

Abstract
To antagonize tumor-derived TGFβ contemporaneously to anticancer immunotherapy, we genetically engineered a fusion protein coupling IL-2 and the ectodomain of TGFβ receptor II (Fusion of Interleukin-2 and Soluble TGFβ receptor – a.k.a. FIST). FIST possesses intriguing gain-of-function properties and induces potent activation of IL2-receptor expressing cells and inhibits tumor-derived angiogenesis. Thus FIST constitutes a first-in-class biological that couples anti-angiogenesis to an immune antitumor response.
Keywords: IL-2, TGFβ Receptor II
TGFβ Role in Tumor Progression
The balance between proliferative and inhibitory signals is essential to maintain immune homeostasis. In late stage tumor progression, immunostimulatory signals provide by proinflammatory cytokines are antagonized by tumor derived immunosuppressive cytokines, such as TGFβ. The members of the Transforming Growth Factor β (TGFβ) family are cytokines involved in essential cellular functions such as proliferation, differentiation, apoptosis, tissue remodeling, angiogenesis, immune response, cell adhesion, and also play a key role in pathophysiology of disease states such chronic inflammatory conditions and cancer.1 The members of this family include the three isoforms of TGFβs, β1, β2, β3; bone morphogenetic proteins (BMPs) and activins. Among them, TGFβ1 is the most frequently overexpressed by carcinomas.2 TGFβ, the most potent immunosuppressive cytokine described to date, exerts severe deleterious effects on several components of the immune system response against cancer cells. TGFβ abolishes the effector functions of macrophages, B cells, cytotoxic T cells, dendritic cells and NK cells, where TGFβ acts as negative regulator of IFNγ production via its mediators SMAD2, SMAD3 and SMAD4. TGFβ act as a prometastatic and proangiogenic factor in late stage cancer by constitutively inducing epithelial to mesenchymal transition (EMT) and tumor associated angiogenesis.1 The strategy of blocking tumor-derived active TGF for therapeutic proposes has been extensively explored. Several therapeutic approaches target TGFβ pathway for the treatment of a number of invasive cancer such as breast cancer and melanoma. Intracellular inhibition of TGFβ receptor I (TβRI) kinase with a small-molecule inhibitors, effectively reduce the number and size of lung metastasis in both orthotropic xenografts and experimental metastasis model of breast carcinoma.3 In addition, antagonists of TGFβ binding to heteromeric receptor, such as a soluble Fc:TGFβ type II receptor fusion protein (Fc:TβRII) used decoy receptor has shown significant reduction of tumor cell motility, intravasation, and metastases in three experimental models of breast cancer. However, these treatment strategies in general do not affect cellular proliferation,4 which indicate that the TGFβ blocking agents in vivo does not target tumor cell proliferation. Therefore, a pro-inflammatory stimulus is required to alarm the immune system against proliferating tumor cells.
The question then arises: among the myriad pro-inflammatory cytokines known to date, which would be best combined to a TGFβ decoy to achieve enhanced anti-cancer immunity?
IL-2 as Partner Cytokine for sTβRII
Proinflammatory cytokines such as IL-2 constitute useful adjuvants for which extensive clinical experience exists for the treatment of cancer. IL-2 is well-known factor for lymphocyte activation and clonal expansion. IL-2 acts as autocrine factor for T cells, supports the development of cytotoxic T cells and stimulates NK cell proliferation and cytotoxicity. However, IL-2 also acts to constrain lymphoid growth and maintain peripheral tolerance. The development of lymphoproliferative disorders in IL-2 knockout mice are tangible evidence that IL-2 can operate as both immunostimulatory and immunosuppressive. As immunosuppressor, IL-2 maintains peripheral tolerance by inducing the generation of regulatory cells.5 For these reasons, IL-2 is considered a double-edge sword. The versatility of its functions is influenced by the cytokine environment and the interaction with other cytokines. Among them, TGFβ play an essential role in sculpting IL-2 dependent signaling pathway.
