

Abstract
Hyaline Fibromatosis Syndrome (HFS) is a human genetic disease caused by mutations in the anthrax toxin receptor 2 (or cmg2) gene, which encodes a membrane protein thought to be involved in the homeostasis of the extracellular matrix. Little is known about the structure and function of the protein or the genotype–phenotype relationship of the disease. Through the analysis of four patients, we identify three novel mutants and determine their effects at the cellular level. Altogether, we show that missense mutations that map to the extracellular von Willebrand domain or the here characterized Ig-like domain of CMG2 lead to folding defects and thereby to retention of the mutated protein in the endoplasmic reticulum (ER). Mutations in the Ig-like domain prevent proper disulphide bond formation and are more efficiently targeted to ER-associated degradation. Finally, we show that mutant CMG2 can be rescued in fibroblasts of some patients by treatment with proteasome inhibitors and that CMG2 is then properly transported to the plasma membrane and signalling competent, identifying the ER folding and degradation pathway components as promising drug targets for HFS.
Keywords: CMG2, conformational disease, protein folding
INTRODUCTION
Hyaline Fibromatosis Syndrome (HFS) is an autosomal recessive disorder triggered by mutations in Capillary Morphogenesis Gene 2 (cmg2) (Dowling et al, 2003; Hanks et al, 2003; Stucki et al, 2001). The most severe form of the disease, called infantile systemic hyalinosis (ISH [MIM 236490]), manifests in the first weeks of life by painful swelling of the skin and the large joints as well as oedema of the skin. Although the disease is named after the accumulation of hyaline material, possibly collagen VI (Glover et al, 1991; Katagiri et al, 1996; Tanaka et al, 2009), in the skin and other organs (Fayad et al, 1987; Keser et al, 1999; Landing & Nadorra, 1986), this might be a secondary consequence of changes in the basal membrane of capillary vessels (Stucki et al, 2001). Over a time of months to a few years, affected individuals develop cutaneous tumours over regions of chronic mechanical stimulation, such as the gingiva, the perianal region, the alae nasi, the external ears and the nuchal region on the skull. In addition, patients suffer from flexion contractures of the joints and osteolytic lesions of the long bones and distal phalanges (Fayad et al, 1987; Keser et al, 1999) (also see http://www.orpha.net/). ISH may lead to death in the first 2 years of life because of chronic malabsorption and diarrhoea or pulmonary infection. Patients suffering from the milder form, juvenile hyaline fibromatosis (JHF [MIM 228600]), usually do not present these complications and do not have the infantile painful phase, but do present cutaneous tumours and debilitating loss of joint mobility.
The ANTXR2/cmg2 gene encodes for a type I membrane protein, ANTXR2/CMG2, which harbours an extracellular von Willebrand A domain (vWA), followed by an uncharacterized immunoglobulin-like domain (Sun & Collier, 2010), a transmembrane domain and finally a 148 residue cytosolic tail (van der Goot & Young, 2009). The exact function of this protein has not been established. The vWA was shown to bind collagen IV and laminin (Bell et al, 2001), suggesting that CMG2—nomenclature further used here—is involved in interacting with the extracellular matrix. In agreement with its initial identification in a gene profiling study during in vitro capillary formation (Bell et al, 2001), it was recently shown that silencing cmg2 leads to a decrease in the proliferation of human umbilical vein endothelial cells (HUVECs) and in the ability to form capillaries in 3D matrices (Reeves et al, 2009). The best-characterized role of CMG2 is, however, to be the receptor for the anthrax toxin (Liu et al, 2009; van der Goot & Young, 2009). CMG2 enables the anthrax toxin to bind to cells, be internalized and reach the cytosol where it exerts its toxic function (van der Goot & Young, 2009).
