

Abstract
TGFβ1 is a regulatory cytokine with a crucial function in the control of T cell tolerance to tumors. Our recent study revealed that T cell-produced TGFβ1 is essential for inhibiting cytotoxic T cell responses to tumors. However, the exact TGFβ1-producing T cell subset required for tumor immune evasion remains unknown. Here we showed that deletion of TGFβ1 from CD8+ T cells or Foxp3+ regulatory T (Treg) cells did not protect mice against transplanted tumors. However, absence of TGFβ1 produced by activated CD4+ T cells and Treg cells inhibited tumor growth, and protected mice from spontaneous prostate cancer. These findings suggest that TGFβ1 produced by activated CD4+ T cells is a necessary requirement for tumor evasion from immunosurveillance.
Keywords: immunosurveillance, immunotherapy, T cell tolerance, TGFβ, tumor immunity
Introduction
In immunocompetent hosts, the adaptive immune system is postulated to respond to and suppress spontaneous tumor development.1,2 In mice and humans, studies have demonstrated that tumor development induces adaptive immune responses.3-7 Deficiency in Rag-1, a gene required for the development of T, B and natural killer T cells, or antibody depletion of CD4+ or CD8+ T cells renders mice more susceptible to spontaneous and carcinogen-induced tumors.8,9 In addition, tumors that grow on immunodeficient Rag2−/− background are rejected upon transplant into immune-intact mice whereas transplant of tumors from wild type mice into wild type mice does not result in tumor rejection.8-10 These findings demonstrate a host-protective role for lymphocytes in tumor development and further show that lymphocytes can also shape the immunogenicity of tumors.
The effector mechanisms that trigger lymphocyte-dependent defense against cancer have begun to be understood. Heterozygosity for the tumor suppressor Trp53 predisposes hosts to tumor development. Trp53+/− mice that are simultaneously deficient in perforin, the pore-forming molecule required for the cytolytic function of CD8+ T and NK cells spontaneously develop B cell lymphomas with an earlier onset than Trp53+/− mice.11 Lymphomas arising in these mice are rejected by a CD8+ T cell-dependent mechanism upon transfer into wild type mice but grow progressively in perforin-deficient recipients.11 These findings indicate a requirement for cytotoxic CD8+ T cell function for tumor rejection and the shaping of tumor cell immunogenicity. IFNγ is an effector cytokine produced predominantly by activated lymphocytes. Treatment of wild type mice with IFNγ neutralization antibodies results in tumor outgrowth.8 Additionally, abrogation of IFNγ signaling either by IFNγ or IFNGR1 deficiency results in spontaneous disseminated lymphomas and increased sensitivity to MCA-induced sarcomas.9,12 Although the specific cell type-produced IFNγ that is required for tumor surveillance remains undefined, these findings demonstrate that IFNγ sensitivity is essential for tumor rejection.
Cancer development is a complex process and the functional outcomes of tumor recognition by T cells differ between tumor types. In a transgenic model of spontaneous cancer where the simian virus 40 T antigen (SV40 Tag) oncogene triggers tumor development in diverse tissues, tumors induce Tag-specific CD8+ T cell responses but these T cells are unable to kill target cells.7 In addition, in the transgenic adenocarcinoma of mouse prostrate (TRAMP) model of cancer, a high proportion of cells that infiltrate prostate tumors are T cells.13 However, tumor burden in TRAMP mice deficient in adaptive immunity are not affected compared with control TRAMP littermates,13 suggesting functional defects or intrinsic inability of adaptive immunity-mediated TRAMP tumor immunosurveillance. TRAMP tumors express a histone H4 peptide self antigen that is recognized by CD8+ T cells.5 When naïve tumor-reactive TCR transgenic (HRC) T cells are transferred into TRAMP mice, the T cells recognize the histone H4 peptide but show limited effector functions.5 Thus, T cell-mediated tumor rejection is not observed in these tumor models. Instead, tumor growth is associated with the functional defects in tumor antigen-specific T cells. Such a phenomenon of T cell tolerance is not restricted to animal models. In melanoma patients, tumor-specific T cell responses can arise de novo, but similar to animal models, these T cells have blunted responses to tumor antigens.3,4 These observations imply that T cell dysfunction is likely a general phenomenon found in spontaneous tumors.
How autochthonous tumors induce T cell tolerance is of interest not only for understanding disease mechanisms but also for cancer immunotherapy. One prominent factor that is often overproduced in animal models of cancer and cancer patients is transforming growth factor β (TGFβ). TGFβ is a family of regulatory cytokines comprising TGFβ1, 2 and 3. The TGFβ ligands elicit their biological activity by binding the serine or threonine kinases TGFβ type I (TGFβRI) and type II (TGFβRII) receptors leading to the phosphorylation and activation of Smad2 and Smad3 transcription factors and subsequent regulation of Smad-dependent genes.14 Among the three isoforms of TGFβ, TGFβ1 is the most highly expressed in the immune system. Tgfb1-null mice manifest severe autoimmune multiorgan pathology that includes lymphadenopathy and myocarditis and die around three to four weeks of age.15,16 Because TGFβ1 regulates the homeostasis of both lymphoid and non-lymphoid tissues, the contribution of T cells to the pathological phenotype of mice harboring germline deletion of the Tgfb1 gene had to be definitively determined. On a genetic background of either MHC class I or class II deficiency, the inflammatory phenotype and severe wasting disease that characterize TGF-β-null mice are greatly ameliorated indicating that disease pathology in these mice is mediated mainly by CD4+ and CD8+ T cells.17,18 In other studies to address the role of TGFβ signaling in T cell tolerance, mice with T cell-specific deletion of TGF-RII alleles were generated.19,20 In these studies, it was shown that abrogation of TGFβ signaling in T cells phenocopies Tgfb1-null mice.19,20 These findings indicate that TGFβ signaling plays a crucial role in the maintenance of peripheral T cell tolerance to self-antigens.
