic bar) into the spectrofluorometer.
Simultaneously record the fluorescence signals F340nm and F380nm with emission wavelength at 510 nm. The running protocol is: BSS, addition of 0.5 μl EGTA (0.25 M stock), addition of 1 μl thapsigargin (TG, 10 mM stock), addition of 4 μl Ca2+(1 M stock).
2. Mn quenching assay in cultured cells
Perform excitation wavelength scanning of Fura-2 in solutions containing various Ca2+ concentration ranging from 0 to 39.8 μM to determine the Ca concentration independent wavelength as the isosbestic point. Usually, the isosbestic point is at or around 360 nm.
KYSE-150 cells are cultured in glass-bottom dishes and loaded with Fura-2 AM.
Set up the perfusion system with 5 different BSS solutions: Ca2+ (2 mM), EGTA/TG (0.5 mM and 5 μM, respectively), Mn2+ (0.5 mM, without addition of EGTA or Ca2+, containing free Ca2+ in μM range), Mn2+/2-APB (75 μM), EGTA/Triton X-100 (0.1%).
Mount the dish on the stage of the microscope and select the "Excitation Ratio" mode with excitation wavelengths at 360 nm and 390 nm and emission at 510 nm.
Simultaneously record the fluorescence signals F360nm and F390nm with Ratio (F360nm/F390nm) displayed in the third window. The running protocol is: Ca2+ for 1 min to stabilize the fluorescence, Mn2+ for 30 sec, EGTA/TG for 10 min, Mn2+ for 2 min, EGTA-Triton X-100 (0.1%) for 1 min to obtain the background reading. Mn2+/2-APB solution can be used instead of Mn2+ to examine the abolished fluorescence quenching, which indicates that the Mn2+ entry is through SOCE pathway.
3. Mn quenching assay in muscle cells
C2C12, a mouse myogenic cell line, is cultured on glass-bottom dishes in 5% CO2 atmosphere at 37°C in DMEM medium containing 10% fetal bovine serum, 10% horse serum.
After the cells reach confluence, the culture medium is changed to differentiation medium (remove fetal bovine serum and reduce horse serum to 2.5%). The C2C12 myoblasts will differentiate into myotubes after 4 days.
Load C2C12 myotubes with a final concentration of 5 μM Fura-2 AM as described in 1.4-1.6.
Add a final concentration of 20 μM N-benzyl-p-toluene sulphonamide (BTS) to inhibit muscle contractions and the motion artifacts caused by them and allow 15 min before initiating the experiment.
Mount the dish on the stage of the microscope connected to the PTI device and load the following BSS solutions into the perfusion system: 2 Ca2+ (2 mM), 0 Ca2+, Mn2+ (0.5 mM); Mn2+/TG (0.5 mM, 20 μM, respectively).
Set up the perfusion system as described above.
Simultaneously record the fluorescence signals F360nm and F390nm with the Ratio (F360nm/F390nm) displayed in the third window. The running protocol is: 2 Ca2+ for 1 min, 0 Ca2+ for 1 min to flush away Ca2+, Mn2+ for 0.5 min, Mn2+/TG for 10 min, and Mn2+/EGTA-Triton X-100 (0.1%).
