

Abstract
The immunosuppressive effects of apoptotic cells involve inhibition of pro-inflammatory cytokine release and establishment of an anti-inflammatory cytokine profile, thus limiting the degree of inflammation and promoting resolution. We report here that this is in part mediated by the release of the anti-inflammatory mediator annexin A1 from apoptotic cells and the functional activation of annexin A1 receptors of the formyl peptide receptor (FPR) family on target cells. Supernatants from apoptotic neutrophils or the annexin A1 peptidomimetic Ac2-26 significantly reduced IL-6 signalling and the release of TNF-α from endotoxin-challenged monocytes. Ac2-26 activated STAT3 in a JAK-dependent manner, resulting in upregulated SOCS3 levels, and depletion of SOCS3 reversed the Ac2-26-mediated inhibition of IL-6 signalling. This identifies annexin A1 as part of the anti-inflammatory pattern of apoptotic cells and links the activation of FPRs to established signalling pathways triggering anti-inflammatory responses.
Keywords: annexins, apoptosis, inflammation, innate immunity, signalling
INTRODUCTION
Apoptotic cells play an important regulatory role in inflammation and immune responses. Clearance of dying cells by phagocytosis protects the tissue at inflammatory sites from exposure to contents released from disintegrated cells. However, the regulatory effect of apoptotic cells goes beyond the safe disposal of unwanted dangerous material. The phagocytic clearance of apoptotic neutrophils, for example, is anti-inflammatory and contributes to the resolution of inflammation by actively regulating the immune response (Fadok et al, 1998; Savill & Fadok, 2000; Voll et al, 1997). Necrotic cells, in contrast, activate pro-inflammatory responses. Indeed, the pathogenesis of autoimmune diseases has been linked to inefficient clearance of apoptotic cells (Gregory & Devitt, 2004).
Apoptotic cells release and present a vast array of different molecules including receptors, soluble mediators and translocated intracellular molecules, which mediate apoptotic cell clearance and provide anti-inflammatory stimuli, thereby limiting the degree of inflammation and promoting resolution (Byrne & Reen, 2002; Fadok et al, 1998; Huynh et al, 2002). A clear body of evidence shows that apoptotic cells can suppress the release of pro-inflammatory mediators from the phagocyting cells and induce an anti-inflammatory cytokine profile (Savill & Fadok, 2000). However, the underlying signalling elicited by the interaction of apoptotic cells with activated monocytes and macrophages that finally leads to the anti-inflammatory response is not well understood (Serhan & Savill, 2005). Here, we identify annexin A1 that is released from apoptotic neutrophils as an important mediator triggering anti-inflammatory circuits in phagocyting cells.
Annexin A1, a glucocorticoid-inducible protein abundantly expressed in neutrophils, has long been known as a potent anti-inflammatory mediator (Perretti & Flower, 2004; Rescher & Gerke, 2004). Annexin A1-deficient mice show an exacerbated inflammatory response and do not respond to glucocorticoid treatment of inflammatory reactions (Hannon et al, 2003; Yang et al, 2004). The anti-inflammatory activity of annexin A1 is exerted by the unique N-terminal part, which can be released from the full-length protein through proteolysis (Glenney et al, 1987). The formyl peptide receptors (FPRs; Migeotte et al, 2006), a class of heptahelical, G-protein coupled pattern recognition receptors (PRRs), have been identified as receptors for the mimetic annexin A1 peptide Ac2-26, which corresponds to the N-terminal part of the molecule and has full anti-inflammatory activity (Ernst et al, 2004; Perretti et al, 2002; Walther et al, 2000). Externalization of annexin A1 by a yet undefined mechanism has been reported to occur during apoptosis (Arur et al, 2003; Gan et al, 2008; Scannell et al, 2007) and has been linked to the phagocytic clearance of apoptotic cells (Arur et al, 2003; Fan et al, 2004; Maderna et al, 2005). Thus, we addressed whether annexin A1 is involved in conveying the well-known anti-inflammatory properties of apoptotic cells. We show that annexin A1 is released from apoptotic neutrophils and that treatment of monocytes with the annexin A1-containing supernatants of apoptotic granulocytes results in a significantly diminished release of pro-inflammatory cytokines when the monocytes are subsequently challenged with endotoxin. This counter-regulatory activity could be abolished by an antagonist of the cell surface annexin A1 receptors. The anti-inflammatory annexin A1 peptidomimetic Ac2-26 also counteracted TLR-induced pro-inflammatory cytokine release. Via phospholipase D (PLD)-dependent MAP kinase activation, Ac2-26 activated the JAK-STAT3 signalling pathway to induce expression of SOCS3, which in turn limited the pro-inflammatory response. Thus, released annexin A1 is part of the apoptotic cell-associated molecular patterns (ACAMPs) that interact with PRRs to convey a switch towards an anti-inflammatory response.
RESULTS
Anti-inflammatory mediators released from apoptotic cells activate the JAK/STAT pathway
We first examined the effects of mediators released by apoptotic human neutrophils on the lipopolysaccharide (LPS, Calbiochem)-induced TNF-α release in monocytes. Human monocytes were pre-treated with culture supernatants obtained from apoptotic neutrophils, challenged with LPS and the levels of released TNF-α were then determined by enzyme-linked immunosorbent assay (ELISA; Immunotools). As expected, a substantial TNF-α release occurred in LPS-stimulated monocytes. However, when monocytes were pre-treated with apoptotic supernatant, a significant decrease in the amount of released TNF-α was observed (Fig 1A).
Figure 1. The anti-inflammatory effects of apoptotic cells are mediated through activation of JAK/STAT3 signalling.
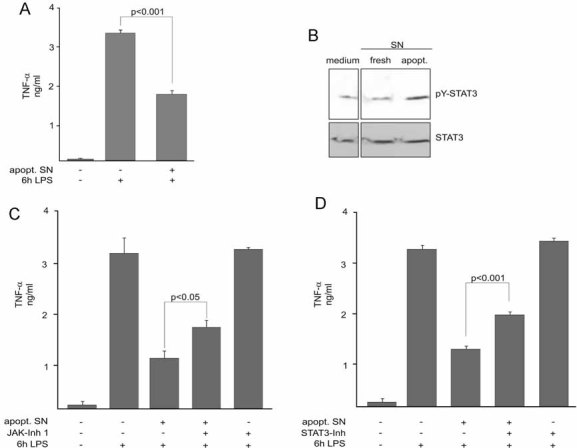
- Apoptotic neutrophil culture supernatants (apopt. SN) suppress LPS-induced TNF-α release. Monocytes were pre-treated for 3 h with apopt. SN and subsequently stimulated with LPS for 6 h. TNF-α contents in the culture media were measured by TNF-α ELISA.
- Activation of STAT3. Monocytes were treated with culture supernatants from either fresh or apoptotic cells for 1 h. STAT3 activation was detected by immunoblotting of cell lysates with the anti-pTYR705-STAT3 antibody. Total STAT3 levels were visualized by reprobing the membrane using a STAT3 antibody.
- Involvement of JAK in the dampening of acute TNF-α release by apoptotic cell supernatants. Monocytes were treated with apopt. SN for 3 h with or without a prior incubation with JAK inhibitor 1 for 15 min. Monocytes were then stimulated with LPS for 6 h and TNF-α levels in the culture media were measured by ELISA.
- STAT3 dependence of the inhibitory effect of apoptotic cell supernatants. Monocytes were pre-treated with the specific STAT3 Inhibitor VI for 30 min prior to treatment with the apoptotic neutrophil supernatant for 3 h and the LPS stimulation for 6 h. ELISA data are presented as the mean ± SEM of four independent experiments. The significance of the differences was evaluated by one-way ANOVA followed by Bonferroni's Multiple Comparison Test. p-values < 0.05 were considered significant. In (B), a representative blot of three independent experiments is shown.