Whether TGFβ suppresses T cell surveillance for malignancy is not completely addressed. In tumor transplantation studies, inhibition of TGFβ signaling in T cells either by expression of a dominant negative mutant of TGFβRII (DNR), or the administration of blocking antibodies or soluble TGFβRII improve T cell responses and inhibits tumor growth.21-26 However these studies fail to recapitulate the physiological function of TGFβ in T cell responses to autochthonous tumors. In a model of cancer where SV40 Tag oncogene induces sporadic tumor development in diverse tissues, studies by Willimsky and Blankenstein showed that tumor growth results in general CTL unresponsiveness. Importantly, CTL dysfunction is also associated with elevated serum concentration of TGFβ1 cytokine.7 These findings corroborate studies in cancer patients where increased TGFβ production is a negative prognostic indicator.21 Altogether, these results reveal that elevated production of TGFβ1 occurs in animal models and human cancers and is associated with T cell unresponsiveness.
However, it remains unknown whether TGFβ production is a consequence or a direct cause of T cell functional defects in tumor-bearing hosts. Recently, we have used TRAMP mice with T cell-specific attenuation of TGFβ signaling via transgenic expression of dominant negative form of TGFβRII (DNR) to show that blockade of TGFβ signaling in DNR-TRAMP mice results in T cell differentiation into IFNγ and GzmB-producing effectors that suppressed tumor development in TRAMP mice.13 In addition, in cases where the tumors did grow, they did not advance to higher histological grades in DNR-TRAMP mice. In adoptive transfer experiments, HRC CD8+ T cells that are specific to histone H4 peptide self-antigen, expressed by TRAMP tumors, are bereft of antitumor activity reflecting tolerance.5 When we crossed HRC mice to DNR mice, we found that DNR-HRC CD8+ T cells are resistant to tolerance induction upon transfer into TRAMP mice as measured by proliferation and GzmB expression in the draining lymph nodes and prostate.13 These observations indicate that TGFβ signaling regulates tumor antigen-specific T cell proliferation and GzmB expression and further suggest that TGFβ production is likely not a consequence but rather a direct cause of T cell unresponsiveness in tumor-bearing hosts.
Virtually all nucleated cells produce TGFβ, and because of the ubiquitous expression of the TGFβ receptors, almost every cell type responds to TGFβ. As a result, which cell type-produced TGFβ mediates the suppression of T cell response to tumors is an important question. One of the earliest indications of the role of TGFβ in tumor development was demonstrated in the early 1990s by overexpression of an active form of TGFβ1 in a highly immunogenic tumor cell line. This modified cell line, but not the parental line, suppressed anti-tumor immune responses.27 This finding formed the basis of the classical notion that tumor cell-produced TGFβ inhibits T cell responses and promotes immune evasion. However, a critical evaluation of data from these studies would argue that they simply indicate that over-production of active TGFβ in tumors can cause T cell hyporesponsiveness without pointing out the precise source of the TGFβ under physiological settings. Thomas and Massague addressed this question more definitively using RNA interference to knock down EL4 tumor-derived TGFβ1.25 When analyzed for tumor immunity, this modified EL-4 tumor did not induce protective antitumor T cell responses whereas tumor expression of soluble TGFβRII enhanced T cell responses, suggesting that TGFβ1 derived from host but not tumor cells might be more important for T cell functional defects.
Understanding the precise source of TGFβ1 that induces T cell dysfunction to tumors is of fundamental interest as it could facilitate the development of more targeted cancer therapies. Earlier, we showed that TGFβ1 produced by T cells controls T cell differentiation and tolerance in autoimmunity settings.28 More recently, we also we demonstrated an important function for T cell-produced TGFβ1 in the control of T cell responses to autochthonous tumors and metastasis.13 We found that deletion of T cell TGFβ1 in TRAMP mice resulted in the differentiation of CD4+ and CD8+ T cells into IFNγ and GzmB-producing effectors in the draining lymph nodes and prostate, and inhibited tumor development in TRAMP mice.13 In metastasis assay, deficiency of TGFβ1 in T cells also prevented lung colonization by B16-OVA tumors. Additionally, chromium release assays revealed that tumor-specific CTL response was specifically mediated by CD8+ T cells in Tgfb1f/n Cd4-cre mice. Altogether, these findings show that T cell tolerance to tumors is controlled by T cell-produced TGFβ1 independent of tumor-derived TGFβ1. However, which precise subpopulation of T cells produce TGFβ1 to suppress T cell responses to tumors remains to be determined.
In this study, we crossed mice carrying floxed/null alleles of Tgfb1 to a series of cre recombinase strains of mice to further delineate the function of TGFβ1 produced by specific subpopulation of T cells in tumor development. We found that deletion of TGFβ1 from either CD8+ T cells or Foxp3+ regulatory T cells alone did not suppress lung colonization by B16-OVA tumors. However, deficiency of TGFβ1 from activated CD4+ T cells and Treg cells inhibited tumor development in TRAMP mice and protected mice from lung colonization by B16-OVA tumors. These results suggest that TGFβ1 production by activated CD4+ T cells is necessary for inhibiting T cell surveillance of tumors.