4. Spatially and temporally resolved SOCE in skinned muscle fibers
Prepare the following solutions. Isotonic Tyrode buffer: 140 mM NaCl, 5 mM KCl, 10 mM HEPES, 2 mM MgCl2, 2.5 mM CaCl2, pH 7.2, 290 mosm Culture medium: DMEM with 2% horse serum and 1% penicillin and streptomycin Wash solution #1: 109.6 mM K-glutamate, 2 mM EGTA-KOH, 6.7 mM MgCl2, 2 mM ATP, 6 mM creatine phosphate (CP), 20 mM N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES)-KOH, pH 7.0 Wash solution #2: 140 mM K-glutamate, 5 mM EGTA-KOH, 6.5 mM MgCl2, 2 mM ATP, 6 mM CP, 20 mM BES-KOH, pH 7.0 Skinning solution: 140 mM K-glutamate, 6.5 mM MgCl2, 2 mM ATP, 6 mM CP, 20 mM BES-KOH, pH 7.0 TT-loading solution: 90.6 mM K-glutamate, 18 mM Na-glutamate, 0.55 mM CaCl2, 2 mM EGTA-KOH, 6.7 mM MgCl2, 5.4 mM ATP, 15 mM CP, 0.0025 mg/ml creatine kinase (CK), 20 mM BES-KOH, 5 μM carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), pH 7.0, pCa 7.0 SR loading solution: 107.8 mM K-glutamate, 0.98 mM CaCl2, 2 mM EGTA-KOH, 6.6 mM MgCl2, 5.4 mM ATP, 15 mM CP, 0.0025 mg/ml CK, 20 mM BES-KOH, 5 μM FCCP, pH 7.0, pCa 6.6 SR depleting solution: 100 mM K-glutamate, 40 mM Na-glutamate, 10 mM EGTA-KOH, 10 mM 1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA), 0.35 mM MgCl2, 0.5 mM ATP, 1 mM CP, 20 mM BES-KOH, 5 μM FCCP, pH 7.0. 25 mM caffeine/20 μM thapsigargin (TG) are added before experiments.
To dissect the Extensor digitorum longus (EDL) muscle, first lay down the mice and arrange the leg at lateral position, then remove the skin from the ankle area up to the knee, cut the superficial muscle layers of tissue to expose the EDL, and sever both the upper and lower tendons to free the intact EDL muscle; it is important to keep tendons as long as possible. The EDL is transferred to a Tyrode solution containing 0 Ca2+ and 0.1 mM EGTA to prevent any contractions.
Under a stereomicroscope, use two tweezers to hold lower insertion tendons and split the EDL muscle into two bundles. Repeat the process to obtain 4 and then 8 bundles. It is critical that tendons remain for each of the 8 bundles at the end of the process. If they are lost from some bundles, discard them.
Under a stereomicroscope, each EDL bundle strip is griped at both tendons and carefully stretched until 3~4 separated single muscle fibers are left intact with tendon on both sides.
Put 1 drop of Ca2+ free tyrode buffer in the middle of the glass-bottom dish, lay down the EDL strips straight on the dish, quickly remove as much as possible of the solution, then tape down both tendons using water resistant scotch tape, and make sure the tape is tight. Wash the fiber first with the 0 Ca2+ solution and then with a 2.5 mM Ca2+ solution 2 times. If the fiber hyper-contracts and damage is noticed, discard the fiber.
If needed, the fiber can be cultured for up to 96 hours, allowing for genetic manipulations. Wash the fiber 3 times with complete culture medium and add 2 ml medium into the dish, place in 5% CO2 and 37°C incubator. Before each experiment, examine muscle fibers and discard those without clear striation or with signs of contamination, or with signs of damage such as contracture of the sarcolemmal membrane. Good fibers respond to KCl, electrical stimulation, and caffeine stimulation.
Single muscle fibers are washed 3 times with the wash solution # 1 and bathed in intracellular-like skinning solution with 500 μM Rhod-5N and 0.5 mM Ca2+ for 5 min.
Using a Tungsten needle attached to a pin holder, mechanically skin the fiber under a stereomicroscope, allowing transverse tubules (TT) to automatically reseal and to trap the Rhod-5N conjugated with Ca2+ in the TT, wash the skinned fibers twice with washing solution #2. The mechanical skinning process is optimal when less than 25% of the cell width is removed during the skinning process.
To load the SR, skinned fibers are incubated with the skinning solution containing 20 μM Fluo-5N AM for 1 h at room temperature, followed by extensive washes using washing solution #2, and incubate for an additional 30 min to allow complete de-esterification of the dye.
After 30 min, skinned fibers are visualized under 60X objective of the confocal microscope. Fluorescence images are acquired at wavelengths of the corresponding dyes (excitation at 543 nm and emission longer than 570 nm for Rhod-5N; excitation at 488 nm and emission at 530-560 nm for Fluo-5N). Regions of interest (ROIs) are selected for each dye loaded areas.