IL-2 and TGFβ1, Dangerous Liaisons Against Cancer Therapy
As suppressor of IL-2 signaling, TGFβ1 inhibits anti-CD3 and anti-CD28 induced IL-2 mRNA and IL-2 protein in a Smad3-dependent manner in T cells. TGFβ1 also inhibits IL-2-induced lymphocyte proliferation through a SMAD3 independent mechanism, which may involve inactivation of phosphatase 2A (PP2A).6
Previous analysis of IL-2Rβ associated kinase activation (JAK1 and JAK3), as well as STAT5 activation by IL-2 in the presence or absence of TGFβ show no evidence of inhibition of Jak/Stat pathway, but instead TGFβ-dependent inhibition occurs at the nuclear level on a subset of IL-2 target genes related to cell proliferation, including c-myc, cyclin D2 and cyclin E. By contrast, the IL-2 mediated induction of genes involved in cell viability such as bcl-xL and bcl-2 was not inhibited by TGFβ. Thus, the combination of TGFβ and IL-2 would predictably generate a unique pattern of gene expression that neither IL-2 nor TGFβ can generate independently.7
This dangerous liaison may lead to dramatic effects in the development of an effective anti-tumor response, which can be severely compromised by regulatory CD4CD25 cells that requires the presence of both cytokines for the induction of Foxp3.5 With this hypothesis in mind, we sought to block the suppressive effects of tumor-derived TGFβ in tandem with a robust immune activation, and therefore engineered a novel recombinant fusion protein comprised of IL-2 fused to the extracellular domain of TGFβ receptor II, sTβRII (FIST). The coupling of these two molecules not only recapitulates each moiety’s function but also gives rise to a new cytokine-like molecule with unheralded cell biological properties.8
FIST, a New Strategy to Overcome Tolerance and Immunosuppression
We have previously demonstrated that the fusion two cytokines with different bioactivities not only recapitulate synergistic effects but also possess unheralded biopharmaceutical properties not seen by the simple combined use of each moiety.9 Similarly, the fusion of IL-2 and sTβRII not only promote activation of IL-2 receptor expressing cells but also blocks tumor derived TGFβ-mediated suppression in these cell compartments (Fig. 1).
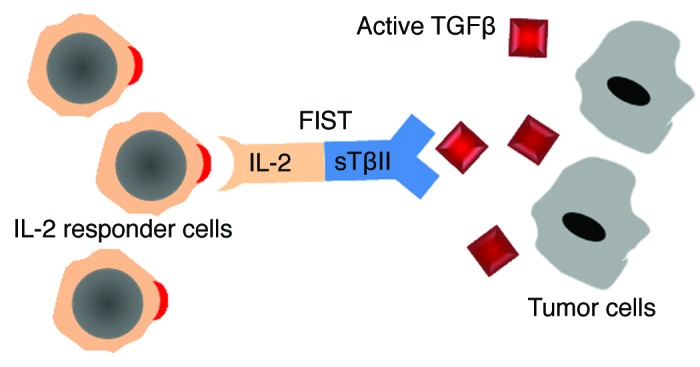
Figure 1. Schematic representation of FIST mechanism of action. Through IL-2 moiety, FIST induces the activation of IL-2 receptor expressing immune cells, whereas the sTβRII moiety functions as decoy receptor blocking tumor-derived TGFβ-mediated suppression on immune cells.
Specifically, the hyperactivation of STAT1 is the landmark of the mechanism underpinning FIST effects. STAT1 is key transcription factor implicated in the development of TH1 cell-mediated immunity against tumor cells. STAT1 is a positive regulator of T-bet, the well-known master regulator of TH1 lineage commitment and IFNγ production. In addition, STAT1 is also considered as the master transcriptional regulator of antigen-specific TH1 cell trafficking in vivo through the induction of IFNγ-inducible chemokines (CXCL9, CXCL10 and CXCL11).10 To complete the array of transcription factors required for an effective anti-tumor response, Smad7 is also overexpressed due to STAT1 hyperactivation. Smad7 operates along with sTβRII moiety to block TGFβ signaling.8
The level of FIST-mediated hyperactivation of STAT proteins is far more potent than the resultant combination of IL-2 receptor engagement and extracellular depletion of active TGFβ suggesting intrinsic gain-of-function properties. Consequently, FIST-stimulated lymphocytes reach a spectacular level of activation and production of pro-inflammatory cytokine and chemokines. In summary, FIST acts as an IFNγ-like cytokine with specificity for IL-2 receptor expressing cells. Through STAT1 activation, FIST involves important transcription factors for TH1 cell-mediated immunity.