Results
TGFβ1 produced by Treg cells and CD8+ T cells is dispensable for the immune tolerance of B16-OVA tumors
TGFβ1 produced by T cells has comprehensive effects in tumor development: it suppresses antitumor T cell function to promote both primary tumor growth and tumor metastasis. However, in Tgfb1f/n Cd4cre mice, the Tgfb1 gene is deleted from all T cells. Thus the precise TGFβ1-producing T cell subpopulation required for the control of tumor immune tolerance remains unknown. To address this question, we employed T cell subpopulation-specific TGFβ1-deficient strains of mice. In an earlier study, we demonstrated that deletion of TGFβ1 from CD4+Foxp3+ regulatory T cells was insufficient to inhibit primary tumor growth in Tgfb1f/n Foxp3cre-TRAMP mice.13 In addition, Treg cell-produced TGFβ1 is dispensable for suppressing the differentiation of effector T cells in TRAMP tumors.13 In this report, we tested the function of Treg cell-derived TGFβ1 in tumor metastasis. We found that lung colonization by B16-OVA tumors was comparable between Tgfb1f/n Foxp3-cre mice and Tgfb1f/n control littermates (Fig. 1A and B). This finding established that production of TGFβ1 by Treg cells is not essential for the induction of host tolerance to primary TRAMP tumors as well as B16-OVA tumors.

Figure 1. Treg cell- or CD8+ T cell-derived TGFβ1 is dispensable for promoting tumor growth (A and B) B16-OVA melanoma cells were injected into age-matched Tgfb1f/n and Tgfb1f/nFoxp3-cre mice and pulmonary metastatic nodules assessed 15– 21 days later. (C) RT-PCR showing the deletion of Tgfb1 gene from FACS-sorted CD4+ and CD8+ T cells isolated from Tgfb1f/nCd8-cre mice and Tgfb1f/n control littermates (D and E) B16-OVA melanoma cells were injected into age-matched Tgfb1f/n and Tgfb1f/nCd8-cre mice and pulmonary metastatic nodules assessed 15–21 days later.
To investigate the effects of CD8+ T cell-produced TGFβ1 on tumor immune tolerance, we generated Tgfb1f/n Cd8cre mice by crossing Tgfb1f/n mice with Cd8-cre transgenic mice. Using RT-PCR, we confirmed that the Tgfb1 gene is efficiently deleted specifically from CD8+ T cells (Fig. 1C). Interestingly, B16-OVA tumor lung colonization was comparable between Tgfb1f/n Cd8cre mice and Tgfb1f/n littermate controls (Fig. 1D and E). In tumor-specific chromium release assays, deficiency of CD8+ T cell-derived TGFβ1 did not improve the ability of splenocytes to kill EL-4 target cells compared with splenocytes from wild type mice (data not shown). These observations reveal a nonessential role for CD8+ T cell-derived TGF-β1 in suppressing B16-OVA tumor lung colonization and EL-4 tumor-specific cytolytic activity.
Deletion of TGF-β1 from activated CD4+ T cells and Treg cells inhibits tumor development
To further narrow down the cellular source of TGFβ1 in tumor immune tolerance, we generated Tgfb1f/n Tnfrsf4 (encoding Ox40)-cre mice by crossing Tgfb1f/n mice with Tnfrsf4-cre transgenic mice. We showed recently that Tnfrsf4-cre induces efficient deletion of the Tgfb1 allele in Treg cells and activated CD4+ T cells with minimal deletion in naïve T cells and activated CD8+ T cells.29 When tested for the effect of TGFβ1 deficiency in activated CD4+ T and Treg cells, we found that Tgfb1f/n Tnfrsf4-cre mice were protected from B16-OVA lung colonization compared with Tgfb1f/n control mice (Fig. 2A and B). Occasionally, tumor nodules were found growing in other organs such as the kidney and the peritoneal cavity. Therefore, we reasoned that lung colonization per se would not be a definitive measure of host protective immunity. When assessed for survival, whereas Tgfb1f/n mice (n = 5) had an average survival of 35 days, one Tgfb1f/n Tnfrsf4-cre mouse lived beyond 56 days, and was sacrificed without any obvious signs of disease. The remaining four Tgfb1f/n Tnfrsf4-cre mice had an average survival of 41 d (data not shown). Because we observed comparable tumor burden between Treg cell-specific TGF-β1-deficient mice and their control littermates(Fig. 1A and B), these findings imply that TGFβ1 produced by activated CD4+ T cells is essential for promoting B16-OVA tumor growth.
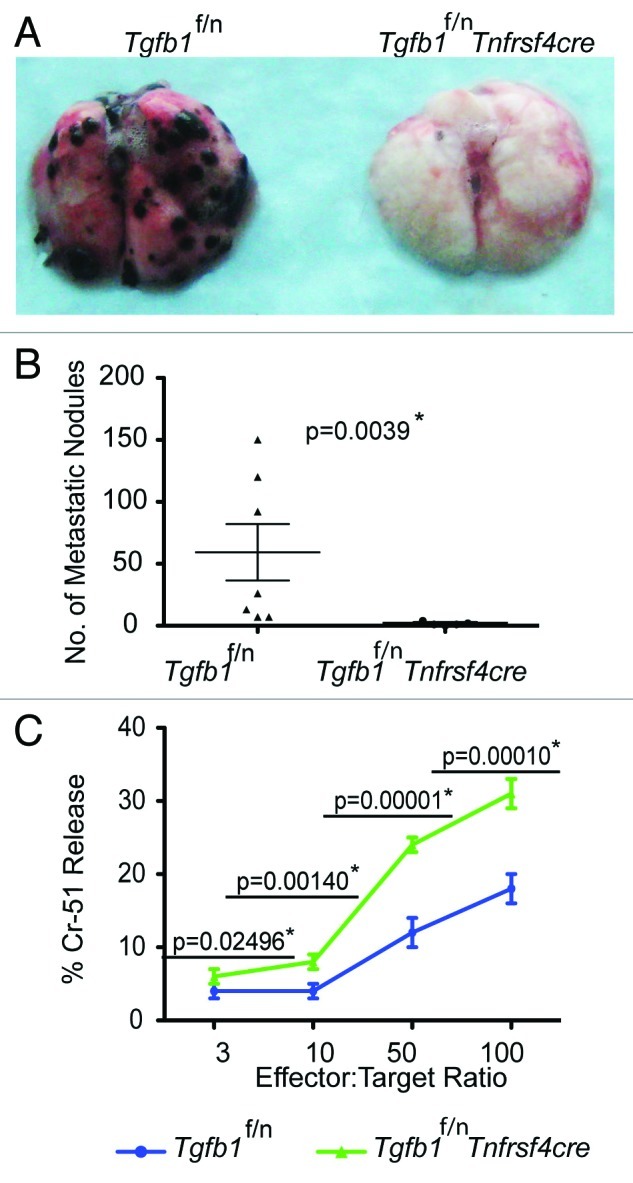
Figure 2. Deficiency of TGFβ1 in activated CD4+ T cells and Treg cells enhances tumor-specific CTL responses (A and B) B16-OVA melanoma cells were injected into age-matched Tgfb1f/n and Tgfb1f/nTnfrsf4-cre mice and pulmonary metastatic nodules assessed 15–21 days later. The p value between two groups of pulmonary metastatic nodules is shown (Students t-test) *Depicts statistically significant difference. (C) Tgfb1f/n and Tgfb1f/nTnfrsf4-cre mice were challenged intraperitoneally with 1 × 106 El-4 tumor cells and the splenocytes were used as effectors against EL-4 targets at the indicated effector:target ratios. Chromium-51 (Cr-51) release assay 10 days after tumor challenge is shown. The p values between the two groups of number of metastatic nodules and percent Chromium-15 release are shown (Students t-test). *Depicts statistically significant difference.
