

Abstract
A recent study from our laboratory demonstrated that epidermal growth factor receptor (EGFR) inhibitors (EGFRIs) augment the expression of class I and class II MHC molecules. This finding provides an additional mechanism through which EGFRIs may exert anti-tumor effects and supports the notion that EGFRIs may influence adaptive immune responses by altering immune gene expression.
Keywords: EGFR, EGFR Inhibitor, Immune Response Modifier, immunotherapy, MHC
Recent and ongoing efforts to develop inhibitors of the human epidermal growth factor receptor (HER) family to treat cancer is fueled by the paradigm that these receptors play a critical role in driving tumor cell proliferation, survival and migration.1 Because it has been implicated in the pathogenesis of several forms of human cancer, enormous efforts have focused on the founding member of the HER family, the epidermal growth factor receptor (EGFR, also known as HER1 and ErbB1). Unfortunately, only a subset of patients respond to EGFR inhibitor (EGFRI) therapy and there is poor correlation between tumor EGFR protein expression and the response to EGFRI therapy.2 Indeed, the complexity of signaling in cancer cells suggests that for most tumor types, targeting a single enzyme or pathway in advanced cancer may be insufficient.3 Despite these challenges, the incorporation of EGFRIs into clinical practice has drawn attention to the immunologic effects that arise in the setting of EGFR inhibition. This has been especially true for immune responses in the skin. In the majority of patients, EGFRIs promote skin inflammation.4 Further, because the presence and severity of EGFRI-induced skin inflammation correlate with survival, there has been increased interest in understanding how EGFRIs influence the expression of immune system genes. This is particularly true for genes (and their encoded proteins) that can influence anti-tumor immune responses. In this point-of-view article, I will summarize our recent studies demonstrating that EGFRIs augment the expression of major histocompatibility complex (MHC) class I (MHCI) and II (MHCII) molecules.5 After this, I will try to place these findings in context with other recent studies that support the notion that EGFRIs may have untapped therapeutic value due to their effects on immune gene expression.
MHCI and MHCII molecules are cell surface glycoproteins that are required for the presentation of peptides to CD8+ and CD4+ T lymphocytes respectively. As the requisite first signal for the activation of T cells, MHC molecules are pivotal to adaptive immune responses including those against tumor cells. Because of this, the expression pattern of MHCI and MHCII molecules in cancer has received much attention. Many studies have demonstrated that the expression of MHC molecules can influence anti-tumor immune responses, prognosis, and the immune response to anti-tumor vaccines.6-8 Thus, new approaches to alter the expression of MHC molecules on tumor cells are being sought.9
The cross-talk that occurs between the IFNγ receptor complex and the EGFR, via the shedding of EGFR ligands, suggested to us that EGFR-mediated signals may influence the induction of MHC molecules by IFNγ (Fig. 1A).10 Prior reports have demonstrated that if keratinocytes are treated simultaneously with an EGFR ligand and IFNγ, there is an attenuation of MHCII induction.11 Based upon this, we hypothesized that EGFRIs would have the opposite effect and would therefore augment MHCII induction by IFNγ. Because MHCII gene expression is largely regulated at the level of transcription, we focused our initial studies on a critical transcriptional regulator of MHCII molecule expression, the MHCII transactivator named CIITA.12 We found that in the presence of an EGFRI, the induction of CIITA by IFNγ was augmented, and that conversely, in the presence of EGFR ligands, CIITA induction was attenuated. Thus, transactivation of the EGFR following IFNγ appears to repress CIITA induction and when EGFR activation is blocked, CIITA induction is increased. As expected, when we examined a key transcriptional target of CIITA, namely the MHCII gene HLA-DR, its induction increased in the presence of EGFR inhibition. To explore the role of cell surface proteases and EGFR ligands, we repeated these experiments using a protease inhibitor and an EGFR-blocking antibody with similar results. Thus, the blockade of EGFR activation can increase MHCII molecules in normal and malignant keratinocytes through a mechanism that likely involves CIITA.
Figure 1.