We next analysed the intracellular mechanisms that are activated by factors released from apoptotic cells and counteract the pro-inflammatory response to endotoxin. Since activation of STAT3 has been associated with anti-inflammatory signalling in monocytic cells and animal models, we assessed whether the STAT3 activation state was affected. Therefore, we studied the amount of STAT3 tyrosine phosphorylation in human monocytes incubated with conditioned media from apoptotic cells by Western blot analysis using specific anti-phosphoTyr705-STAT3 antibodies. Addition of apoptotic supernatant induced a transient STAT3 activation not observed for STATs 1, 2 or 5 (not shown). The exact onset of activation differed from experiment to experiment, probably reflecting differences in the different donors of both monocytes and granulocytes, but was always detected upon 60–90 min of incubation. Media alone or culture supernatants collected from non-apoptotic granulocytes did not provoke STAT3 tyrosine phosphorylation (Fig 1B). STAT phosphorylation typically occurs through kinases of the JAK family (Kisseleva et al, 2002), but crosstalk with other pathways has also been demonstrated to result in STAT3 phosphorylation and activation (Galdiero et al, 2006; Leaman et al, 1996). To determine whether JAK-mediated activation of STAT3 by factors released from apoptotic cells impacts LPS-induced TNF-α release, we used pharmacologic inhibitors, namely JAK-Inhibitor 1, a potent cell-permeable JAK selective inhibitor (Thompson, 2005), and STAT3-Inhibitor VI, a cell-permeable compound that inhibits STAT3 activation by binding to the STAT3-SH2 domain (Lin et al, 2009; Siddiquee et al, 2007). JAK-Inhibitor 1 was applied at a concentration (5 µM) that did not affect the phosphorylation patterns of other kinases like p38 and Erk1/2 (data not shown). As shown in Fig 1C, inhibition of JAK did not affect TNF-α release in monocytes treated with LPS alone. However, monocytes pre-treated with apoptotic supernatants in the presence of the inhibitor released significantly higher amounts of TNF-α upon LPS challenge than control cells treated with apoptotic supernatant alone (Fig 1C). Furthermore, the same impact on LPS-induced TNF-α release in monocytes pre-incubated with apoptotic supernatants was observed when monocytes were treated with STAT3-Inhibitor VI (Fig 1D). These results strongly suggest that JAK-mediated STAT3 activation is part of the anti-inflammatory signals elicited by endogenous factors released from apoptotic cells.
Activated STAT3 molecules form a homodimer which translocates into the nucleus, where it recognizes specific DNA elements and activates transcription. To assess STAT3 activation, nuclear translocation and DNA binding, we treated monocytes with apoptotic cell culture supernatant, prepared cytosolic and nuclear extracts and probed for tyrosine-phosphorylated STAT3 by Western blotting. As shown in Fig 2A, increased amounts of tyrosine-phosphorylated STAT3 were detected in the nuclear, but not in the cytosolic fraction upon 60–90 min addition of apoptotic cell culture supernatant. Furthermore, electrophoretic mobility shift assay (EMSA) analysis revealed that nuclear extracts from monocytes treated for 60 min with apoptotic cell culture supernatant contained more STAT3 bound to its cognate DNA probe than extracts from untreated control cells (Fig 2B, lanes 1 and 3). The observed STAT3 binding was highly specific, since binding to the DNA was completely inhibited in the presence of a 100-fold molar excess of unlabelled probe (Fig 2B, lanes 2 and 4).
Figure 2. Apoptotic cell supernatants induce DNA binding of phospho-STAT3 and an upregulation of SOCS3 expression.
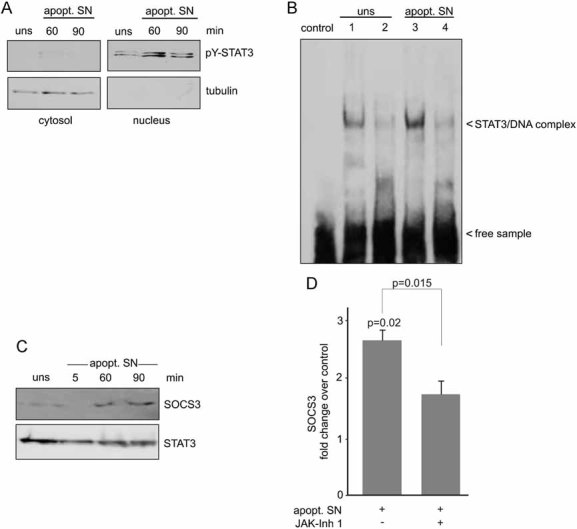
- Nuclear appearance of tyrosine-phosphorylated STAT3 following treatment with apoptotic cell supernatants. Monocytes were either left unstimulated (uns) or stimulated with apopt. SN for the indicated periods of time and cytosolic (20 µg) as well as nuclear protein fractions (5 µg) were probed by immunoblotting with a pY-STAT3 antibody. The cytosolic versus nuclear fractionation was confirmed by analysing the distribution of the cytosolic marker tubulin. A representative result of four independent experiments is shown.
- EMSA analysis of STAT3 DNA binding activity in the nuclear fractions of either control cells (lane 1) or cells treated with apopt. SN (lane 3). The specificity of STAT3 binding in the complexes was verified by competition with a 100-fold molar excess of unlabelled specific dsDNA probe (lanes 2 and 4). Arrowheads indicate the position of the STAT3/DNA complex. The control lane shows the DNA probe without the addition of protein. A representative result of four independent experiments is shown.
- Supernatants from apoptotic neutrophils induce an increase in SOCS3 protein levels. Monocytes were stimulated for the indicated periods of time with apopt. SN and SOCS3 protein levels were analysed by Western blotting. Membranes were reprobed with a STAT3 antibody to ensure equal loading. A representative blot of three independent experiments is shown.
- Apoptotic cell supernatants induce an upregulation of SOCS3 mRNA. Total RNA was extracted from monocytes incubated with apopt. SN with or without prior treatment with JAK Inhibitor 1 and subjected to qPCR. Levels of SOCS3 mRNA were calculated as change over control (unstimulated cells). Data are expressed as means ± SEM of at least three independent experiments. The significance of the differences was evaluated by two-sided t-tests and respective p-values are given.
To further relate the relevance of our findings to inflammatory scenarios, we analysed the capacity of the observed STAT3 activation to interfere with the inflammatory activation of monocytes. A well-known target gene of active STAT3 is SOCS3, which is induced via the JAK/STAT pathway and acts as a negative feedback regulator of the IL-6 type cytokines that signal through the shared receptor subunit gp130. As shown in Fig 2C, augmented SOCS3 protein levels were detected in lysates obtained from monocytes stimulated with apoptotic cell culture supernatant for 60–90 min (Fig 2C). In addition, quantitative real time PCR data revealed that the upregulation of SOCS3 expression induced by apoptotic cell culture supernatant was, at least in part, JAK dependent (Fig 2D).
Annexin A1 is released from apoptotic PMNs and suppresses LPS-induced TNF-α release in human monocytes via activation of formyl peptide receptors
Since activation of the G protein-coupled FPRs through endogenous ligands, such as the anti-inflammatory mediator annexin A1, has been shown to be a main axis of anti-inflammation, we next asked whether activation of the FPRs is also involved in the observed STAT3 activation elicited by mediators released from apoptotic cells. As shown in Fig 3A, pre-treatment of monocytes with the pan-FPR antagonist Boc2 substantially impaired the level of STAT3 tyrosine phosphorylation induced by conditioned media, arguing for a role of FPRs in transducing signals of factors released from apoptotic neutrophils.
Figure 3. Annexin A1 is released from apoptotic cells and mediates STAT3 activation and a reduction of pro-inflammatory cytokine secretion via agonistic activation of formyl peptide receptors.
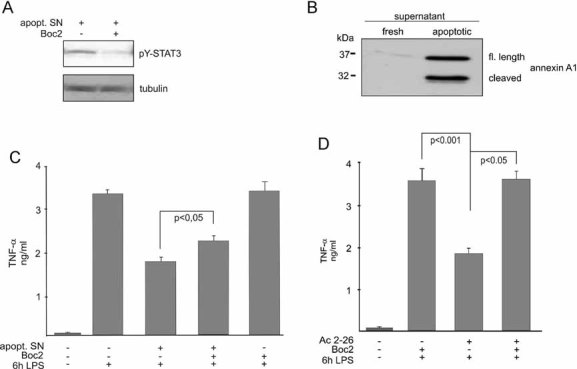
- Involvement of FPRs in the apoptotic cell-mediated activation of STAT3. Monocytes were treated with the supernatant of apoptotic neutrophils (apopt. SN) with or without the addition of the pan-FPR inhibitor Boc2 and STAT3 activation was revealed by immunoblotting of monocyte extracts with the anti-pTYR705-STAT3 antibody. Reprobing the membrane with a tubulin antibody served as a control for equal loading.
- Release of annexin A1 from apoptotic neutrophils. Supernatants of PMNs, which were either freshly isolated or apoptotic, were probed for the presence of annexin A1 by Western blotting with a polyclonal antibody that recognizes both the intact (upper band) and the cleaved protein (lower band).
- Participation of FPRs in the regulation of pro-inflammatory cytokine release. Human monocytes were treated with the apopt. SN with or without the addition of the pan-FPR inhibitor Boc2 for 3 h before stimulation with LPS for 6 h. TNF-α levels in the culture media were measured by ELISA.