The experimental protocol is as following: TT-loading solution for 120 s, SR-loading solution for 120 s, SR-depletion solution for 400-750 s to deplete the SR Ca2+ store. During this process, changes in Rhod-5N intensity (TT Ca2+) and Fluo-5N intensity (SR Ca2+) are recorded, creating a time course determination of relative fluorescence changes in the TT and SR. Mean values of fluorescence intensity from multiple fibers are further analyzed. The maximal loading intensity at the end of TT/SR-loading is normalized to 100%.
5. Representative Results
We examined SOCE activity in KYSE-150 cells using intracellular Ca2+ measurement (Fig 2.). Using RFP as reporter, we could select the individual cells transfected with plasmid containing specific shRNA against orai1, a gene encoding the SOCE channel. Compared with cells transfected with plasmids containing scramble shRNA (black trace), the knocking down of Orai1 protein results in decreased SOCE activity (red trace).
SOCE in KYSE-150 cells was also confirmed with the Mn2+ quenching assay (Fig 3.). The excitation wavelength of 360 nm reports the isosbestic point of Fura-2, where the fluorescence is independent of the Ca2+ concentration. After TG completely depleted ER Ca2+ stores, the perfusion of Mn2+ resulted in a significant fluorescence decrease. The overall SOCE activity can be measured by the decrease rate of Fura-2 fluorescence intensity with a steeper slope indicating a more active SOCE, while a shallower slope meaning a less active SOCE. The slope of fluorescence decrease in 2-APB treated cells appeared to be much shallower, which indicated that SOCE activity was blocked by this compound. The Mn2+ quenching rates were determined from the record of the initial 10 seconds, where the quenching was still in linear range without saturation.
A similar Mn2+ quenching assay was performed in the muscle (Fig 4.). In this case Mn2+ was applied together with TG. While TG was depleting the SR Ca2+ stores, the fluorescence quenching slope gradually changed until it reached its maximal rate. The sigmoidal curve indicates the graded activation of SOCE under these experimental conditions.
Healthy intact single muscle fibers in culture showed clear and uniform striation, no signs of contaminations, and no signs of contraction-induced damage. These fibers were able to contract in response to electrical stimulation or depolarizing solutions. Under confocal imaging, skinned fiber trapping of Rhod-5N dye in the TT compartment showed the characteristic mammalian doublet pattern (visualized in the red channel). After loading of the SR with Fluo-5N AM, the typical punctuated SR pattern was visible (in green channel). Upon perfusion of the skinned fiber with TT/SR loading solution, fluorescence intensity of both Rhod-5N and Fluo-5N increased; upon perfusion with SR depletion solution, fluorescence levels in the SR compartment and the TT compartment started to decrease, indicating tight coupling between SR Ca2+ store depletion and SOCE activation, respectively (Fig 5.).
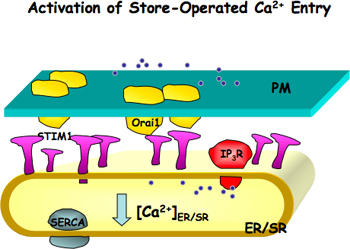 Figure 1. Activation of store-operated Ca2+ entry (SOCE). Orai1, a channel pore forming unit, is located on the plasma membrane (PM) and STIM1, a Ca2+ sensor, is located on the endoplasmic or sarcoplasmic reticulum (ER/SR) membranes. When ER/SR Ca2+ stores are reduced either due to blocking of ER/SR Ca2+ pump (SERCA) or Ca2+ release through IP3 receptor or ryanodine receptor, STIM1 is activated. Activated STIM1 molecules form patches and induce the aggregation of Orai1, which further leads the activation of SOCE.
Figure 1. Activation of store-operated Ca2+ entry (SOCE). Orai1, a channel pore forming unit, is located on the plasma membrane (PM) and STIM1, a Ca2+ sensor, is located on the endoplasmic or sarcoplasmic reticulum (ER/SR) membranes. When ER/SR Ca2+ stores are reduced either due to blocking of ER/SR Ca2+ pump (SERCA) or Ca2+ release through IP3 receptor or ryanodine receptor, STIM1 is activated. Activated STIM1 molecules form patches and induce the aggregation of Orai1, which further leads the activation of SOCE.