Given that B16 tumors secrete TGFβ1,30 protection against B16-OVA lung colonization in Tgfb1f/n Tnfrsf4-cre mice suggests that absence of TGFβ1 from activated CD4+ T cells and Treg cells is sufficient for generation and maintenance of antitumor immunity independent of tumor-derived TGFβ1. To further test this hypothesis and also determine if the inhibitory function of TGFβ1 from activated CD4+ T cells is applicable to other tumor types, we utilized EL-4 thymoma, another tumor that secretes TGFβ1.22 We injected the mice intraperitoneally and ten days later, assessed EL-4 tumor-specific cytolytic activity in a chromium release assay. We found that splenocytes from Tgfb1f/n-Tnfrscre mice showed significantly higher EL-4-specific cytolytic activity than splenocytes from Tgfb1f/n mice (Fig. 2C). This observation strongly implies that TGFβ1 produced by activated CD4+ T and Treg cells suppresses the generation of tumor-specific T cell effector activity.
Transplanted tumor-induced immune responses could be fundamentally different from those of spontaneous tumors.31 Thus we needed to further substantiate, in a more physiological way, the acquisition of antitumor immunity due to the absence of TGFβ1 derived from activated CD4+ T cells and Treg cells. To this end, we bred Tgfb1f/n Tnfrsf4-cre mice to TRAMP mice to obtain Tgfb1f/n Tnfrsf4cre-TRAMP mice. When evaluated at 8 mo of age, Tgfb1f/n Tnfrsf4cre-TRAMP mice had substantially lower tumor burden than Tgfb1f/n-TRAMP mice (Fig. 3A). Analogous to TRAMP mice with attenuated T cell TGF-β signaling and total T cell-specific deletion of TGFβ1,13 CD4+ and CD8+ T cells in Tgfb1f/n Tnfrsf4cre-TRAMP mice differentiated into producers of IFNγ in the tumor-draining lymph nodes (Fig. 3B). Compared with the tumor draining lymph nodes, the CD8+ T cells that migrated into the prostates further upregulated GzmB production in Tgfb1f/n Tnfrsf4cre-TRAMP mice compared with Tgfb1f/n-TRAMP controls (Fig. 3B). Because Tgfb1f/n Foxp3cre-TRAMP mice are not protected from tumor development,13 these findings suggest that the inhibition of T cell responses in TRAMP mice has a specific requirement for TGFβ1 produced by activated CD4+ T cells.

Figure 3. Deficiency of TGF-β1 in activated CD4+ T cells and Treg cells inhibits tumor development (A) Tgfb1f/n-TRAMP and Tgfb1f/nTnfrsf4-cre-TRAMP mice were evaluated for tumor development at 8 months of age. The weights (Wt) of urogenital tracts (UG) normalized to body Wt ± s.e.m of Tgfb1f/n TRAMP (n = 4) and Tgfb1f/nTnfrsf4-cre TRAMP (n = 3) mice are shown. The p value between two groups of tumor burden is shown (Students t-test). *Depicts statistically significant difference. (B) Flow cytometric analysis examining the expression of interferon gamma (IFNγ), granzyme B (GzmB) and PD-1 proteins in CD4+ and CD8+ T cells in the draining lymph nodes and prostates of Tgfb1f/n TRAMP and Tgfb1f/nTnfrsf4cre-TRAMP mice. For IFNγ expression, T cells were re-stimulated in vitro for five hours with phorbol 12-myristate 13-acetate (PMA), ionomycin and GolgiStop. Representatives of two independent experiments are shown.
The functional outcome of T cell responses to tumors depends on the integration and balance of antitumor responses and inhibitory signals. The expression of programmed death 1 (PD-1) on activated T cells limits effector T-cell responses. PD-1 expression on tumor infiltrating T cells plays an important role in the induction and maintenance of T-cell unresponsiveness in tumors.32,33 In our recent report, we found that blockade of TGFβ signaling in T cells or deletion of TGFβ1 from T cells in TRAMP mice led to diminished PD-1 expression in tumor-infiltrating CD8+ T cells compared with CD8+ T cells from control Tgfb1f/n-TRAMP littermates.13 Consistent with this observation, analysis of Tgfb1f/n Tnfrsf4cre-TRAMP mice showed that protection against tumor development was associated with reduced PD-1 expression in tumor-infiltrating CD8+ T cells (Fig. 3B). Altogether, these findings suggest that augmented anti-tumor immunity in Tgfb1f/n Tnfrsf4cre-TRAMP mice is associated with increase in cytotoxic molecule expression with concomitant reduction in immunoinhibitory signals in tumor-infiltrating CD8+T cells.