We have recently shown that HFS mutations mapping to the vWA result in a loss of function due to retention of the protein in the endoplasmic reticulum (ER; Deuquet et al, 2009). Here, we have performed a genetic analysis of four new HFS patients and analysed the consequences of these mutations at the molecular level. Three of the patients were homo- or heterozygous for frame-shift mutations in exon 13. We show that these frame-shift mutations lead to a decrease in the mRNA levels of cmg2, presumably due to degradation via the nonsense-mediated mRNA decay (NMD) pathway (Rebbapragada & Lykke-Andersen, 2009). We also identified three novel missense mutations (p.C39F, p.V310F and p.C315W), which did not affect the corresponding mRNA levels but had a severe impact at the protein level. Residues Val-310 and Cys-315 localize to the Ig-like domain of CMG2, for which we now provide a model. We found that mutations in the Ig-like domain affect folding and disulphide bond formation in the ER, thereby leading to ER retention. Interestingly, depending on the location of the mutation (vWA or Ig-like domain), CMG2 was differentially targeted to the ER-associated degradation (ERAD) pathway. Using patient-derived fibroblasts, we show that partial rescue of CMG2 and targeting of a functional protein to the cell surface can be achieved through treatment with proteasome inhibitors such as MG132 or bortezomib (BZ; Santa Cruz), a substance currently in clinical trials (Phuphanich et al, 2010), indicating that these compounds are potential therapeutic drugs to treat HFS.
RESULTS
Description and genetic analysis of the HFS patients
We performed the molecular diagnosis of four patients from four different families; the neonatal presentation of all index patients initially raised suspicion of the most severe form of the disease ISH (see Supplementary Information for more detailed description of each patient, Table 1). In particular, painful contractures were the major clinical sign in each case at or shortly after birth, leading to progressive joint limitations and delay of motor development; intellectual development was normal. Typical skin eruptions and mucocutaneous lesions became apparent early in life. Because of feeding difficulties, gingivectomy was performed in at least three of the patients. Despite the similar initial clinical phenotype, the disease followed a clearly different course for each patient after birth (Supplementary Information). The most severe disease manifestation was observed in Patient 3 who showed the full-blown picture of ISH with recurrent, intractable diarrhoea and episodes of intermittent systemic inflammatory activity (SIRS), causing acute fatal circulatory failure in infancy. Clinical evidence of multiorgan involvement was also observed in Patient 4 with the diagnosis of protein-losing enteropathy at the age of 10 months. His general condition, however, remained stable under supportive therapy, and no instance of SIRS or generalized bacterial infection has occurred up to his current age of 3 years. In contrast, neither Patient 1 nor Patient 2, who showed the most favourable clinical course, developed further complications of ISH. Our patients thus collectively represent a continuum of phenotypes associated with HFS, ranging from a fatal multisystem disorder to a milder, yet disabling, oligosymptomatic connective tissue disease. We were interested in finding molecular explanations for these clinical differences with the aim of identifying genotype–phenotype correlations.
Table 1.
Clinical features of HFS patients
| Age at genetic diagnosis | Painful contractions in infancy | Skin eruptions, gingival hypertrophy | Failure to thrive, short stature | Intractable, recurrent diarrhoea | Systemic inflammatory reaction, recurrent infections | |
|---|---|---|---|---|---|---|
| Patient 1 (individual II.1, family 1) | 2 years | ++ | ++ | ++ | − | − |
| Patient 2 (individual II.2, family 2) | 14 years | + | ++ | + | − | − |
| Patient 3 (individual II.2, family 3) | 6 weeks | +++ | + | +++ | ++ | +++ |
| Patient 4 (individual II.2, family 4) | 3 years | +++ | ++ | +++ | +++ | − |
Note that the age of genetic diagnostics differs from the age of symptom onset. For example, some symptoms were apparent shortly after or at birth for Patients 1, 2 and 3. Patient 3 died before the age of 2, as often for the infantile form of HFS.
In the least affected families 1 and 2, different compound heterozygous mutations of cmg2 were detected (Table 2). Patient 1 carries a c.116G>T transversion predicted to cause a novel p.C39F amino acid substitution in the amino terminus of CMG2. In the second allele, a previously described (Hanks et al, 2003) c.1074delT single nucleotide deletion in exon 13 was found, which modifies the open reading frame by a frame shift leading to a change in the cytosolic tail of the protein and a premature stop (Fig 1). Patient 2 carries a missense mutation resulting in a c.928G>T transversion, leading to substitution of valine 310 in the ectodomain with a phenylalanine (Fig. 1). In the second allele, a single base insertion (c.1073_1074insC) was found again in exon 13, also leading to a frame shift and a premature stop. Both cases of severe HFS in families 3 and 4 proved to be associated with homozygous mutations. Patient 3 carried a biallelic novel c.945T>G transversion, leading to the change of cysteine 315 to tryptophan (Fig 1). Patient 4 is homozygous for the same c.1073_1074insC insertion detected in Patient 2 (Fig 1). The presence of insertions or deletions in exon 13 for three out of the four patients supports the previous observation that a GC-rich stretch in exon 13 is a mutational hot spot (Dowling et al, 2003; El-Kamah et al, 2010; Hanks et al, 2003; Lee et al, 2005).