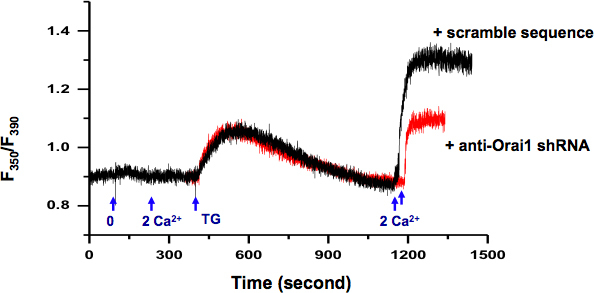 Figure 2. Measurement of intracellular Ca2+ in KYSE-150 cells. Exchange of extracellular solution from 0 Ca2+ (0.5 mM EGTA) to 2 mM Ca2+ did not induce any change in intracellular Ca2+. 5 μM TG in 0 Ca2+ bath solution induced passive ER Ca2+ release. After ER Ca2+ stores were depleted (>10 min), addition of extracellular Ca2+ (2 mM) activated a sustained intracellular Ca2+ elevation through SOCE, which can be blocked by SOCE inhibitors, e.g. skf-96365 and 2-APB (data not show). Cells transfected with plasmids containing shRNA specifically against orai1 (red) demonstrated significantly reduced SOCE than cells transfected with scramble sequence (black).
Figure 2. Measurement of intracellular Ca2+ in KYSE-150 cells. Exchange of extracellular solution from 0 Ca2+ (0.5 mM EGTA) to 2 mM Ca2+ did not induce any change in intracellular Ca2+. 5 μM TG in 0 Ca2+ bath solution induced passive ER Ca2+ release. After ER Ca2+ stores were depleted (>10 min), addition of extracellular Ca2+ (2 mM) activated a sustained intracellular Ca2+ elevation through SOCE, which can be blocked by SOCE inhibitors, e.g. skf-96365 and 2-APB (data not show). Cells transfected with plasmids containing shRNA specifically against orai1 (red) demonstrated significantly reduced SOCE than cells transfected with scramble sequence (black).
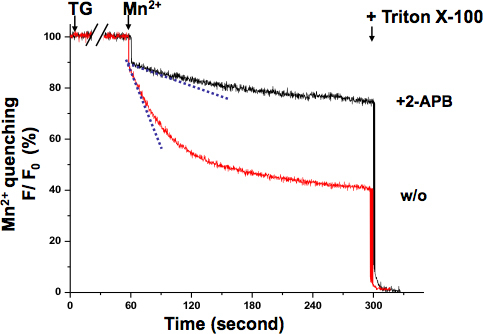 Figure 3. Mn2+ quenching assay of SOCE in KYSE-150 cells. The quenching of Fura-2 fluorescence by Mn2+ (0.5 mM) was measured at the Ca2+-independent excitation wavelength of Fura-2 (360 nm). Cells were treated with 5 μM TG for 10 min to completely deplete ER Ca2+ stores. The decay slope of Fura-2 fluorescence upon Mn2+ addition (dash lines, within the first 10 sec) represented the activation of SOCE, which was expressed as percent decrease in fluorescence per unit time (the initial value as 100%). The maximally quenched fluorescence signal was established at the end of the experiment by lysing the cells with 0.1% Triton X-100 (as 0%). Cells treated with 2-APB (75 μM) demonstrated a much shallower slope, suggesting SOCE is blocked by this compound in KYSE-150 cells.
Figure 3. Mn2+ quenching assay of SOCE in KYSE-150 cells. The quenching of Fura-2 fluorescence by Mn2+ (0.5 mM) was measured at the Ca2+-independent excitation wavelength of Fura-2 (360 nm). Cells were treated with 5 μM TG for 10 min to completely deplete ER Ca2+ stores. The decay slope of Fura-2 fluorescence upon Mn2+ addition (dash lines, within the first 10 sec) represented the activation of SOCE, which was expressed as percent decrease in fluorescence per unit time (the initial value as 100%). The maximally quenched fluorescence signal was established at the end of the experiment by lysing the cells with 0.1% Triton X-100 (as 0%). Cells treated with 2-APB (75 μM) demonstrated a much shallower slope, suggesting SOCE is blocked by this compound in KYSE-150 cells.