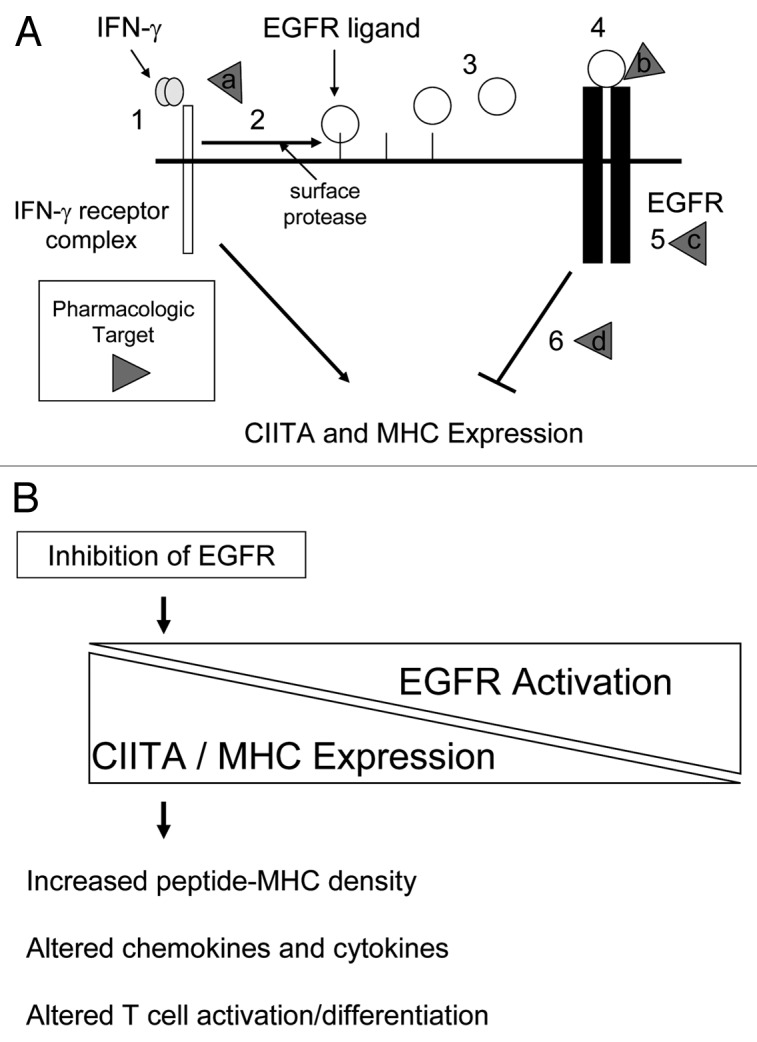
The impact of EGFR activation on MHC expression.
In addition to regulating MHCII molecules, CIITA can regulate the expression of MHCI molecules.13 Therefore, we expanded our analysis to determine whether MHCI expression is influenced by EGFR inhibition. Unlike MHCII molecules that are not expressed by most epithelial cell types in the absence of cytokines, MHCI molecules are constitutively expressed on most nucleated cells. Therefore, we measured the induction of MHCI molecules by IFN-γ in the presence of EGFRIs and saw an increase in MHCI molecule induction in this setting. When we examined MHCI protein levels in the absence of IFNγ, we found EGFRIs alone could augment MHCI induction in primary and malignant human keratinocytes. Thus, EGFR inhibition, even without inflammatory cytokines, can alter MHCI molecule expression. This suggests that there is a balance between EGFR-mediated signals and MHCI and MHCII expression. As EGFR activity is decreased, MHCI and MHCII expression increases (Fig. 1B). In vivo confirmation of the above findings were obtained by examining the expression of MHCI and MHCII protein and RNA levels from skin biopsies of patients taken before and during EGFRI therapy. We found that there was an increase in the expression of MHCI and MHCII molecules on epidermal keratinocytes during EGFRI therapy. However, these changes did not necessarily correlate with clinical or microscopic inflammation. This suggests that while an increase in MHC expression alone is not sufficient to induce clinical inflammation; it may prime the skin to react to immunologic challenge.
The increased levels of MHCI and MHCII protein expression that we observed would be expected to influence T cell biology since the density of peptide-MHC (pMHC) complexes can influence T cell activation and differentiation.14-16 This hypothesis is supported by reports demonstrating that EGFRIs can alter T cell-driven processes. For example, EGFRI therapy has been reported to exacerbate psoriasis, a T cell-mediated autoimmune disease. Additionally, the cutaneous eruption induced by EGFRI therapy has been reported to be provoked by physical trauma (an effect known as the Koebner phenomenon), which is seen in several T cell-mediated autoimmune skin disorders including psoriasis, lichen planus and vitiligo.17,18 Finally, there have been reports that pre-malignant actinic keratoses, whose progression to carcinoma is at least partially prevented by immunosurveillance, can respond to the EGFRI erlotinib.19 Animal studies support the above observations in humans and the notion that EGFRIs can influence T-cell driven immune responses and photocarcinogenesis.