Table 2.
HFS mutations analysed in the present work
| Patient | HFS | Zygocity | DNA | Protein | Exon | References |
|---|---|---|---|---|---|---|
| 1 | Juvenile | Heterozygote | c.116G>T | p.C39F | 1 | This study |
| c.1074delT | p.A359HfsX50 | 13 | ||||
| 2 | Juvenile | Heterozygote | c.928G>T | p.V310F | 11 | |
| c.1073-1074insC | p.A359CfsX13 | 13 | ||||
| 3 | Infantile | Homozygote | c.945T>G | p.C315W | 11 | |
| 4 | Infantile | Homozygote | c.1073-1074insC | p.A359CfsX13 | 13 | |
| 5 | Infantile | Heterozygote | c.566T>C | p.I189T | 7 | (Hanks et al, 2003) |
| c.1073-1074insCC | p.A359LfsX51 | 13 | ||||
| n.a. | Juvenile | Homozygote | c.876-877insCAA | p.insQ293 | 11 | |
| n.a. | Infantile | Homozygote | c.652T>C | p.C218R | 13 | |
| n.a. | Juvenile | Homozygote | c.986 T>G | p.L329R | 12 | (Dowling et al, 2003) |
| n.a. | n.a. | n.a. | c.149 A>C | p.D50A | 1 | (Scobie & Young, 2005) |
Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to the nomenclature proposed by the Human Genome Variation Society (http://www.hgvs.org/mutnomen). The initiation codon is codon 1.
n.a. = not applicable.
Figure 1. Clinical presentation, pedigrees of HFS families and molecular characterization.

Abridged pedigrees of four HFS families are shown, numbers of individuals correspond to Table 1. Symbol notations: squares, males; circles, females; open symbols, unaffected individuals; filled symbols, affected mutation carrier of the nucleotide and predicted amino acid change indicated on top; index patients are marked with a red arrow. Electropherograms of index patients are shown to the right of each pedigree, altered nucleotides are marked in pink in both reference sequence (top) and actual read (bottom); underlined nucleotides indicate deletions or the site of insertions in exon 13. Inserted nucleotides in patients' sequences are also underlined, predicted changes in the amino acid sequence given above. All mutations segregated with the disease phenotype, showing compound homozygosity or homozygosity in patients, heterozygosity in parents, and wild-type alleles or heterozygous mutations in unaffected siblings.
The HFS mutations lead to drastic reduction of CMG2 protein levels
We made use of a newly generated monoclonal antibody (2F6) to analyse CMG2 in patient-derived fibroblasts. The 2F6 monoclonal antibody was obtained by genetic immunization of rats with a CMG2-expressing plasmid. Characterization of the antibody indicated that 2F6 specifically binds CMG2 on western blots of total cell extracts from different cell types, that it preferentially recognizes the non-reduced form and that it labels CMG2 by immunofluorescence staining but fails to recognized the improperly folded ER precursor forms (Supplementary Fig 1).
Based on the mutations identified in the patients, the following bands were expected to be revealed the patient fibroblasts: two bands of, respectively, 55 and 40 kDa for heterozygous Patient 1; two bands of 55 and 37 kDa for heterozygous Patient 2, a single ≍50 kDa full-length form for the homozygous Patient 3, and a single 37 kDa band for homozygous Patient 4. Surprisingly, CMG2 was undetectable using the 2F6 antibody in cell extracts of Patients 2–4, weakly detectable for Patient 1 and as expected readily detectable in control fibroblasts (Supplementary Fig 2). Enrichment of CMG2 by immunoprecipitation, however, allowed the detection in all patients but at very low levels (Fig 2A). Note that migration of full length CMG2 varied between Patients P1–P3 and the control. As will become apparent below, this is due to the fact that SDS–PAGE was performed under non-reducing conditions and that the patient mutations affect disulphide bonding of CMG2.