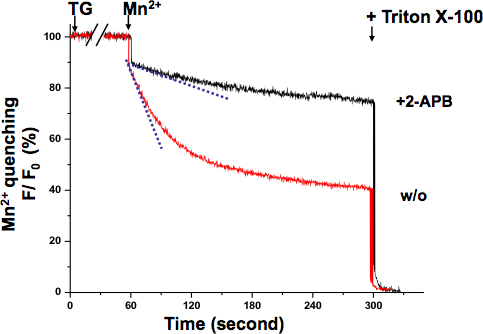 Figure 4. Graded activation of SOCE in muscle cells revealed by Mn2+ quenching assay. Activation of SOCE could be registered while TG was depleting SR Ca2+ stores. Simultaneous application of Mn2+ (0.5mM) and TG (20 μM) induced a graded- and sigmoidal decrease of intracellular Fura-2 fluorescence (cyan dash line to show the fastest quenching point), which was distinct from initial Mn2+ quenching rate (blue dash line). Mn2+ quenching slope remained almost the same as basal level in cells treated 2-APB (75 μM), suggesting that SOCE was inhibited.
Figure 4. Graded activation of SOCE in muscle cells revealed by Mn2+ quenching assay. Activation of SOCE could be registered while TG was depleting SR Ca2+ stores. Simultaneous application of Mn2+ (0.5mM) and TG (20 μM) induced a graded- and sigmoidal decrease of intracellular Fura-2 fluorescence (cyan dash line to show the fastest quenching point), which was distinct from initial Mn2+ quenching rate (blue dash line). Mn2+ quenching slope remained almost the same as basal level in cells treated 2-APB (75 μM), suggesting that SOCE was inhibited.
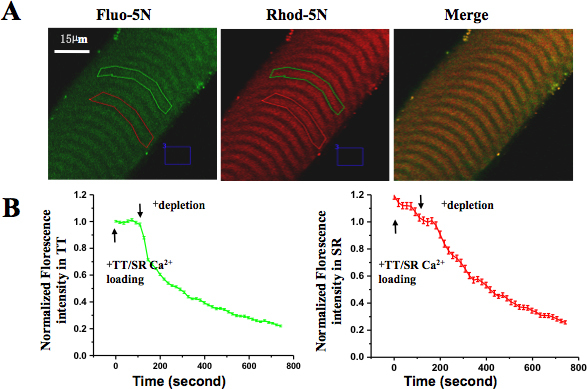 Figure 5. Spatially and temporally resolved SOCE in skinned muscle fiber. (A) Rhod-5N salt was loaded into TT and Fluo-5N AM was loaded into SR. (B) After applying Ca2+ depletion solution, the fluorescence in both TT and SR compartments decreased. The loss of Fluo-5N fluorescence indicated rapidly released SR Ca2+ content and the loss of Rhod-5N fluorescence suggested activation of SOCE.
Figure 5. Spatially and temporally resolved SOCE in skinned muscle fiber. (A) Rhod-5N salt was loaded into TT and Fluo-5N AM was loaded into SR. (B) After applying Ca2+ depletion solution, the fluorescence in both TT and SR compartments decreased. The loss of Fluo-5N fluorescence indicated rapidly released SR Ca2+ content and the loss of Rhod-5N fluorescence suggested activation of SOCE.
Discussion
Although the excitation wavelengths for Ca2+-binding and Ca2+ free Fura-2 are 340 nm and 380 nm, respectively, the best ratio dynamic range of Fura-2 for Ca2+ concentration measurement may occur at other wavelengths in a particular microscope system. Such shifts in wavelength are usually due to changes in the optical path with the addition of various optical components. In this study, the excitation wavelengths for Fura-2 were determined as 350 nm and 390 nm by performing spectral analysis of Fura-2 fluorescence.
The intracellular Ca2+ level in the cytosol results from a balance of several sources, including intracellular Ca2+ release, extracellular Ca2+ entry, as well as Ca2+ exclusion mechanism at the ER and plasma membrane. To isolate the unidirectional SOC-mediated Ca2+ influx from intracellular Ca2+ release and Ca2+ extrusion, the Mn2+ quenching assay can be used. Mn2+ is known to be able to permeate into cells via SOCE but is impervious to surface membrane extrusion or ER uptake by Ca2+ pumps. Therefore, fluorescence quenching represents a measurement of unidirectional Mn2+ flux into cells that estimates the degree of activation of SOCE. Mn2+ quenching assay is performed at the isosbestic point, at such wavelength Fura-2 fluorescence intensity is independent of Ca2+ concentration. The isosbestic point for each system should be determined by spectrum scanning. Alternatively, Mn2+ quenching can be recorded by adjusted fluorescence at any two wavelengths in such a way that the final value is independent of Ca2+ concentration13.