- The annexin A1 peptidomimetic Ac2-26 decreases LPS-induced cytokine release in a FPR-dependent manner. Monocytes were treated with Ac2-26 with or without the addition of the pan-FPR inhibitor Boc2 for 3 h prior to stimulation with LPS for 6 h. TNF-α levels in the culture media were then measured by ELISA. Data are expressed as mean values ± SEM of four independent experiments. The significance of the differences was evaluated by one-way ANOVA followed by Bonferroni´s Multiple Comparison Test and respective p-values are given. p-values < 0.05 were considered significant.
Next we assessed whether the FPR ligand annexin A1 participates in mediating the immunosuppressive effects of apoptotic cells on cytokine release. Therefore, we first investigated whether apoptotic neutrophils released annexin A1. In contrast to non-apoptotic neutrophils, annexin A1 was readily detectable in the supernatants from cells undergoing spontaneous apoptosis (Fig 3B). In line with previous findings (Rescher et al, 2006; Vong et al, 2007), a considerable amount of the externalized protein corresponds to a truncated form of 32 kD, the annexin A1 core protein. This indicates that the N-terminal part of the protein, which harbors the anti-inflammatory activity (Cirino et al, 1993), had been liberated by proteolysis. Since the high resolution crystal structure of annexin A1 revealed that the respective N-terminal region is partially buried in the core domain (Rosengarth et al, 2001), proteolytic release of the N-terminal part following annexin A1 externalization might be required to convert the N-terminal annexin A1 domain from the inaccessible, inactive state to an accessible form with full agonistic activity. To specifically assess the activity of N-terminal annexin A1 derivatives released from apoptotic polymorphonuclear leukocytes (PMNs), we next incubated monocytes with either the conditioned medium of apoptotic neutrophils (as before, Fig 1) or the annexin A1 peptidomimetic Ac2-26, which corresponds to the N-terminal domain of annexin A1. This peptide has full physiological activity and acts agonistically on all three members of the human FPR family (Ernst et al, 2004) expressed in human monocytes (Le et al, 2002). The concentration of Ac2-26 was optimized in titration experiments (data not shown) and proved to match the concentrations shown to elicit anti-inflammatory effects in different inflammation models (Gavins et al, 2003; Lim et al, 1998; Perretti et al, 1993). As shown in Fig 3C and D, both the pre-incubation of monocytes with supernatants from apoptotic PMNs and with Ac2-26 significantly suppressed LPS-induced TNF-α release. To analyse a possible contribution of the annexin A1-mediated activation of FPRs to the observed inhibitory effect of apoptotic cell supernatants on TNF-α release, we incubated monocytes with the pan-FPR antagonist Boc2 in combination with apoptotic cell supernatants prior to LPS stimulation. While the antagonist alone did not significantly affect TNF-α release, blocking the FPRs partially but significantly reversed the inhibitory effect of apoptotic granulocyte supernatants on TNF-α secretion (Fig 3C), arguing for a contribution of annexin A1 and its cognate receptors of the FPR family to the anti-inflammatory properties of apoptotic cells. Consistently, the inhibitory effect of the annexin A1 mimetic Ac2-26 was completely blocked by Boc2 (Fig 3D).
Annexin A1 induces activation of STAT3
To determine directly whether annexin A1 is involved in the process of anti-inflammatory activation, we incubated monocytes for various periods of time with the Ac2-26 peptidomimetic and monitored STAT3 activation. The annexin A1 N-terminal peptide transiently activated STAT3 phosphorylation in a dose- and time-dependent manner. Phosphorylation of STAT3 tyrosine 705 was robustly detected upon 60 min of Ac2-26 stimulation and returned to basal levels at later time points (Fig 4A), thus again mirroring the effect of total apoptotic PMN supernatants. No evidence for activation of STATs 1, 2 or 5 was found in the samples (not shown). To rule out that the observed STAT3 activation was due to a contamination with bacterial endotoxins (Benkhart et al, 2000; Carl et al, 2004), activation with either LPS or Ac2-26 was carried out in the presence of the LPS inhibitor polymyxin B. As shown in Fig 4B, polymyxin B completely inhibited the LPS-induced STAT3 tyrosine phosphorylation, whereas STAT3 activation by Ac2-26 was unaffected, confirming that activation by the annexin A1 peptide was specific.
Figure 4. The annexin A1 peptidomimetic Ac2-26 triggers STAT3 phosphorylation in a FPR-dependent manner.
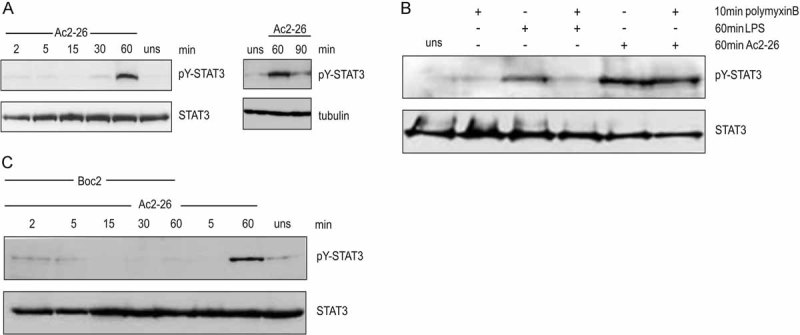
- Activation of STAT3 by Ac2-26. Monocytes were treated with Ac2-26 for the indicated times and tyrosine phosphorylation of STAT3 was detected by immunoblotting with the anti-pTYR705-STAT3 antibody. Probing for total STAT3 and tubulin served to visualize equal loading.
- To ensure the specificity of annexin A1 activation, monocytes were incubated for 10 min with the LPS antagonist polymyxin B prior to stimulation with either LPS or Ac2-26.
- Involvement of FPRs in the Ac2-26-mediated STAT3 activation. STAT3 phosphorylation in response to Ac2-26 was investigated in the presence of the pan-FPR inhibitor Boc2 by Western blot analysis of lysates of the appropriately treated monocytes. Representative immunoblots of at least three independent experiments are shown.
Next, we determined whether the annexin A1 receptors (FPRs) are involved in transducing the Ac2-26-elicited signal that leads to activation of STAT3. As shown in Fig 4C, pre-incubation of monocytes with the Boc2 antagonist almost completely inhibited Ac2-26 induced tyrosine phosphorylation of STAT3. In contrast, STAT3 phosphorylation induced by IL-6 remained unchanged in the presence of Boc2 (not shown), excluding an unspecific effect of this inhibitor and clearly indicating that STAT3 activation was dependent on agonistic signalling of Ac2-26 through FPRs.
PLD and ERK act upstream of STAT3 in the anti-inflammatory signalling induced by Ac2-26
To further dissect the signalling cascade that is elicited by Ac2-26-mediated activation of FPRs and leads to STAT3 activation, we investigated whether STAT3 phosphorylation was dependent on MAPK activation. Pre-incubation with UO126, a specific inhibitor of Mek1/2 kinase module upstream of Erk1/2, resulted in a marked reduction of Ac2-26-mediated STAT3 tyrosine phosphorylation (Fig 5A), indicating that the MAPK pathway contributes to the Ac2-26-triggered STAT3 tyrosine phosphorylation.
Figure 5. Ac2-26-mediated STAT3 activation is PLD- and ERK-dependent.
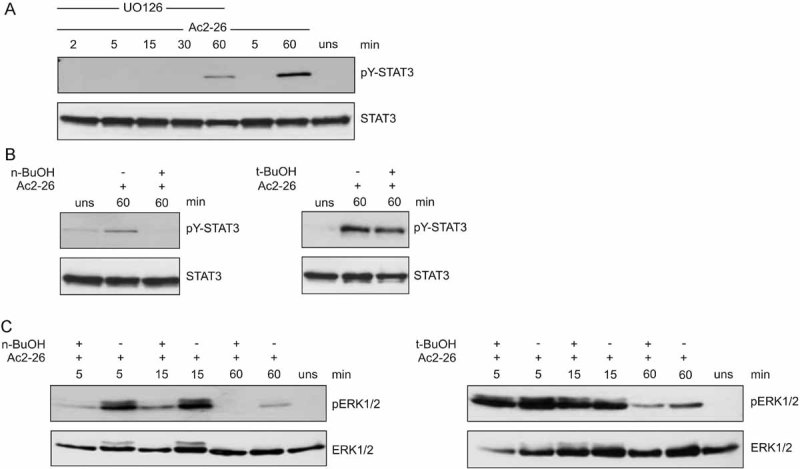
-
MEK1/2 activity is required for Ac2-26-triggered STAT3 activation. Monocytes were pre-treated with medium alone or the MEK1/2 inhibitor UO126 and then stimulated with Ac2-26 for the indicated periods of time. Levels of pY-STAT3 and total STAT3 in the cellular lysates were detected by immunoblotting. A representative blot of at least three independent experiments is shown.(B and C) PLD is required upstream of ERK1/2 for Ac2-26-mediated STAT3 activation.