Figure 2. CMG2 protein and mRNA levels in HFS patient fibroblasts.
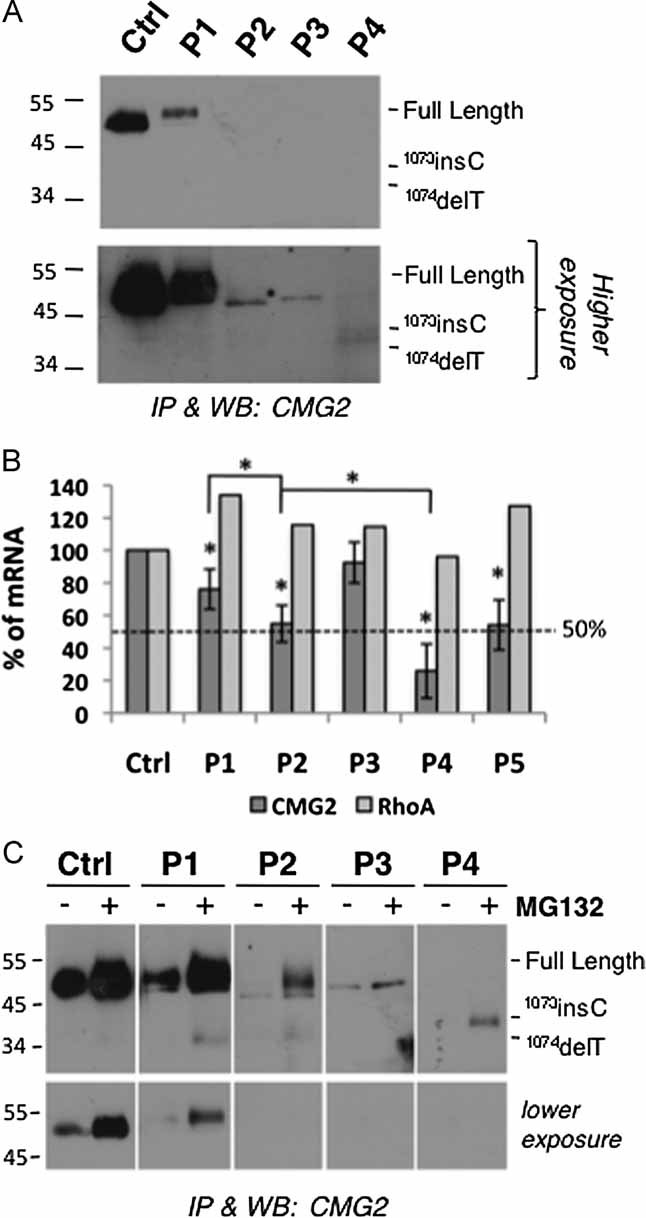
- CMG2 was immunoprecipitated from 300 µg of cell lysates from patient-derived fibroblasts and analysed by SDS–PAGE under non-reducing conditions and Western blotting using the 2F6 anti-hCMG2 antibody.
- mRNA levels of cmg2 in patient fibroblasts were analysed by quantitative real-time RT-PCR. Patient 5 has been previously characterized and harbours the I189T mutation in the vWA domain and a frame-shift mutation in exon 13 (Table 2; Deuquet et al, 2009; Dowling et al, 2003). The cmg2 mRNA levels in this patient was 54 ± 15% of the control (n = 5). Levels and number of experiments for other patients are mentioned in the main text. mRNA levels of the unrelated gene RhoA are shown for comparison. Errors bars represent standard deviations. Paired t-test analysis between control cells and patient-derived cells was performed showing that the observed differences are statistically significant (*p < 0.05).
- Patient-derived fibroblasts were incubated with 10 µM MG132 or not for 16 h. CMG2 was immunoprecipitated from 300 µg of cell lysates and analysed by SDS–PAGE under non-reducing conditions and Western blotting using the 2F6 anti-hCMG2 antibody.