Discussion
Although T cell unresponsiveness in nonvirus-induced tumors is a well-known phenomenon, the underlying mechanisms remain poorly understood. Production of TGFβ1 cytokine is commonly associated with T cell dysfunction and tumor progression in cancer patients and experimental models of cancer. We showed recently using a model of oncogene-induced prostate cancer that T cell-specific attenuation of TGFβ signaling resulted in T cell differentiation into potent effectors that blocked tumor development, suggesting that cell-intrinsic TGFβ signaling mediates T cell tolerance and promotes the development of oncogene-induced tumors.13 Additionally, we demonstrated that T cells themselves are the major in vivo source of TGFβ1 cytokine that suppresses the capacity of T cells to survey for malignancy.13 Here in this study, we have further demonstrated that deletion of TGFβ1 from activated CD4+ T cells and Treg cells but not CD8+ T cells or Treg cells alone protected mice from tumor development. This finding established a specific requirement for TGFβ1 from activated CD4+ T cells and provided insight into the mechanism of tolerance mediated by TGFβ in tumor development.
T cell tolerance could occur anywhere between the tumor-draining lymph nodes, where naïve tumor-specific T cells are primed, and the tumor sites after T cell infiltration. In the current view, TGFβ is thought to exert its suppressive effects on T cells at the effector phase. Once at the tumor site, T cells experience large quantities of TGFβ secreted by tumors that render T cells ineffective at killing target cells. This notion has held sway partly because it is an appealing model although direct evidence for it is lacking. In our recent findings, we provided an alternative mechanistic insight into TGFβ-mediated negative regulation of T cell responses to tumors (Fig. 4). We showed that tumor development triggered substantially higher phosphorylation of Smad2 and Smad3 in the tumor draining lymph nodes of 8-mo-old TRAMP mice than in other tissues including the tumor.13 This was an intriguing finding and it revealed that higher T cell TGFβ signaling is coincident with T cell priming to tumor antigens. By attenuating T cell intrinsic TGFβ signaling using DNR-TRAMP mice and deleting T cell TGF-β1 using Tgfb1f/n Cd4cre-TRAMP (TKO-TRAMP) mice, we inhibited T cell Smad phosphorylation in the tumor-draining lymph nodes resulting in blockade of tumor development.13

Figure 4. Model of TGFβ-mediated inhibition of antitumor T cell response. In the tumor-draining lymph nodes, CD4+ T cells become activated in response to tumor-associated antigens. Activated CD4+ T cells in the tumor draining lymph nodes secrete TGFβ1 that suppresses the activation, proliferation and differentiation of naïve tumor-specific CD8+ T cells into IFNγ-secreting T cells. Consequently, activated CD8+ T cells that migrate to the tumor site as effectors fail to elaborate CTL functions including GzmB expression. In addition, effector CD4+ T cells at the tumor site might also produce TGFβ1 to suppress the effector activities of CD8+ T cells. The net result is CTL dysfunction, which allows tumors to grow.
Because use of Cd4 promoter to delete the Tgfb1 allele in Tgfβ1f/n Cd4-cre mice results in nonselective loss of TGFβ1 from all T cells in Tgfβ1f/n Cd4cre-TRAMP mice, one crucial question could not be addressed by this approach, that is the contribution of TGFβ1 produced by specific T cell subpopulations to the observed inhibitory effect of TGFβ1 in T cell antitumor responses. Because of the pleiotropic function of the TGFβ1 cytokine, it is possible that TGFβ1 produced by different subpopulations of T cells could prohibit T cell responses in one context of immune responses but not another. Distinguishing between such subtleties in TGFβ1 function is important for understanding the TGFβ-mediated mechanism of immune regulation. Indeed, when we crossed Tgfβ1f/n mice to different Cre recombinase strains of mice, we found that lung colonization by B16-OVA, a cell line that produces TGFβ1,30 was inhibited in Tgfβ1f/n Tnfrsf4-cre mice but not Tgfβ1f/n Cd8-cre or Tgfβ1f/n Foxp3-cre mice. Additionally, when tested for protective tumor immunity against EL-4, another tumor that produces substantial amounts of TGFβ1,22 absence of TGFβ1 in activated CD4+ T cells and Treg cells but not CD8+ or Foxp3+ Treg cells alone resulted in the induction of high tumor-specific cytolytic activity. Altogether, these findings suggest that TGFβ1 produced by activated CD4+ T cells is required for negative regulation of tumor-specific CTL responses in these transplanted tumor models (Fig. 4).
To investigate whether TGFβ1 produced by activated CD4+ T cells regulates T cell responses to spontaneous tumors, we generated Tgfb1f/n Tnfrs4cre-TRAMP mice by crossing TRAMP mice to Tgfb1f/n Tnfrs4-cre mice. Similar to DNR-TRAMP and Tgfb1f/n Cd4cre-TRAMP mice, we found that Tgfb1f/n Tnfrs4cre-TRAMP mice were protected from tumor development compared with control Tgfb1f/n-TRAMP littermates. This observation suggests that TGFβ1 produced by activated CD4+ T cells promotes TRAMP tumor development. Because tumorigenesis is driven by the same rat probasin promoter activity, it is reasonable to assume equivalent SV40 Tag-induced transformation and tumor initiation events between control Tgfb1f/n-TRAMP and Tgfb1f/n Tnfrs4cre-TRAMP mice. Therefore, this difference in tumor burden that we observed is likely to be direct consequences of immune intervention via the deficiency of TGFβ1 from activated CD4+ T cells. Indeed, protective tumor immunity was associated with enhanced GzmB and IFNγ expression in the tumor-draining lymph nodes and prostate of Tgfb1f/n Tnfrs4cre-TRAMP mice and enhanced EL-4 tumor-specific cytolytic activity in Tgfb1f/n Tnfrs4-cre mice. In our recent findings, we showed that although the T cells in wild type TRAMP mice that trafficked to tumor tissue had significantly dissipated TGFβ-induced Smad phosphorylation, CD8+ T cells failed to elaborate GzmB.13 TGFβ has been shown to directly inhibit GzmB transcription in a Smad-dependent manner.25 It is possible that TGFβ regulates GzmB expression through Smad-independent pathways in tumors. Alternatively, low GzmB expression in CD8+ T cells in the tumor might be a long-lasting footprint of T cell TGFβ signaling in the tumor draining lymph nodes.