- Monocytes were pre-treated with either n- or tert-butanol and then stimulated with Ac2-26 for 60 min. Levels of pY-STAT3 and total STAT3 in the cellular lysates were detected by immunoblotting.
- Monocytes were pre-treated with either n- or tert-butanol and then stimulated with Ac2-26 for the indicated periods of time. Levels of p-ERK1/2 and total ERK1/2 in the cellular lysates were detected by immunoblotting. Representative blots of at least three independent experiments are shown in each case.
GPCRs have been shown to activate PLD in various signalling pathways (McDermott et al, 2004). PLD generates phosphatidic acid (PA), which can function as an intracellular lipid messenger. To assess the potential contribution of PLD to the regulation of Ac2-26/FPR-induced STAT3 activation, we inhibited PLD-mediated PA production by n-butanol. In the presence of such primary alcohol, PLD transfers the phosphatidyl group of the substrate phosphatidylcholine in a transphosphatidyl reaction onto the primary alcohol, thereby generating phosphatidylbutanol instead of PA. The tertiary isomer tert-butanol cannot be used as transphosphatidylation substrate and therefore served as control. As shown in Fig 5B, inhibition of PA production by n-butanol completely inhibited the tyrosine phosphorylation of STAT3 induced by Ac2-26 (left panel), whereas the control tert-butanol had no significant effect (right panel). We next investigated whether PLD acts upstream of the ERK module. Figure 5 shows that Ac2-26-induced ERK activation, which occurred already at 5 min of stimulation, was abrogated in monocytes pre-treated with n-butanol, but not tert-butanol, suggesting that the annexin A1 peptide-induced PLD activation occurs upstream of Erk1/2. This in turn is essentially required for the STAT3 phosphorylation (see above). The identical contribution of PLD and Erk1/2 signalling was also observed when monocytes were treated with apoptotic PMN supernatant strengthening the conclusion that annexin A1 is a component of apoptotic cells that contributes to the anti-inflammatory activation of monocyes (data not shown).
Ac2-26 triggers SOCS3 induction via the JAK pathway
To elucidate the effect of Ac2-26-induced STAT3 activation on the nuclear translocation of activated STAT3, we prepared cytoplasmic and nuclear extracts from annexin-treated monocytes and subjected them to immunoblotting with the phospho-STAT3-specific antibody. As shown in Fig 6A, pY-STAT3 was found in the nuclear fraction in response to Ac2-26. We next investigated whether Ac2-26-induced tyrosine phosphorylation of STAT3 correlated with STAT3 DNA binding activity. Therefore, monocytes were stimulated with Ac2-26 for 60 min, and the DNA binding activity of STAT3 was determined by EMSA. Whereas unstimulated cells showed relatively few STAT3/DNA complexes (Fig 6B, lane 1), STAT3 DNA-binding was readily observed in Ac2-26-treated monocytes (Fig 6B, lane 3). Collectively, these data indicate that Ac2-26 induces STAT3 phosphorylation, nuclear translocation and DNA binding via agonistic activation of the FPRs.
Figure 6. The Ac2-26-mediated STAT3 activation is JAK-dependent and induces nuclear translocation and DNA binding of phosphorylated STAT3 as well as increased SOCS3 expression.
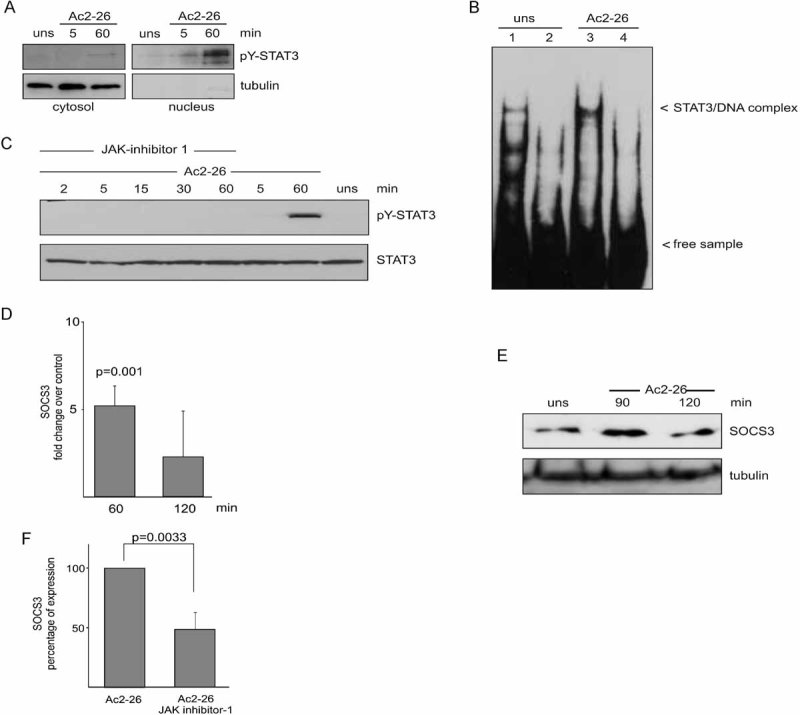
- Ac2-26 triggers nuclear appearance of phosphorylated STAT3. Cytosolic and nuclear fractions of monocytes stimulated with Ac2-26 for the indicated periods of time were immunoblotted to detect the subcellular distribution of pY-STAT3. The cytosolic versus nuclear fractionation was verified by analysing the distribution of the cytosolic marker tubulin, which was absent from the nuclear fractions. A representative result of three independent experiments is shown.
- Ac2-26 induces DNA binding of STAT3. EMSA analysis of STAT3 DNA binding activity in the nuclear fractions of either control (lane 1) or Ac2-26-treated monocytes (lane 3). The specificity of STAT3 binding in the complexes was ensured by competition with a 100-fold molar excess of unlabelled dsDNA probe (lanes 2 and 4). Arrowheads indicate the position of the STAT3/DNA complex. A representative result of three independent experiments is shown.
- The Ac2-26-mediated STAT3 activation is JAK-dependent. Monocytes were pre-treated with JAK Inhibitor 1 and then stimulated with Ac2-26 for the indicated periods of time. Levels of pY-STAT3 and total STAT3 in the cellular lysates were detected by immunoblotting. A representative blot of three independent experiments is shown.
- Ac2-26 induces an increase in SOCS3 mRNA expression. Total RNA was extracted from monocytes stimulated with Ac2-26 for 60 or 120 min and subjected to SOCS3-specific qPCR. Levels of SOCS3 mRNA present following Ac2-26 stimulation were calculated as change over control (unstimulated cells).
- SOCS3 protein levels are increased following Ac2-26 treatment. Monocytes were stimulated for the indicated periods of time with Ac2-26 and SOCS3 protein present in the cellular extracts was identified by Western blotting. Membranes were reprobed with a tubulin antibody to ensure equal loading. A representative blot of three independent experiments is shown.
- The Ac2-26-induced upregulation of SOCS3 requires JAK activity. Monocytes were stimulated with Ac2-26 for 60 min in the absence or presence of JAK Inhibitor 1 and levels of SOCS3 mRNA were determined by qPCR. Data in (D) and (F) are expressed as means ± SEM of at least three independent experiments. The significance of the differences was evaluated by two-sided t-tests and respective p-values are given.
To determine whether the Ac2-26-triggered STAT3 activation was mediated by JAK, we treated monocytes prior to annexin A1 stimulation with JAK inhibitor I. Figure 6 shows that the Ac2-26 induced tyrosine phosphorylation of STAT3 was completely abrogated in cells pre-treated with the JAK inhibitor, while the levels of STAT3 protein remained unchanged. This indicates that the Ac2-26-mediated STAT3 tyrosine phosphorylation occurs through activation of the JAK pathway.
Next, we investigated whether Ac2-26-mediated FPR activation has an effect on SOCS3 expression. Quantitative real time PCR revealed a significant Ac2-26-induced upregulation of SOCS3 mRNA upon 60 min of stimulation (Fig 6D). Furthermore, we were able to detect augmented SOCS3 protein levels in Ac2-26-treated monocytes (Fig 6E). Interestingly, a robust increase in cellular SOCS3 protein was seen after 90 min, possibly reflecting the time between increased mRNA production and protein synthesis. At later time points (120 min), the extent of SOCS3 induction already decreased. The Ac2-26-induced upregulation of SOCS3 expression was, at least in part, JAK dependent (Fig 6F). In contrast to SOCS3, the expression levels of SOCS1, 2, 4–6 did not show any significant changes following Ac2-26 treatment (not shown). Thus, all anti-inflammatory effects elicited by apoptotic cell supernatants in human monocytes can be faithfully reproduced by the purified annexin A1 peptide.