Low abundance of 2F6 detectable CMG2 protein could be due to lower mRNA levels in patients. To test this possibility, we performed quantitative PCR on RNA extracts from patient-derived fibroblasts. Normalization was performed to three house-keeping genes (see Materials and Methods Section). In addition, we analysed an unrelated gene, RhoA, and found that its mRNA level was identical in all patients (Fig 2B). Levels of cmg2 mRNA varied greatly amongst patients, with the lowest, 26 ± 17% (n = 7), observed for Patient 4, homozygous for the c.1073-1074insC frame-shift mutation. The two heterozygous Patients 1 and 2, carrying a nonsense mutation on one allele and a frame-shift mutation on the other, had intermediate mRNA levels. These observations suggest that the nonsense mutations lead to mRNAs recognized by the NMD pathway (Rebbapragada & Lykke-Andersen, 2009). Interestingly, the mRNAs containing a premature stop codon varied in susceptibility to the NMD pathway. Indeed, the cmg2 mRNA level of Patient 2, carrying the c.1073-1074insC mutation, was significantly lower (55 ± 11% of that of controls, n = 7) than that in Patient 1 (76 ± 12%, n = 4) carrying the c.1074delT—even more so considering that both patients also express a cmg2 mRNA from one allele that is not prematurely degraded.
While the amount of cmg2 mRNA can explain the very low level of truncated CMG2 protein in Patient 4, this is not the case for the other patients suggesting that CMG2 is affected at the protein level. To test this hypothesis, we investigated whether inhibition of the proteasome with MG132 would affect CMG2 protein levels in patient cells. MG132 led to an increase in the CMG2 levels in patient-derived fibroblasts indicating that the protein was partly degraded by the proteasome. Degradation by other pathways, such as lysosomes or autophagy, may also occur since the levels of CMG2 observed for example in Patient 3 upon MG132 treatment by no means match the levels predicted by the mRNA. Alternatively, CMG2 levels might be increased but undetectable using the 2F6 antibody. MG132 also led to a marked increase in CMG2 levels in cells from the control patient. This could be due to various reasons. Firstly, normal folding of wild-type (WT) CMG2 could be rather inefficient leading to significant degradation of newly synthesized protein by the ERAD pathway and MG132 would increase the amount of CMG2 that exits the ER. Secondly, MG132 could have an effect on the turnover rate of CMG2 at the plasma membrane and lead to an increase of its half-life.
Model of the CMG2 extracellular Ig-like domain
Our subsequent aim was to understand the mechanisms leading to the premature degradation of CMG2 mutants in patients. Mutation p.C39F found in Patient 1 maps to the stem of the vWA domain (Fig 3). Upon expression of the vWA domain in Escherichia coli, this cysteine forms a disulphide bridge with Cys-218 (Lacy et al, 2004). The two other mutations, p.V310F (Patient 2) and p.C315W (Patient 3) map to the Ig-like domain, the structure of which is unknown. We therefore modelled this domain using a combination of fold recognition (Biegert et al, 2006; Soding, 2005) and manual modelling (Guex & Peitsch, 1997). The model shows an Ig-like fold (Fig 3B) where, quite remarkably, four cysteine residues are positioned in a manner compatible with the formation of two disulphide bridges: C230-C315 and C255-C279. In further support of the model, the two potential N-glycosylation sites, at least one of which is known to be modified (Deuquet et al, 2009), localize to the solvent exposed surface of the domain.
Figure 3. Model of the ectodomain of CMG2.
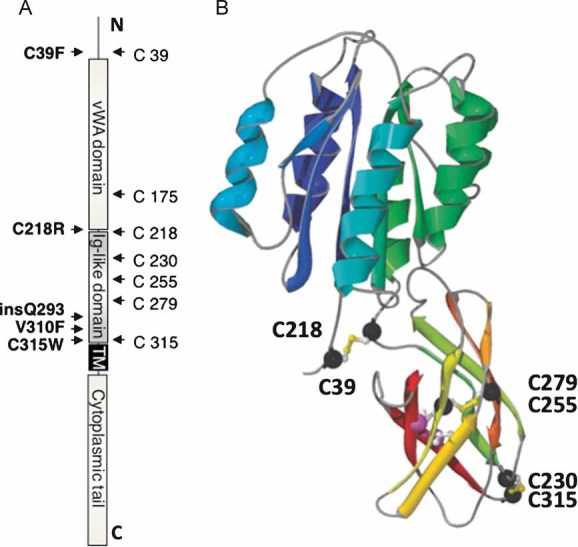
- Basic representation of the CMG2 protein and its domains, highlighting the positions of the seven cysteines and HFS mutations analysed in this study.