Our findings contradict earlier work by several groups showing that tumor-produced TGFβ promotes tumor escape from immune surveillance.23,27,34,35 One possible explanation for these different findings is that mechanisms of TGFβ-mediated T cell tolerance differ between primary tumors and the transplanted tumor models used in the aforementioned studies. Because of the broad distribution of the TGFβ receptors,36 tumor cells and other cells in the immediate vicinity of the tumor will most likely consume TGFβ secreted by tumors before it reaches the draining lymph nodes to regulate T cell differentiation. Thus, tumor-derived TGFβ1 probably makes only a minor contribution to the observed TGFβ-mediated T cell tolerance. Nonetheless, a definitive function of tumor cell-produced TGFβ1 in control of tumor immunosurveillance would require the generation of tumor cell-specific TGFβ1-deficient mice.
Immune response to tumors is a complex battlefield with both tumor-suppressing and tumor-promoting pathways vying for dominance. The integration and balance of these signals ultimately determines the outcome of tumor immune recognition. Expression of the inhibitory co-receptor PD-1 in activated CD8+ T cells has been linked with the induction and maintenance of T-cell dysfunction in tumors.32,33,36-39 In TRAMP mice with deletion of TGFβ1 from activated CD4+ T cells, host-protective immunity was associated with diminished PD-1 expression in tumor-infiltrating CD8+ T cells. This observation is consistent with PD-1 expression on CD8+ T cells that infiltrate the prostates of DNR-TRAMP and Tgfb1f/n Cd4cre-TRAMP mice that we reported recently.13 TGFβ might directly promote PD-1 expression in tumor-infiltrating T cells. In melanoma patients, defective tumor-specific CD8+ T cell responses occur in response to chronic antigen stimulation in association with the induction of PD-1 expression.32 Thus, there remains the possibility that the reduced PD-1 expression in CD8+ T cells is secondary to the tumor protection. The exact mechanism by which TGFβ regulates PD-1 expression, and the molecular targets downstream of TGFβ that antagonize antitumor T cell activity are open for future investigation.
TGFβ1 is secreted as an inactive form that needs to be liberated from the constraints of the latency-associated protein.40 Among other mechanisms, dendritic cell-expressed αvβ8 integrin is required for the activation of latent TGFβ1 and for the regulation of T cell responses.41,42 Therefore, the selectively enhanced TGFβ signaling in tumor-draining lymph node T cells reported in our recent studies13 may be due to the specific requirement of dendritic cells to prime naïve T cells. It also remains to be determined whether the observed requirement of T cell TGFβ1 for enhanced Smad phosphorylation in the tumor-draining lymph node T cells is specifically dependent on TGFβ1 from activated CD4+ T cells. At what time point activated CD4+ T cells secrete TGFβ1 in sufficiently large quantities to regulate CD8+ T cell effector activity is unknown. Does T cell tolerance induction require some threshold of tumor burden to allow high amounts of tumor-associated antigens to trigger T cell TGFβ1 secretion as a result of chronic antigen stimulation in the tumor-draining lymph nodes? In a tumor model where tumorigenesis is triggered by stochastic Tag oncogene activation, Willimsky and Blankenstein demonstrated that high TGFβ1 levels are already present in mice with premalignant lesions (defined by presence of anti-Tag IgG in the serum without visible tumors) and this phenomenon is associated with the induction of CTL unresponsiveness.43 This observation underscores increased TGFβ1 levels in tumor-bearing hosts as an early event. In that study, no evidence was found linking high TGFβ1 levels to tumor cells. Thus, it is likely that T cell secretion of TGFβ1 to inhibit T cell tumor immunity is a very early event that starts when the T cells first encounter tumor-associated antigens.
The emerging insights from our study provide basis for a more informed understanding of the role of TGFβ in suppressing adaptive immune control of cancer. Blockade of TGFβ-dependent immune suppression represents a clinically relevant approach for cancer therapy. Indeed, various strategies to modulate TGFβ signaling are already in different stages of clinical evaluation including neutralizing antibodies (Genzyme, GC-1008), silencing oligonucleotides (Antisense Pharma, AP12009) and small molecule inhibitors (Lilly Research Laboratories, LY364947). However, owing to the pleiotropic properties of TGFβ, important caveats relating to both safety and efficacy of TGFβ modulators raise reasonable concerns. TGFβ has a dual function in tumor development; not only does it promote cancer progression but it can also suppress tumor development posing a great challenge to the development of TGFβ-targeted cancer therapies. In fact, the contribution of the anti-proliferative effect of TGFβ to suppressing tumor growth has recently been documented in animal models of spontaneous cancer. When prostate epithelial cells were specifically attenuated for TGFβ signaling via transgenic expression of dominant negative TGFβRII, TRAMP tumor growth was accelerated compared with wild type controls.44 The goal of TGFβ-targeted therapy is to abolish the tumor-promoting effect of TGFβ and preserve its tumor suppressor function simultaneously. Thus, the therapeutic success of TGFβ antagonism will require a better understanding of the mechanisms by which TGFβ promotes tumor development. TGFβ blocking strategies via neutralizing antibodies, small molecule inhibitors and silencing oligonucleotides lack specificity and consequently target TGFβ signaling systemically without preserving the benefits of its cytostatic effects on tumor cells. The data discussed here reveal previously unrecognized cellular mechanism of TGFβ-mediated immunosuppression that should affect and inspire the design of specific TGFβ-targeting therapies for cancer. Our study provides rationale for design strategies that block the paracrine TGFβ signaling pathway specifically in T cells. This approach will not only awaken anti-tumor immunity but will also ensure the preservation of the cytostatic effect of TGFβ on tumor cells for cancer eradication.