SOCS3 regulates the Ac2-26-mediated inhibition of IL-6 signalling
To investigate whether Ac2-26-induced SOCS3 expression negatively regulates gp130 cytokines signalling, we first analysed the effect of Ac2-26 on IL-6 induced STAT3-activation. It is well known that IL-6 together with the signal transducer gp130 activates STAT3. As expected, IL-6 stimulation rapidly and robustly increased pY-STAT3 levels, reaching a maximum already 15 min after stimulation. This is in clear contrast to Ac2-26-elicited STAT3 phosphorylation, which occurred much later (Fig 7A, lanes 2 and 3). To test whether Ac2-26 elicited STAT3 activation is perturbed by pre-treatment with IL-6, monocytes were first stimulated with IL-6. After 90 min, when IL-6 induced STAT3 phosphorylation was already back to basal levels (not shown), Ac2-26 was given as a second stimulus and Ac2-26-elicited STAT3 phosphorylation was analysed. As presented in Fig 7A (lane 4), the STAT3 phosphorylation signal obtained in these cells was almost equivalent to the signal seen in cells stimulated with Ac2-26 only. This shows that STAT3 activation in response to Ac2-26 is not inhibited by pre-treatment with IL-6. Next, we tested for cross-inhibition of IL-6 signalling by Ac2-26 pre-treatment. Monocytes were pre-cultivated for 90 min in the presence of Ac2-26. At this time point, annexin-mediated STAT3 activation was back to basal levels (see Fig 4A). As shown in Fig 7A (lane 5), a subsequent 15 min stimulation with IL-6 resulted in decreased STAT3 phosphorylation as compared to treatment with IL-6 alone (Fig 7A, lane 2). Ac2-26 inhibited the IL-6-mediated STAT3 phosphorylation for up to 30 min of IL-6 application (not shown). To test whether the Ac2-26-induced upregulation of SOCS3 expression documented above (Fig 6) is the cause of impaired IL-6 signalling in the Ac2-26-treated monocytes, we performed stimulation experiments in monocytes, which had been treated with antisense phosphorothioate oligodeoxynucleotides (ODN; Sigma-Genosys) complementary to SOCS3 mRNA. A sense phosphorothioate ODN served as control. As expected, the response pattern of sense ODN treated monocytes was identical to that of control cells. In marked contrast, Ac2-26 was not able to interfere with IL-6-induced STAT3 activation in antisense ODN-treated monocytes (Fig 7B). These findings demonstrate that Ac2-26 negatively regulates IL-6 signalling via upregulation of SOCS3.
Figure 7. Ac2-26 downregulates IL-6 signalling via upregulation of SOCS3.
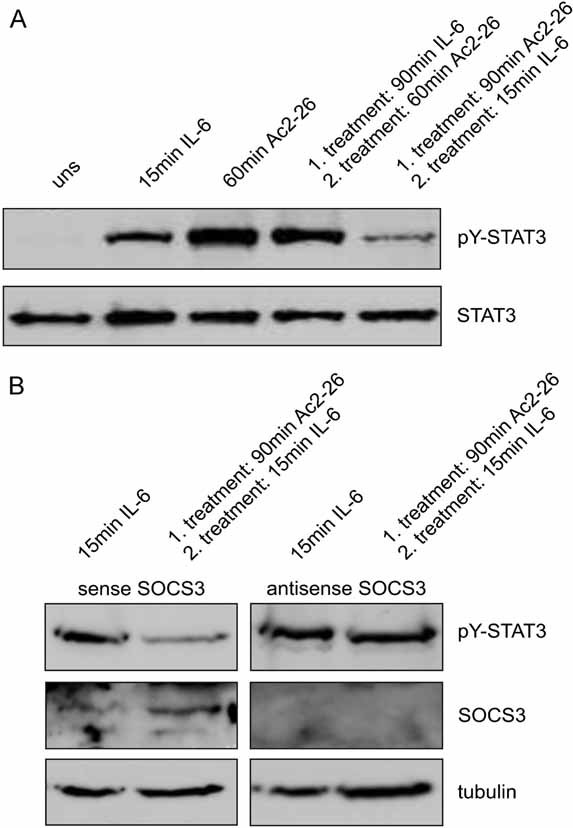
- Ac2-26 treatment dampens IL-6-induced STAT3 activation. Cell lysates of monocytes, which were stimulated for the indicated periods of time with either IL-6, Ac2-26 or a combination of both (90 min Ac2-26 followed by 15 min IL-6 or 90 min IL-6 followed by 60 min Ac2-26 as a control for the sensitivity of the cells towards activation), were analysed for the amount of pY-STAT3 by immunoblotting. Membranes were reprobed with a tubulin antibody to ensure equal loading.
- SOCS3 is required for mediating the inhibitory effects of Ac2-26 on IL-6 signalling. Monocytes depleted of cellular SOCS3 by phosphorothioate ODN complementary to SOCS3 mRNA (antisense) or treated with sense ODN employed as a control were treated with either IL-6 or Ac2-26/IL-6 as above. The cellular amounts of pY-STAT3 and SOCS3 were analysed by immunoblotting. Tubulin was used as an internal control for equal loading. Representative blots of at least three independent experiments are shown.
DISCUSSION
Apoptotic cells present conserved specific sets of molecules, the apoptotic cell-ACAMPs, which in addition to functioning as ‘eat-me’ signals for efficient apoptotic clearance counteract inflammation. Similar to the recognition of pathogen-associated molecular patterns (PAMPs) by PRRs on the cell surface, ACAMPs are recognized by the engulfing cell (Gregory & Devitt, 2004). However, the receptors involved are not well defined and the signal transduction pathways elicited by ACAMPs that lead to anti-inflammatory modulation are mostly unknown. Cytokine receptor signalling, on the other hand, which crucially depends on the JAK-STAT pathway, is a well understood signal transduction cascades (Jove, 2000). Activated JAKs phosphorylate the receptor domains to allow subsequent binding and phosphorylation of the STAT transcription factors, which in turn translocate into the nucleus to activate gene expression. Activation of STAT3 mediates the anti-inflammatory effects of IL-10 and, accordingly, expression of constitutively active STAT3 has been reported to mimic the suppressive effect of IL-10 (Williams et al, 2007). Among the STAT3-regulated genes is SOCS3, which functions as a feedback inhibitor of the IL-6 receptor by binding to gp130 (Croker et al, 2008; Lang et al, 2003). Our results demonstrate that factors released from apoptotic cells induce activation of the JAK/STAT3 pathway and subsequent upregulation of SOCS3 expression and that this in part involves PRRs of the FPR family, the bona fide annexin A1 receptors.
We hypothesized that the anti-inflammatory protein annexin A1, which is released during neutrophil apoptosis through yet undefined mechanisms, could function as an ACAMP to actively counter-regulate the inflammatory activation of monocytes. In line with previous findings showing that apoptotic cells release annexin A1 (Arur et al, 2003; Fan et al, 2004; Gan et al, 2008; Maderna et al, 2005; Scannell et al, 2007), we could detect annexin A1 in supernatants of apoptotic granulocytes, with a considerable amount of the released protein being processed by proteolytic cleavage. Although we cannot definitively rule out that some of the released annexin A1 may arise from activated, non-apoptotic PMNs, this appears very unlikely, for significant release of annexin A1 from freshly isolated granulocytes was previously shown to occur only upon adhesion of PMNs to endothelial cells or, to a minor extent, upon priming with cytochalasin B and subsequent chemokine stimulation of PMN in the absence of an endothelial monolayer (Rescher et al, 2006). Since the anti-inflammatory properties of annexin A1 are retained in the N-terminal peptide, which can be generated in vivo from the full-length protein by neutrophil proteases (Rescher et al, 2006; Vong et al, 2007), proteolytic release of this domain might be required to obtain full functional activity. Evidence for the presence of annexin A1 peptide derivatives in apoptotic cell supernatants has also come from nanoelectrospray liquid chromatography mass spectrometry analysis (Scannell et al, 2007). However, direct detection of the actual agonistic N-terminal fragment has remained elusive, since common immunological methods such as Western blotting and ELISA fail to yield reliable signals due to the lack of antibodies of high specificity against that region. The peptidomimetic of annexin A1, the N-terminal peptide Ac2-26, which corresponds to the sequence that can be released by the proteolytic cleavage, was therefore used in many studies including ours. It was able to negatively regulate LPS-induced TNF-α release through activation of STAT3 and in all our assays shows the same effects as total apoptotic cell supernatants. Since annexin A1 derivatives are part of these supernatants and since antagonists of the annexin A1 receptors (FPRs) partially block the anti-inflammatory effect of the supernatants we conclude that annexin A1 is a component of the ACAMPs. It has to be noted, however, that the block by FPR antagonists is only partial indicating that apoptotic cell supernatants contain in addition to annexin A1 other components contributing to their anti-inflammatory activity.