- Structural model of the extracellular domains of CMG2. The vWA domain has been taken from the crystal structure 1tzn, chain ‘a’, and the predicted Ig-like model (this work) has been grafted onto the crystal structure of the vWA domain (1tzn, chain a). To better show the overall fold, the successive secondary structure elements each have a distinct colour from dark-blue (N-terminal) to red (C-terminal). Cysteines participating in predicted disulphide bridges are marked as black spheres.
We next sought experimental evidence for the existence of the predicted disulphide bonds. Single cysteine to alanine mutations were generated for each of the seven ectodomain cysteine residues (Fig 3B) as well as double cysteine mutants corresponding to the predicted pairs. Expression was analysed in two cell lines, human Hela cells and Chinese Hamster Ovary (CHO) cells, since these two cell types vary in their protein glycosylation and/or folding capacity, leading to differences in the CMG2 migration pattern on SDS gels (Deuquet et al, 2009). In CHO cells, CMG2 shows a very distinct pattern with a single sharp band, corresponding to the glycosylated ER precursor form, and a broad higher molecular weight smear corresponding to mature CMG2, in which the N-linked sugars have been modified by Golgi enzymes into more complex oligosaccharides (Deuquet et al, 2009). Hela cells have the advantage of being easily transfected at high rates; however, the migration pattern of CMG2 in these cells does not show a clear boundary between the precursor and mature forms, although both are present (Deuquet et al, 2009).
Single mutants of the vWA domain cysteines did not drastically affect the migration pattern, i.e. both the precursor and the mature form were observed (Fig 4A), indicating that a significant proportion of the protein was able to exit the ER. Immunofluorescence analysis and surface biotinylation experiments further showed that mutation of Cys-39 or Cys-218 to alanine does not significantly affect targeting of CMG2 to the cell surface (Fig 4B and E).
Figure 4. Mutations of Ig-like domain cysteines lead to ER retention of CMG2.
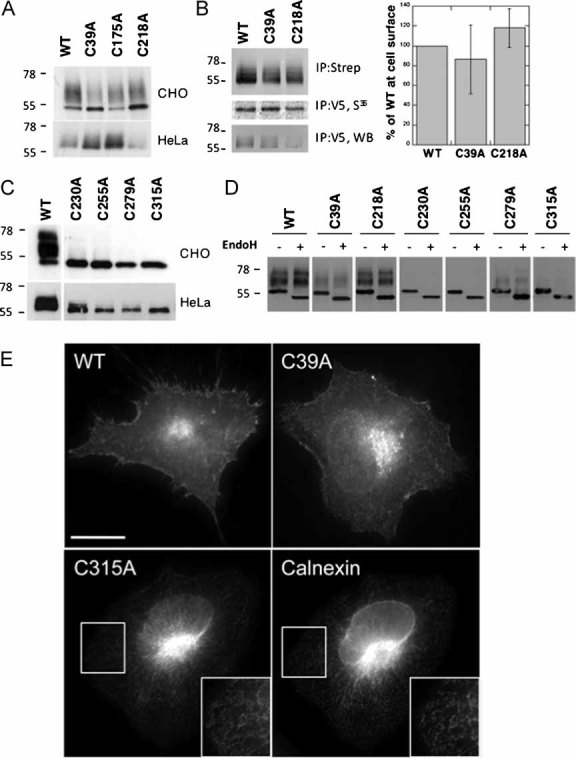
- A,C. HeLa or CHO cells were transfected for 48 h with WT or mutant CMG2-V5 constructs. Cell extracts were analysed by SDS–PAGE and Western blotting with an anti-V5 antibody.
- B. HeLa cells were transfected for 48 h with WT or mutant CMG2-V5 cDNAs. Confluent cells were pulsed with [35S]-methionine and subsequently incubated with 0.2 mg/ml NHS-SS-biotin. Biotinylated CMG2 proteins and expression levels were quantified using the Typhoon scanner. For each experiment, biotinylated CMG2 values were normalized to the synthesis levels and expressed as a percentage of WT. Error bars represent the standard deviation (n = 3).