FIST, a Novel Angiostatic Factor
The formation of new blood vessels is essential for tumors to growth more 2 mm2 in volume and progress to metastasis. We have found that FIST disrupts the harmonic regulation of angiogenesis by two mechanisms: first, by sequestering active TGFβ through TβRII moiety, FIST may reduce the availability of active TGFβ for its receptors on endothelial cells. Second, by inducing the production of the angiostatic chemokine CXCL10 by NK cells via STAT1 hyperactivation, FIST alters the formation and/or stability of blood vessels.8 Thus FIST targets tumor derived angiogenesis at different checkpoints, which make this molecule an effective angiostatic compound for cancer therapy.
Conclusions
FIST is characterized by inhibiting TGFβ canonical pathway simultaneously with a distinctive STAT1 hyperactivation via IL-2 receptor on immune cells. Thus FIST conveys a robust upregulation of STAT1 target genes including key factors essential for an effective TH1 cell-mediated immunity. This is the first biological agent with the ability to effectively couple anti-angiogenesis to an immune antitumor response, resulting in potent anticancer properties. We propose that the strategy of coupling functionally distinct cytokine/receptor pathways into a single novel fusion cytokine-like molecule may provide a rich and fertile source of novel biological compounds for cancer immunotherapy.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/18458
References
- 1.Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–57. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- 2.Derynck R, Goeddel DV, Ullrich A, Gutterman JU, Williams RD, Bringman TS, et al. Synthesis of messenger RNAs for transforming growth factors alpha and beta and the epidermal growth factor receptor by human tumors. Cancer Res. 1987;47:707–12. [PubMed] [Google Scholar]
- 3.Bandyopadhyay A, Agyin JK, Wang L, Tang Y, Lei X, Story BM, et al. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 2006;66:6714–21. doi: 10.1158/0008-5472.CAN-05-3565. [DOI] [PubMed] [Google Scholar]
- 4.Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, et al. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest. 2002;109:1551–9. doi: 10.1172/JCI15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–6. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 6.Petritsch C, Beug H, Balmain A, Oft M. TGF-beta inhibits p70 S6 kinase via protein phosphatase 2A to induce G(1) arrest. Genes Dev. 2000;14:3093–101. doi: 10.1101/gad.854200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson BH, Martyak TP, Thompson LJ, Moon JJ, Wang T. Uncoupling of promitogenic and antiapoptotic functions of IL-2 by Smad-dependent TGF-beta signaling. J Immunol. 2003;170:5563–70. doi: 10.4049/jimmunol.170.11.5563. [DOI] [PubMed] [Google Scholar]
- 8.Penafuerte C, Bautista-Lopez N, Bouchentouf M, Birman E, Forner K, Galipeau J. Novel TGF-beta antagonist inhibits tumor growth and angiogenesis by inducing IL-2 receptor-driven STAT1 activation. J Immunol. 2011;186:6933–44. doi: 10.4049/jimmunol.1003816. [DOI] [PubMed] [Google Scholar]
- 9.Williams P, Galipeau J. GM-CSF-based fusion cytokines as ligands for immune modulation. J Immunol. 2011;186:5527–32. doi: 10.4049/jimmunol.1003699. [DOI] [PubMed] [Google Scholar]
- 10.Mikhak Z, Fleming CM, Medoff BD, Thomas SY, Tager AM, Campanella GS, et al. STAT1 in peripheral tissue differentially regulates homing of antigen-specific Th1 and Th2 cells. J Immunol. 2006;176:4959–67. doi: 10.4049/jimmunol.176.8.4959. [DOI] [PubMed] [Google Scholar]