Annexin A1 has been originally identified as a mediator of the inhibitory actions of glucocorticoids on the inflammatory response and its potential to counteract inflammation has been shown in a number of experimental model systems (Perretti & Flower, 2004). However, a molecular explanation of the beneficial effects of annexin A1 and its peptidomimetics on inflammation was provided only recently by the identification of the FPRs as receptors for annexin A1 (Ernst et al, 2004; Perretti et al, 2002; Walther et al, 2000). The three heptahelical G-protein coupled human FPRs, FPR1, FPR2/ALX and FPR3, are a class of PRRs with a broad spectrum of ligands ranging from formylated bacterial peptides to viral peptides as well as endogenous ligands (Le et al, 2002; Migeotte et al, 2006). Recognition of host ligands such as ACAMPs through PRRs is emerging as a common theme, suggesting that signalling through PRRs might not be limited to the detection of invading microbes but be part of a complex scenario responsible for the balanced innate immune response (Franc et al, 1999; Gregory & Devitt, 2004). Correspondingly, the FPRs have been implicated in counter-regulation of the immune response via recognition of their endogenous ligands, one such scenario identified here being the annexin A1-FPR axis.
Whereas PAMPs activate their cognate PRRs to elicit pro-inflammatory signalling, ACAMPs binding to PRRs induces anti-inflammatory responses (Jeannin et al, 2008). To mechanistically understand the counter-regulatory consequences of the Ac2-26 interaction with FPRs on inflammation, we analysed the involvement of the JAK-STAT-SOCS signalling modules. We found cross-inhibition of IL-6 induced STAT3 activation by the anti-inflammatory annexin peptide, whereas STAT3 activation induced by the annexin peptide is not affected by prior IL-6 challenge.
Recent data suggest that IL-6/gp130 signalling through STAT3 also controls leukocyte infiltration in inflammatory arthritis (Nowell et al, 2009), and SOCS3 has been shown to negatively regulate IL-6 signalling in vivo (Croker et al, 2003). Thus, although both ligand-bound IL-10R and IL-6R can activate SOCS3 in a STAT3-dependent manner, IL-6R is incapable of mounting the anti-inflammatory response probably due to different sensitivities of the respective cytokine receptors towards SOCS3, that is, IL-6R is itself SOCS3 controlled whereas IL-10R is not (Murray, 2007). The results of our study suggest that annexin A1 has the ability to impair subsequent IL-6 signalling activity through the upregulation of the negative feedback inhibitor SOCS3. Blocking the JAK/STAT3 pro-inflammatory axis could therefore define an important mechanism that contributes to the anti-inflammatory properties of annexin A1.
Activation of the JAK-STAT signalling pathway is not unique to cytokines, but also initiated by growth factors and GPCRs, although the activation mechanism is still poorly described (Sun et al, 2007). FPR2/ALX activation with the synthetic WKYMVm peptide was shown to cause STAT3 serine, but not tyrosine, phosphorylation, via PLD-mediated ERK-activation, without concomitant JAK activation (Jo et al, 2004). The annexin A1 peptide is the only endogenous pan-FPR agonist known to date. Thus, in the context of apoptotic cell clearance, cleaved annexin A1 will also activate FPR2. Our observation that the Ac2-26-mediated and MAP kinase-dependent JAK activation results in STAT3 tyrosine phosphorylation thus points to different ligand textures of the FPR agonists, resulting in different activation patterns. Alternatively, parallel activation of the three FPR family members could give rise to alternative signalling patterns, possibly because of the formation of heterodimers. A broad range of FPR agonists have been identified in recent years (Bürli et al, 2006; Edwards et al, 2005; Nanamori et al, 2004; Schepetkin et al, 2007; Wan et al, 2007). However, elucidation of the relative contribution of the different receptor subtypes (FPR1, 2 or 3) is hampered by the lack of subtype-selective agonists and antagonists and thus remains to be clarified.
It had been reported previously that suppression of TNF-α release is caused by the paracrine action of the anti-inflammatory cytokine TGF-β which is produced upon ingestion of apoptotic cells by macrophages. Release of TGF-β seems to require ligation of the macrophage phosphatidylserine receptor by phosphatidylserine exposed on the cell surface of apoptotic cells (Fadok et al, 1998; Huynh et al, 2002; Stuart et al, 2002). Consistent with these observations, we could not detect elevated levels of TGF-β following exposure of monocytes to apoptotic cell culture supernatants which did not contain whole cells (not shown). Previous work has suggested that annexin A1 is involved in facilitating the interaction with the engulfing cells (Gregory & Devitt, 2004). Our results suggest that the annexin A1-mediated counter-regulation of the LPS-driven inflammatory response can be functionally separated from tethering apoptotic cells to the phagocyte surface since the anti-inflammatory signalling can be elicited by cell-free conditioned media or the Ac2-26 annexin A1 N-terminal peptide. In conclusion, our findings identify the key anti-inflammatory mediator annexin A1 as part of the counter-regulatory circuits elicited by apoptotic cells and couple its anti-inflammatory action to well-defined intracellular signalling cascades.
MATERIALS AND METHODS
Isolation of human leukocytes
Human peripheral blood monocytes and PMNs were isolated from buffy coats as described (Ernst et al, 2004; Lange et al, 2007). Monocytes were cultured overnight in McCoy's medium supplemented with 30% fetal calf serum (FCS), 100 Units/ml penicillin–streptomycin, 2 mM l-glutamine and 1% non-essential amino acids (Cambrex) in Teflon bags or Teflon-coated lumox cell culture dishes (Greiner Bio-One) to prevent firm adhesion. PMNs were isolated by dextran sedimentation, and remaining erythrocytes were removed by hypotonic lysis. To obtain supernatants of apoptotic PMNs, 5 × 106 cells/ml were cultured overnight in serum-free RPMI supplemented with 100 Units/ml penicillin–streptomycin, 2 mM l-glutamine and 1% non-essential amino acids. Supernatants were cleared by centrifugation and used directly.
Cytokine production and inhibitors
Monocytes were treated with either supernatants from apoptotic PMNs, LPS (10ng/ml, Calbiochem), annexin A1 N-terminal peptide Ac2-26 (50µM, Cambridge Research Biomedicals), JAK-Inhibitor 1 (5µM, Calbiochem) STAT3 Inhibitor VI (20 µM, Calbiochem) or vehicle, respectively, for different periods of time. Monocyte supernatants were obtained after the indicated periods of time, cleared by centrifugation and stored at −70°C. The release of TNF-α was determined by ELISA according to the manufacturer's protocol.
Monocyte stimulation for Western blot analysis
Monocytes (2.5 × 106 cells/200 µl reaction) were resuspended in Dulbecco's Modified Eagle's Medium (DMEM) (PAA Laboratories) supplemented with 100 Units/ml penicillin–streptomycin, 2 mM l-glutamine and 1% non-essential amino acids and either pre-incubated with inhibitors, or vehicle at 37°C. Cells were then stimulated with either the annexin A1 N-terminal peptide Ac2-26, 10 µm SB203580 (Merck Biosciences), 10 µM UO126 (Promega), 0.25% n- or tert-butanol (AppliChem) or vehicle for different periods of time. Cells were lysed with hot SDS sample buffer, resolved by SDS–PAGE and transferred to PVDF membrane (Millipore). Membranes were incubated overnight at 4°C with the following primary antibodies: rabbit polyclonal anti-phospho-ERK1/2 (p44/42), rabbit polyclonal anti-pTYR705-STAT3 (all from Cell Signalling) or rabbit polyclonal anti-SOCS3, 1:200 (Santa Cruz). To ensure equal loading, blots were stripped and reprobed for total Erk1/2 and total STAT3 (Cell Signalling). Protein bands were visualized according to standard ECL protocols.
Nuclear protein extraction and electrophoretic mobility shift assay
Cells were lysed in hypotonic buffer (10 mM KCl, 10 mM HEPES, pH 7.9; 0.1 mM EDTA, pH 7.4; 0.1 mM EGTA, pH 8.0; 1 mM DTT, 0.025% NP-40, protease inhibitor mix, Roche), and nuclei were harvested by centrifugation (1000 × g for 10 min). Nuclear fractions were prepared as described (Medvedev et al, 1999). In brief, nuclear proteins were extracted by incubation in extraction buffer (25% glycerol, 20 mM HEPES, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, protease inhibitor mix) for 30 min on ice and a subsequent centrifugation (10,000 × g, 10 min). Protein concentration was determined using Bradford assay. The extracted proteins were stored at −20°C.