Materials and Methods
Mice
TRAMP, Foxp3-cre, Tnfrsf4-cre mice, and mice with floxed and null alleles (f/n) of Tgfb1 gene have previously been described.28,29,45 Cd8-Cre mice were obtained from the Jackson Laboratory. Tgfb1f/n Tnfrsf4-cre TRAMP mice were produced by crossing Tgfb1f/n Tnfrsf4-cre mice to wild-type TRAMP mice. All mice were maintained under specific pathogen-free conditions, and animal experimentation was conducted in accordance with institutional guidelines.
Tumor cell lines and injections
Ovalbumin-expressing B16 melanoma cells and EL-4 cells were cultured in vitro in DMEM and RPMI-1640 media respectively supplemented with 10% fetal calf serum, 0.1 mM glutamine and 10 U/ml penicillin. B16 melanoma tumor cells were collected by incubation in 0.25% trypsin. Tumor cells were washed two times in endotoxin-free PBS, and 1.0 × 105 or 1.0 × 106 cells injected via tail vein (B16) or intraperitoneally (EL-4) respectively in a volume of 0.2 ml PBS. Cell viability was at least 90% as determined by trypan blue exclusion.
Isolation of tumor-infiltrating lymphocytes
Tumor tissues from sacrificed mice were prepared by mechanical disruption followed by 1 h treatment with 0.5 mg/ml collagenase Type D at 37°C in a shaker. Digested tissues were mashed between glass slides, layered on a percoll gradient and centrifuged at 3000 rpm for 30 min. The separated tumor-infiltrating lymphocyte (TIL) fraction was washed two times in PBS before use.
Flow cytometry
Fluorescently labeled antibodies against CD4, CD8, TCR-β, CD45, GzmB, PD-1 and IFNγ markers were purchased from eBiosciences. Splenocytes and lymph node cells were depleted of erythrocytes by hypotonic lysis. Splenocytes, lymph node cells and TILs were incubated with specific antibodies for 20 min on ice in the presence of 2.4G2 mAb to block FcgR binding. IFNγ and GzmB staining were performed with the nuclear protein or the intracellular cytokine staining kits from eBiosciences and BD Biosciences. For intracellular cytokine staining, cells were stimulated with 50 ng/ml phorbol 12-myristate 13-acetate (Sigma, PMA), 1 mM ionomycin (Sigma) and GolgiStop (BD Biosciences) for 4–5 h. After stimulation, cells were stained with cell surface marker antibodies, fixed and permeabilized with a Cytofix/Cytoperm kit (BD Biosciences). All samples were acquired using LSR II flow cytometer (Becton Dickinson) and analyzed with FlowJo software (Tree Star).
Antitumor cytolytic assay
Freshly isolated splenocytes were depleted of erythrocytes by hypotonic lysis and evaluated for their anti-EL-4 lytic activity as described.22 Chromium-51 (Cr-51)-labeled target EL-4 cells were incubated with effector splenocytes at different effector:target ratios for 4 h. The release of radioactive chromium was measured using a g-counter (Perkin Elmer) and the percentage of specific Cr-51 release was calculated by the formula: [(CPMexperimental – CPMspontaneous) / (CPMmaximum – CPMspontaneous)] × 100%; where CPMexperimental is Cr-51 release by target cells incubated with effector cells, CPMspontaneous is Cr-51 release by equivalent number of targets without effector cells, and CPMmaximum is total Cr-51 release after addition of 2% Triton X-100 to an equivalent number of target cells.
Pulmonary nodule enumeration
Dissected lungs from sacrificed animals were fixed in Bouin’s fixative and the number of metastases counted with a dissecting microscope.
Statistical analysis
Student’s t-test was used to calculate statistical significance for difference in a particular measurement between groups. The Wilcoxon rank-sum test was used to assess differences between samples that did not meet the assumptions of normality. A p value of < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by an American Cancer Society grant (M.O.L), and a CRI predoctoral fellowship (M.K.D.). M.O.L. is a Rita Allen Foundation Scholar.