Murine studies have demonstrated that in addition to systemic therapy, the topical application of EGFRIs can alter cutaneous immune responses. Specifically, the topical application of an EGFRI has been reported to enhance the elicitation phase of contact hypersensitivity, a T cell-driven process.20 Further, the immunosuppressive effect of UV radiation (UVR), which is derived from the generation of regulatory T cells, can be eliminated by the topical application of an EGFRI.21,22 Such an effect would be expected to be protective against UVR-induced skin cancers since, as eluded to above, they are highly immunogenic and controlled by immunosurveillance. This too has been reported. Specifically, mice treated topically or systemically with as little as a single dose of an EGFRI developed fewer and smaller UVR-induced skin tumors than control mice. Importantly, the effects of the EGFR inhibitor on EGFR activity were short-lived (lasting roughly one day), yet the protection lasted for over 10 weeks.23 While several potential mechanisms may be responsible, these findings suggest an immune-related mechanism is involved. Any changes in MHC expression that are induced by EGFRIs are likely complemented by alterations in the expression of chemokines and cytokines that promote immune cell recruitment and/or activation. Indeed, EGFRIs are known to alter the expression of several chemokines that play a critical role in immune cell recruitment and in controlling tumor cell proliferation.20,24 Taken together, the aforementioned studies suggest that EGFRIs can profoundly influence adaptive immune responses in the skin at least in part by influencing immune cell recruitment and the expression or relevant immune system genes. It will be of interest to determine if similar effects occur in other epithelial tissues.
The relationship between aberrant EGFR signaling and immune gene expression is important since it links a common oncogenic event, namely the acquisition of aberrant EGFR activity, to the regulation of immune system genes that may influence the generation of an anti-tumor immune response. Thus, events that promote tumorigenesis may also influence how tumor cells interact with components of the immune system and facilitate immune evasion. This concept also applies to tumors with other signaling abnormalities, for example those with alterations in Ras or Raf, since these events may also influence immune gene expression. It is important to note that while the recruitment of immune cells into a tumor can be profoundly destructive, this is not always the case. Indeed, the growth of some tumors can be supported by some elements of the immune system.25
In summary, there is growing evidence that EGFRIs may alter tumor:host interactions by both direct effects on tumor cells and through effects on elements of the immune system, both of which may be therapeutically exploitable (Fig. 2). Conceptualizing EGFRIs in a broader therapeutic context is supported by additional studies demonstrating that EGFRIs can augment the release of hematopoietic stem cells, and that these agents have anti-viral activity.26-28 It will be important to determine if the EGFRI-induced changes in immune gene expression can be exploited to combat cancer. For example, can topical or systemic EGFRIs be used to prevent skin cancer in patients at high risk? In addition, can EGFRIs be used to target and/or potentiate the effects of immune-based therapies in melanoma or other cancers29,30? Future studies will continue to define how best to utilize EGFRIs as targeted anti-cancer agents, however, the immune-related effects of EGFRIs may also hold therapeutic promise in the treatment of cancer and warrant further study.
Figure 2.

How the impact of the EGFR Inhibitors on tumor cells and normal tissue may influence anti-tumor immune responses.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/18073
References
- 1.Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77:400–10. doi: 10.1159/000279388. [DOI] [PubMed] [Google Scholar]
- 2.Gusterson BA, Hunter KD. Should we be surprised at the paucity of response to EGFR inhibitors? Lancet Oncol. 2009;10:522–7. doi: 10.1016/S1470-2045(09)70034-8. [DOI] [PubMed] [Google Scholar]
- 3.Arbiser JL. Why targeted therapy hasn't worked in advanced cancer. J Clin Invest. 2007;117:2762–5. doi: 10.1172/JCI33190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peréz-Soler R, Saltz L. Cutaneous adverse effects with HER1/EGFR-targeted agents: is there a silver lining? J Clin Oncol. 2005;23:5235–46. doi: 10.1200/JCO.2005.00.6916. [DOI] [PubMed] [Google Scholar]
- 5.Pollack BP, Sapkota B, Cartee TV. Epidermal growth factor receptor inhibition augments the expression of MHC class I and II genes. Clin Cancer Res. 2011;17:4400–13. doi: 10.1158/1078-0432.CCR-10-3283. [DOI] [PubMed] [Google Scholar]
- 6.Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer. 2010;127:249–56. doi: 10.1002/ijc.25270. [DOI] [PubMed] [Google Scholar]