The paper explained
PROBLEM
Inflammation is a complex biological response of the immune system to infection and tissue damage. The first line of the protection is assured by innate immune cells, which are able to recognize and defeat pathogens. Apoptosis of neutrophils has been recognized in recent years as an element of the protective part of the immune system. Apoptotic cells release multiple soluble mediators and translocated intracellular molecules that not only mediate apoptotic cell clearance but also provide anti-inflammatory stimuli, thereby limiting the degree of inflammation and promoting resolution. Annexin A1 is the important endogenous, glucocorticoid-inducible anti-inflammatory protein that operates during the resolution of inflammation. This protein is strongly expressed in neutrophils and externalized during apoptosis. The effects of this released annexin A1 and, more generally, the action of factors released from apoptotic cells on their prime target cells of the innate immune system, monocytes and macrophages, are poorly understood.
RESULTS
Annexin A1 externalization from apoptotic neutrophils was accompanied by cleavage of the protein and the release of N-terminal peptides. Supernatants from apoptotic neutrophils or the purified N-terminal annexin A1 peptidomimetic Ac2-26 significantly reduced IL-6 signalling and the release of TNF-α from endotoxin-challenged monocytes. Ac2-26 activated STAT3 in a JAK-dependent manner, resulting in upregulated SOCS3 levels, and depletion of SOCS3 reversed the Ac2-26-mediated inhibition of IL-6 signalling.
IMPACT
Our data highlight the significance of Annexin A1 as part of immunosuppressive effects of apoptotic cells and couple its anti-inflammatory action to JAK/STAT3/SOCS3 signalling cascades.
Electrophoretic mobility shift assays were performed using an EMSA-Kit (Panomics, Redwood, CA). Briefly, 5 µg of nuclear extract were used for protein/DNA binding reactions. A biotinylated, double-stranded oligonucleotide (dsDNA) was used to detect STAT3. The DNA-binding reactions were performed in a volume of l0 µl binding buffer provided by the manufacturer containing poly d(I–C) at 15°C. After 30 min incubation, samples were separated through a 6% non-denaturing polyacrylamide gel for 90 min at 120 V and transferred for 30 min at 300 mA to a Biodyne B-Nylon membrane (Pall Life Sciences). After cross-linking under UV light, bands were visualized by ECL detection.
Real time quantitative PCR
Total cellular RNA of stimulated monocytes (107 cells/sample) was isolated using the RNeasy Kit (Qiagen, Germany). Reverse transcription was performed using 1 µg of isolated total RNA and the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Germany). The cDNA preparations (200 ng/sample) were subjected to real-time PCR using an ABI Prism 7900 HT RT-PCR system provided by the Integrated Functional Genomic unit of the University of Muenster Medical School. Primers for the genes of interest were obtained from Qiagen (Quantitect Primer Assay Kit, Qiagen Germany). Real time PCR analysis was performed using the 2−ddCT method (Livak & Schmittgen, 2001) and employed 1XPlatinum SYBR Green qPCR Supermix-UDG with 6-carboxy-X-rhodamine reference dye (Invitrogen, Germany). The PCR was set up as follows: 50°C for 2 min, 95°C for 2 min, then 40 cycles: 95°C for 15 s, 60°C for 60 s. All reactions were carried out in duplicate. GAPDH (Quantitect Primer Assay Kit) was used as the housekeeping gene to normalize expression levels of the genes of interest.
Antisense experiments
Knockdown of SOCS3 was achieved with phosphorothioate-modified antisense ODN. Monocytes were treated for 24 h with 1 µM of the specific antisense (5′-C*G*GGAAACTTGCTGTGGGTGACC*A*T-3′) oligonucleotide in DMEM or McCoy's medium supplemented with 10% FCS. The sense sequence (5′-A*T*GGTCACCCACAGCAAGTTTCC*C*G-3′) served as control. After treatment, the cells were resuspended in DMEM supplemented with 100 Units/ml penicillin–streptomycin, 2 mM l-glutamine and 1% non-essential amino acids. ODN-treated monocytes (2.5 × 106 cells/200 µl reaction) were stimulated with Ac2-26 for indicated periods of time. STAT3 phosphorylation and SOCS3 expression were analysed with specific antibodies by Western Blot.
Statistical analysis
Statistical analysis of data was performed by either two-sided t-test or one-way ANOVA, followed by Bonferroni post-test for multiple comparisons. p < 0.05 was considered statistically significant.
Acknowledgments
We thank Frauke Brinkmann and Andreas Wilbers for technical assistance. The project was supported by the Interdisciplinary Clinical Research Centre (IZKF, RE2/033/04), the fund ‘Innovative Medical Research’ (RE120522) of the University of Münster Medical School, and the German Research Foundation (DFG, RE2611-1). We also thank the IFG (Muenster/Germany) for providing the ABI Prism 7900 HT RT-PCR system.
The authors declare that they have no conflict of interest.
Author contributions
UR designed the research project. DP, JG and DJK designed and performed experiments, analysed data and prepared the figures; UR and VG wrote and edited the manuscript.
For more information
Institute of Medical Biochemistry:
References
- Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, Mohler W, Han DK. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell. 2003;4:587–598. doi: 10.1016/s1534-5807(03)00090-x. [DOI] [PubMed] [Google Scholar]
- Benkhart EM, Siedlar M, Wedel A, Werner T, Ziegler-Heitbrock HW. Role of STAT3 in lipopolysaccharide-induced IL-10 gene expression. J Immunol. 2000;165:1612–1617. doi: 10.4049/jimmunol.165.3.1612. [DOI] [PubMed] [Google Scholar]
- Bürli RW, Xu H, Zou X, Muller K, Golden J, Frohn M, Adlam M, Plant MH, Wong M, McElvain M, Regal K, Viswanadhan VN, Tagari P, Hungate R. Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorg Med Chem Lett. 2006;16:3713–3718. doi: 10.1016/j.bmcl.2006.04.068. [DOI] [PubMed] [Google Scholar]
- Byrne A, Reen DJ. Lipopolysaccharide induces rapid production of IL-10 by monocytes in the presence of apoptotic neutrophils. J Immunol. 2002;168:1968–1977. doi: 10.4049/jimmunol.168.4.1968. [DOI] [PubMed] [Google Scholar]
- Carl VS, Gautam JK, Comeau LD, Smith MF., Jr Role of endogenous IL-10 in LPS-induced STAT3 activation and IL-1 receptor antagonist gene expression. J Leukoc Biol. 2004;76:735–742. doi: 10.1189/jlb.1003526. [DOI] [PubMed] [Google Scholar]
- Cirino G, Cicala C, Sorrentino L, Ciliberto G, Arpaia G, Perretti M, Flower RJ. Anti-inflammatory actions of an N-terminal peptide from human lipocortin 1. Br J Pharmacol. 1993;108:573–574. doi: 10.1111/j.1476-5381.1993.tb12843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–422. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BS, Bologa C, Young SM, Balakin KV, Prossnitz ER, Savchuck NP, Sklar LA, Oprea TI. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol Pharmacol. 2005;68:1301–1310. doi: 10.1124/mol.105.014068. [DOI] [PubMed] [Google Scholar]
- Ernst S, Lange C, Wilbers A, Goebeler V, Gerke V, Rescher U. An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J Immunol. 2004;172:7669–7676. doi: 10.4049/jimmunol.172.12.7669. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Krahling S, Smith D, Williamson P, Schlegel RA. Macrophage surface expression of annexins I and II in the phagocytosis of apoptotic lymphocytes. Mol Biol Cell. 2004;15:2863–2872. doi: 10.1091/mbc.E03-09-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franc NC, White K, Ezekowitz RA. Phagocytosis and development: back to the future. Curr Opin Immunol. 1999;11:47–52. doi: 10.1016/s0952-7915(99)80009-0. [DOI] [PubMed] [Google Scholar]
- Galdiero M, Vitiello M, D'Isanto M, Raieta K, Galdiero E. STAT1 and STAT3 phosphorylation by porins are independent of JAKs but are dependent on MAPK pathway and plays a role in U937 cells production of interleukin-6. Cytokine. 2006;36:218–228. doi: 10.1016/j.cyto.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Gan H, Lee J, Ren F, Chen M, Kornfeld H, Remold HG. Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat Immunol. 2008;9:1189–1197. doi: 10.1038/ni.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavins FN, Yona S, Kamal AM, Flower RJ, Perretti M. Leukocyte antiadhesive actions of annexin 1: ALXR- and FPR-related anti-inflammatory mechanisms. Blood. 2003;101:4140–4147. doi: 10.1182/blood-2002-11-3411. [DOI] [PubMed] [Google Scholar]