To gain the spatial and temporal information of activity of SOCE in skeletal muscles, we employed a dual-dye method, which allows simultaneous measurement of SOCE activity and SR Ca2+ content in skinned adult mammalian EDL muscle fibers. The correlation between changes in SR Ca2+ content and SOCE activity can be plotted to indicate the threshold and sensitivity of SOCE activation to depletion of the SR Ca2+ storage. The TT-loading solution is optimized to additionally load the T-tubules with Ca2+ and for priming of the T-tubules and the SR-loading solution is designed to promote maximal SR Ca2+ loading and to additionally load the T-tubules with ~500 μM Ca2+. FCCP is included in the TT/SR-loading solution and the SR-depleting solution to eliminate the effects from the mitochondria.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank Dr. Noah Weisleder for reading and editing of this manuscript. This work was supported by Research Grants UMDNJ Foundation 62-09 to ZP, American Heart Association SDG2630086 to XZ, 0535555N to MB, and National Institutes of Health RC2AR058962-01 to MB.
References
- Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol. Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Ma J, Pan Z. Retrograde activation of store-operated calcium channel. Cell Calcium. 2003;33:375–384. doi: 10.1016/s0143-4160(03)00050-2. [DOI] [PubMed] [Google Scholar]
- Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol. Rev. 2009;231:189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB. Store-operated CRAC channels: function in health and disease. Nat. Rev. Drug Discov. 2010;9:399–410. doi: 10.1038/nrd3136. [DOI] [PubMed] [Google Scholar]
- Pan Z. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat. Cell Biol. 2002;4:379–383. doi: 10.1038/ncb788. [DOI] [PubMed] [Google Scholar]
- Shin DW. A retrograde signal from calsequestrin for the regulation of store-operated Ca2+ entry in skeletal muscle. J. Biol. Chem. 2003;278:3286–3292. doi: 10.1074/jbc.M209045200. [DOI] [PubMed] [Google Scholar]
- Ma J, Pan Z. Junctional membrane structure and store operated calcium entry in muscle cells. Front Biosci. 2003;8:d242–d255. doi: 10.2741/977. [DOI] [PubMed] [Google Scholar]
- Pan Z, Damron D, Nieminen AL, Bhat MB, Ma J. Depletion of intracellular Ca2+ by caffeine and ryanodine induces apoptosis of chinese hamster ovary cells transfected with ryanodine receptor. J. Biol. Chem. 2000;275:19978–19984. doi: 10.1074/jbc.M908329199. [DOI] [PubMed] [Google Scholar]
- Hirata Y. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys. J. 2006;90:4418–4427. doi: 10.1529/biophysj.105.076570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. Enhanced resistance to fatigue and altered calcium handling properties of sarcalumenin knockout mice. Physiol. Genomics. 2005;23:72–78. doi: 10.1152/physiolgenomics.00020.2005. [DOI] [PubMed] [Google Scholar]
- Zhao X. Azumolene inhibits a component of store-operated calcium entry coupled to the skeletal muscle ryanodine receptor. J. Biol. Chem. 2006;281:33477–33486. doi: 10.1074/jbc.M602306200. [DOI] [PubMed] [Google Scholar]
- Launikonis BS, Barnes M, Stephenson DG. Identification of the coupling between skeletal muscle store-operated Ca2+ entry and the inositol trisphosphate receptor. Proc. Natl. Acad. Sci. U.S.A. 2003;100:2941–2944. doi: 10.1073/pnas.0536227100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth TJ. Temporal relationships between Ca2+ store mobilization and Ca2+ entry in an exocrine cell. Cell. Calcium. 1994;15:457–466. doi: 10.1016/0143-4160(94)90110-4. [DOI] [PubMed] [Google Scholar]