Glossary
Abbreviations:
- TGFβ
transforming growth factor beta
- TKO
Tgfb1f/n Cd4-cre
- DNR
dominant negative TGFβ receptor 2
- TRAMP
transgenic adenocarcinoma of mouse prostate
- HRC
histone-H4 reactive TCR transgenic
- GzmB
granzyme B
- IFNγ
interferon gamma
- PD-1
programmed death-1
- CTL
cytotoxic T lymphocyte
- Tnfrs4
tumor necrosis factor receptor superfamily member 4
- UG
urogenital tract
Disclosure of Potential Conflicts of Interest Statement
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/18481
References
- 1.Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. 1970;13:1–27. doi: 10.1159/000386035. [DOI] [PubMed] [Google Scholar]
- 2.Klein G, Klein E. Immune surveillance against virus-induced tumors and nonrejectability of spontaneous tumors: contrasting consequences of host versus tumor evolution. Proc Natl Acad Sci USA. 1977;74:2121–5. doi: 10.1073/pnas.74.5.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boon T, Coulie PG, Van den Eynde BJ, van der Bruggen P. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 4.Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. 1999;5:677–85. doi: 10.1038/9525. [DOI] [PubMed] [Google Scholar]
- 5.Savage PA, Vosseller K, Kang C, Larimore K, Riedel E, Wojnoonski K, et al. Recognition of a ubiquitous self antigen by prostate cancer-infiltrating CD8+ T lymphocytes. Science. 2008;319:215–20. doi: 10.1126/science.1148886. [DOI] [PubMed] [Google Scholar]
- 6.Schietinger A, Philip M, Schreiber H. Specificity in cancer immunotherapy. Semin Immunol. 2008;20:276–85. doi: 10.1016/j.smim.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–6. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 8.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450:903–7. doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 9.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 10.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–71. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 11.Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192:755–60. doi: 10.1084/jem.192.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Street SE, Trapani JA, MacGregor D, Smyth MJ. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J Exp Med. 2002;196:129–34. doi: 10.1084/jem.20020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donkor MK, Sarkar A, Savage PA, Franklin RA, Johnson LK, Jungbluth AA, et al. T cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-beta1 cytokine. Immunity. 2011;35:123–34. doi: 10.1016/j.immuni.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–9. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–4. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi S, Yoshida K, Ward JM, Letterio JJ, Longenecker G, Yaswen L, et al. Beta 2-microglobulin-deficient background ameliorates lethal phenotype of the TGF-beta 1 null mouse. J Immunol. 1999;163:4013–9. [PubMed] [Google Scholar]
- 18.Letterio JJ, Geiser AG, Kulkarni AB, Dang H, Kong L, Nakabayashi T, et al. Autoimmunity associated with TGF-beta1-deficiency in mice is dependent on MHC class II antigen expression. J Clin Invest. 1996;98:2109–19. doi: 10.1172/JCI119017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–71. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–54. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Buck MB, Fritz P, Dippon J, Zugmaier G, Knabbe C. Prognostic significance of transforming growth factor beta receptor II in estrogen receptor-negative breast cancer patients. Clin Cancer Res. 2004;10:491–8. doi: 10.1158/1078-0432.CCR-0320-03. [DOI] [PubMed] [Google Scholar]
- 22.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7:1118–22. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 23.Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, et al. Tumor evasion of the immune system by converting CD4+CD25- T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. J Immunol. 2007;178:2883–92. doi: 10.4049/jimmunol.178.5.2883. [DOI] [PubMed] [Google Scholar]
- 24.Nam JS, Terabe M, Mamura M, Kang MJ, Chae H, Stuelten C, et al. An anti-transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 2008;68:3835–43. doi: 10.1158/0008-5472.CAN-08-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–80. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Q, Yang X, Pins M, Javonovic B, Kuzel T, Kim SJ, et al. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive CD8+ T cells: eradication of autologous mouse prostate cancer. Cancer Res. 2005;65:1761–9. doi: 10.1158/0008-5472.CAN-04-3169. [DOI] [PubMed] [Google Scholar]
- 27.Torre-Amione G, Beauchamp RD, Koeppen H, Park BH, Schreiber H, Moses HL, et al. A highly immunogenic tumor transfected with a murine transforming growth factor type beta 1 cDNA escapes immune surveillance. Proc Natl Acad Sci USA. 1990;87:1486–90. doi: 10.1073/pnas.87.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–91. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 29.Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity. 2011;34:396–408. doi: 10.1016/j.immuni.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wojtowicz-Praga S, Verma UN, Wakefield L, Esteban JM, Hartmann D, Mazumder A. Modulation of B16 melanoma growth and metastasis by anti-transforming growth factor beta antibody and interleukin-2. J Immunother Emphasis Tumor Immunol. 1996;19:169–75. doi: 10.1097/00002371-199605000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Willimsky G, Blankenstein T. The adaptive immune response to sporadic cancer. Immunol Rev. 2007;220:102–12. doi: 10.1111/j.1600-065X.2007.00578.x. [DOI] [PubMed] [Google Scholar]
- 32.Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175–86. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kao JY, Gong Y, Chen CM, Zheng QD, Chen JJ. Tumor-derived TGF-beta reduces the efficacy of dendritic cell/tumor fusion vaccine. J Immunol. 2003;170:3806–11. doi: 10.4049/jimmunol.170.7.3806. [DOI] [PubMed] [Google Scholar]
- 35.Friese MA, Wischhusen J, Wick W, Weiler M, Eisele G, Steinle A, et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004;64:7596–603. doi: 10.1158/0008-5472.CAN-04-1627. [DOI] [PubMed] [Google Scholar]
- 36.Massagué J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 38.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–82. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamaguchi T, Sakaguchi S. Regulatory T cells in immune surveillance and treatment of cancer. Semin Cancer Biol. 2006;16:115–23. doi: 10.1016/j.semcancer.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 40.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 41.Lacy-Hulbert A, Smith AM, Tissire H, Barry M, Crowley D, Bronson RT, et al. Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc Natl Acad Sci USA. 2007;104:15823–8. doi: 10.1073/pnas.0707421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449:361–5. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willimsky G, Czeh M, Loddenkemper C, Gellermann J, Schmidt K, Wust P, et al. Immunogenicity of premalignant lesions is the primary cause of general cytotoxic T lymphocyte unresponsiveness. J Exp Med. 2008;205:1687–700. doi: 10.1084/jem.20072016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pu H, Collazo J, Jones E, Gayheart D, Sakamoto S, Vogt A, et al. Dysfunctional transforming growth factor-beta receptor II accelerates prostate tumorigenesis in the TRAMP mouse model. Cancer Res. 2009;69:7366–74. doi: 10.1158/0008-5472.CAN-09-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, et al. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci USA. 1995;92:3439–43. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]