- Glenney JR, Jr, Tack B, Powell MA. Calpactins: two distinct Ca++-regulated phospholipid- and actin-binding proteins isolated from lung and placenta. J Cell Biol. 1987;104:503–511. doi: 10.1083/jcb.104.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory CD, Devitt A. The macrophage and the apoptotic cell: an innate immune interaction viewed simplistically? Immunology. 2004;113:1–14. doi: 10.1111/j.1365-2567.2004.01959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon R, Croxtall JD, Getting SJ, Roviezzo F, Yona S, Paul-Clark MJ, Gavins FN, Perretti M, Morris JF, Buckingham JC, et al. Aberrant inflammation and resistance to glucocorticoids in annexin 1-/- mouse. FASEB J. 2003;17:253–255. doi: 10.1096/fj.02-0239fje. [DOI] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeannin P, Jaillon S, Delneste Y. Pattern recognition receptors in the immune response against dying cells. Curr Opin Immunol. 2008;20:530–537. doi: 10.1016/j.coi.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Jo EJ, Lee HY, Kim JI, Kang HK, Lee YN, Kwak JY, Bae YS. Activation of formyl peptide receptor-like 1 by WKYMVm induces serine phosphorylation of STAT3, which inhibits its tyrosine phosphorylation and nuclear translocation induced by hydrogen peroxide. Life Sci. 2004;75:2217–2232. doi: 10.1016/j.lfs.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Jove R. Preface: STAT signaling. Oncogene. 2000;19:2466–2467. doi: 10.1038/sj.onc.1203549. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/s0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ. SOCS3 regulates the plasticity of gp130 signaling. Nat Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- Lange C, Starrett DJ, Goetsch J, Gerke V, Rescher U. Transcriptional profiling of human monocytes reveals complex changes in the expression pattern of inflammation-related genes in response to the annexin A1-derived peptide Ac1-25. J Leukoc Biol. 2007;82:1592–1604. doi: 10.1189/jlb.0307158. [DOI] [PubMed] [Google Scholar]
- Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23:541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- Leaman DW, Leung S, Li X, Stark GR. Regulation of STAT-dependent pathways by growth factors and cytokines. FASEB J. 1996;10:1578–1588. [PubMed] [Google Scholar]
- Lim LH, Solito E, Russo-Marie F, Flower RJ, Perretti M. Promoting detachment of neutrophils adherent to murine postcapillary venules to control inflammation: effect of lipocortin 1. Proc Natl Acad Sci USA. 1998;95:14535–14539. doi: 10.1073/pnas.95.24.14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Amin R, Gallicano GI, Glasgow E, Jogunoori W, Jessup JM, Zasloff M, Marshall JL, Shetty K, Johnson L, et al. The STAT3 inhibitor NSC 74859 is effective in hepatocellular cancers with disrupted TGF-β signalling. Oncogene. 2009;28:961–972. doi: 10.1038/onc.2008.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Maderna P, Yona S, Perretti M, Godson C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac(2-26) J Immunol. 2005;174:3727–3733. doi: 10.4049/jimmunol.174.6.3727. [DOI] [PubMed] [Google Scholar]
- McDermott M, Wakelam MJ, Morris AJ. Phospholipase D. Biochem Cell Biol. 2004;82:225–253. doi: 10.1139/o03-079. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Blanco JC, Qureshi N, Vogel SN. Limited role of ceramide in lipopolysaccharide-mediated mitogen-activated protein kinase activation, transcription factor induction, and cytokine release. J Biol Chem. 1999;274:9342–9350. doi: 10.1074/jbc.274.14.9342. [DOI] [PubMed] [Google Scholar]
- Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006;17:501–519. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- Nanamori M, Cheng X, Mei J, Sang H, Xuan Y, Zhou C, Wang MW, Ye RD. A novel nonpeptide ligand for formyl peptide receptor-like 1. Mol Pharmacol. 2004;66:1213–1222. doi: 10.1124/mol.104.004309. [DOI] [PubMed] [Google Scholar]
- Nowell MA, Williams AS, Carty SA, Scheller J, Hayes AJ, Jones GW, Richards PJ, Slinn S, Ernst M, Jenkins BJ, et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J Immunol. 2009;182:613–622. doi: 10.4049/jimmunol.182.1.613. [DOI] [PubMed] [Google Scholar]
- Perretti M, Flower RJ. Annexin 1 and the biology of the neutrophil. J Leukoc Biol. 2004;76:25–29. doi: 10.1189/jlb.1103552. [DOI] [PubMed] [Google Scholar]
- Perretti M, Ahluwalia A, Harris JG, Goulding NJ, Flower RJ. Lipocortin-1 fragments inhibit neutrophil accumulation and neutrophil-dependent edema in the mouse. A qualitative comparison with an anti-CD11b monoclonal antibody. J Immunol. 1993;151:4306–4314. [PubMed] [Google Scholar]
- Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, Solito E, Serhan CN. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med. 2002;8:1296–1302. doi: 10.1038/nm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescher U, Gerke V. Annexins—unique membrane binding proteins with diverse functions. J Cell Sci. 2004;117:2631–2639. doi: 10.1242/jcs.01245. [DOI] [PubMed] [Google Scholar]
- Rescher U, Goebeler V, Wilbers A, Gerke V. Proteolytic cleavage of annexin 1 by human leukocyte elastase. Biochim Biophys Acta. 2006;1763:1320–1324. doi: 10.1016/j.bbamcr.2006.08.041. [DOI] [PubMed] [Google Scholar]
- Rosengarth A, Gerke V, Luecke H. X-ray structure of full-length annexin 1 and implications for membrane aggregation. J Mol Biol. 2001;306:489–498. doi: 10.1006/jmbi.2000.4423. [DOI] [PubMed] [Google Scholar]
- Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- Scannell M, Flanagan MB, deStefani A, Wynne KJ, Cagney G, Godson C, Maderna P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. 2007;178:4595–4605. doi: 10.4049/jimmunol.178.7.4595. [DOI] [PubMed] [Google Scholar]
- Schepetkin IA, Kirpotina LN, Khlebnikov AI, Quinn MT. High-throughput screening for small-molecule activators of neutrophils: identification of novel N-formyl peptide receptor agonists. Mol Pharmacol. 2007;71:1061–1074. doi: 10.1124/mol.106.033100. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- Siddiquee K, Zhang S, Guidad WC, Blaskovich MA, Greedy B, Lawrence HR, Yip MLR, Joveh R, McLaughlin MM, Lawrence NJ, et al. Selective chemical probe inhibitor of STAT3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart LM, Lucas M, Simpson C, Lamb J, Savill J, Lacy-Hulbert A. Inhibitory effects of apoptotic cell ingestion upon endotoxin-driven myeloid dendritic cell maturation. J Immunol. 2002;168:1627–1635. doi: 10.4049/jimmunol.168.4.1627. [DOI] [PubMed] [Google Scholar]
- Sun Y, McGarrigle D, Huang XY. When a G protein-coupled receptor does not couple to a G protein. Mol Biosyst. 2007;3:849–854. doi: 10.1039/b706343a. [DOI] [PubMed] [Google Scholar]
- Thompson JE. JAK protein kinase inhibitors. Drug News Perspect. 2005;18:305–310. doi: 10.1358/dnp.2005.18.5.904198. [DOI] [PubMed] [Google Scholar]
- Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- Vong L, D'Acquisto F, Pederzoli-Ribeil M, Lavagno L, Flower RJ, Witko-Sarsat V, Perretti M. Annexin 1 cleavage in activated neutrophils: a pivotal role for proteinase 3. J Biol Chem. 2007;282:29998–30004. doi: 10.1074/jbc.M702876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther A, Riehemann K, Gerke V. A novel ligand of the formyl peptide receptor: annexin I regulates neutrophil extravasation by interacting with the FPR. Mol Cell. 2000;5:831–840. doi: 10.1016/s1097-2765(00)80323-8. [DOI] [PubMed] [Google Scholar]
- Williams LM, Sarma U, Willets K, Smallie T, Brennan F, Foxwell BM. Expression of constitutively active STAT3 can replicate the cytokine-suppressive activity of interleukin-10 in human primary macrophages. J Biol Chem. 2007;282:6965–6975. doi: 10.1074/jbc.M609101200. [DOI] [PubMed] [Google Scholar]
- Wan HX, Zhou C, Zhang Y, Sun M, Wang X, Yu H, Yang X, Ye RD, Shen JK, Wang MW. Discovery of Trp-Nle-Tyr-Met as a novel agonist for human formyl peptide receptor-like 1. Biochem Pharmacol. 2007;74:317–326. doi: 10.1016/j.bcp.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Yang YH, Morand EF, Getting SJ, Paul-Clark M, Liu DL, Yona S, Hannon R, Buckingham JC, Perretti M, Flower RJ. Modulation of inflammation and response to dexamethasone by annexin 1 in antigen-induced arthritis. Arthritis Rheum. 2004;50:976–984. doi: 10.1002/art.20201. [DOI] [PubMed] [Google Scholar]