- 7.Lotem M, Machlenkin A, Hamburger T, Nissan A, Kadouri L, Frankenburg S, et al. Autologous melanoma vaccine induces antitumor and self-reactive immune responses that affect patient survival and depend on MHC class II expression on vaccine cells. Clin Cancer Res. 2009;15:4968–77. doi: 10.1158/1078-0432.CCR-08-3320. [DOI] [PubMed] [Google Scholar]
- 8.Mortara L, Castellani P, Meazza R, Tosi G, De Lerma Barbaro A, Procopio FA, et al. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin Cancer Res. 2006;12:3435–43. doi: 10.1158/1078-0432.CCR-06-0165. [DOI] [PubMed] [Google Scholar]
- 9.Lampen MH, van Hall T. Strategies to counteract MHC-I defects in tumors. Curr Opin Immunol. 2011;23:293–8. doi: 10.1016/j.coi.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 10.Burova E, Vassilenko K, Dorosh V, Gonchar I, Nikolsky N. Interferon gamma-dependent transactivation of epidermal growth factor receptor. FEBS Lett. 2007;581:1475–80. doi: 10.1016/j.febslet.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Mitra RS, Nickoloff BJ. Epidermal growth factor and transforming growth factor-alpha decrease gamma interferon receptors and induction of intercellular adhesion molecule (ICAM-1) on cultured keratinocytes. J Cell Physiol. 1992;150:264–8. doi: 10.1002/jcp.1041500207. [DOI] [PubMed] [Google Scholar]
- 12.Boss JM, Jensen PE. Transcriptional regulation of the MHC class II antigen presentation pathway. Curr Opin Immunol. 2003;15:105–11. doi: 10.1016/S0952-7915(02)00015-8. [DOI] [PubMed] [Google Scholar]
- 13.Martin BK, Chin KC, Olsen JC, Skinner CA, Dey A, Ozato K, et al. Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity. 1997;6:591–600. doi: 10.1016/S1074-7613(00)80347-7. [DOI] [PubMed] [Google Scholar]
- 14.González PA, Carreno LJ, Coombs D, Mora JE, Palmieri E, Goldstein B, et al. T cell receptor binding kinetics required for T cell activation depend on the density of cognate ligand on the antigen-presenting cell. Proc Natl Acad Sci USA. 2005;102:4824–9. doi: 10.1073/pnas.0500922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leignadier J, Labrecque N. Epitope density influences CD8+ memory T cell differentiation. PLoS ONE. 2010;5:e13740. doi: 10.1371/journal.pone.0013740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corse E, Gottschalk RA, Allison JP. Strength of TCR-peptide/MHC interactions and in vivo T cell responses. J Immunol. 2011;186:5039–45. doi: 10.4049/jimmunol.1003650. [DOI] [PubMed] [Google Scholar]
- 17.Gerber PA, Enderlein E, Homey B. The Koebner-phenomenon in epidermal growth factor receptor inhibitor-induced cutaneous adverse effects. J Clin Oncol. 2008;26:2790–2. doi: 10.1200/JCO.2007.16.0077. [DOI] [PubMed] [Google Scholar]
- 18.Zorzou MP, Stratigos A, Efstathiou E, Bamias A. Exacerbation of psoriasis after treatment with an EGFR tyrosine kinase inhibitor. Acta Derm Venereol. 2004;84:308–9. doi: 10.1080/00015550410024634. [DOI] [PubMed] [Google Scholar]
- 19.Hermanns JF, Pierard GE, Quatresooz P. Erlotinib-responsive actinic keratoses. Oncol Rep. 2007;18:581–4. [PubMed] [Google Scholar]
- 20.Mascia F, Mariani V, Girolomoni G, Pastore S. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. 2003;163:303–12. doi: 10.1016/S0002-9440(10)63654-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao Y, Wolverton JE, Zhang Q, Marathe GK, Al-Hassani M, Konger RL, et al. Ultraviolet B radiation generated platelet-activating factor receptor agonist formation involves EGF-R-mediated reactive oxygen species. J Immunol. 2009;182:2842–8. doi: 10.4049/jimmunol.0802689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarz T. Photoimmunosuppression. Photodermatol Photoimmunol Photomed. 2002;18:141–5. doi: 10.1034/j.1600-0781.2002.180307.x. [DOI] [PubMed] [Google Scholar]
- 23.El-Abaseri TB, Fuhrman J, Trempus C, Shendrik I, Tennant RW, Hansen LA. Chemoprevention of UV light-induced skin tumorigenesis by inhibition of the epidermal growth factor receptor. Cancer Res. 2005;65:3958–65. doi: 10.1158/0008-5472.CAN-04-2204. [DOI] [PubMed] [Google Scholar]
- 24.Pivarcsi A, Muller A, Hippe A, Rieker J, van Lierop A, Steinhoff M, et al. Tumor immune escape by the loss of homeostatic chemokine expression. Proc Natl Acad Sci USA. 2007;104:19055–60. doi: 10.1073/pnas.0705673104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaidi MR, Davis S, Noonan FP, Graff-Cherry C, Hawley TS, Walker RL, et al. Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature. 2011;469:548–53. doi: 10.1038/nature09666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryan MA, Nattamai KJ, Xing E, Schleimer D, Daria D, Sengupta A, et al. Pharmacological inhibition of EGFR signaling enhances G-CSF-induced hematopoietic stem cell mobilization. Nat Med. 2010;16:1141–6. doi: 10.1038/nm.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–95. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang H, Kim SK, Kim M, Reche PA, Morehead TJ, Damon IK, et al. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J Clin Invest. 2005;115:379–87. doi: 10.1172/JCI23220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119–27. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]