- D. CHO cell lysates were submitted to immunoprecipitation with an anti-V5 antibody, treated with or without EndoH and analysed by SDS–PAGE and Western blotting with an anti-V5 antibody.
- E. HeLa cells were transfected with WT or mutant CMG2-V5 cDNA for 24 h prior to fixation and permeabilization and labelling with anti-V5 monoclonal as well as anti-calnexin polyclonal antibodies. The inserts illustrate an enlargement of a specific region. Bar: 10 µm.
In marked contrast, mutation of any cysteines in the Ig-like domain led to an almost complete disappearance of mature CMG2 (Fig 4C) and rendered the protein fully sensitive to Endoglycosidase H (EndoH; Fig 4D), an enzyme that can only remove non-complex oligosaccharides, suggesting that transport of CMG2 to the Golgi was impaired. Immunofluorescence analysis of the mutants revealed a typical reticulate ER staining (illustrated in Fig 4E for C315A).
To establish the exact disulphide pairing, extracts from cells expressing the single cysteine mutants were treated with N-ethylmaleimide (NEM) to block any free cysteines and analysed by SDS–PAGE under non-reducing conditions. Migration of WT CMG2 was very similar to that observed under reducing conditions (Fig 5A). A higher ≍114 kDa molecular weight form, corresponding to a dimer (see below and Fig 5B), was, however, observed for the two single, C39A and C218A, cysteine mutants (Fig 5A and C). The presence of this disulphide-linked dimer implies that, upon the single cysteine to alanine mutation, another cysteine in the protein became available for inter-molecular disulphide bond formation and thus, that in the WT protein, these two cysteines are bonded. The absence of the dimeric band for the double C39A–C218A mutant confirms the pairing of these residues.
Figure 5. CMG2 cysteine mutants form higher order homo-complexes.
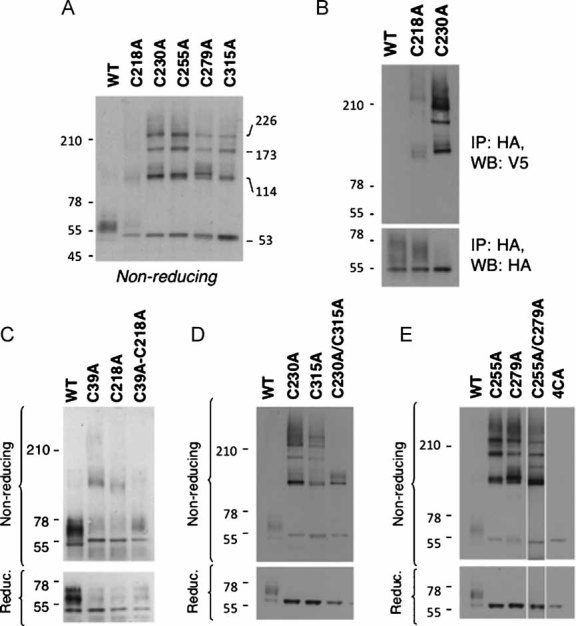
- A,C-E. CHO cells were transfected for 48 h with WT or mutants CMG2-V5 constructs. Cell lysates were subjected to immunoprecipitation with an anti-V5 antibody and analysed by SDS–PAGE—under reducing or non-reducing conditions—and Western blotting with an anti-V5 antibody.
- B. HeLa cells were co-transfected for 48 h with mutant CMG2-V5 or CMG2-HA constructs. Cell lysates were subjected to immunoprecipitation with an anti-HA antibody and analysed by SDS–PAGE under non-reducing (WB: V5) and reducing (WB: HA) conditions and Western blotting with anti-V5 or anti-HA antibodies.
The pattern for the Ig-like domain single cysteine mutants was more complex, with higher order complexes consistent with CMG2 trimers and tetramers (Fig 5A). The presence of higher order complexes for all single cysteine mutants not only indicates that all four Ig-like domain cysteines are involved in disulphide bond formation but that more than one cysteine must have become available for inter-molecular disulphide bond formation. This in turn implies that disruption of one bridge in the Ig-like domain impairs formation of the other.
The migration pattern of the C230A–C315A double mutant was simpler than that of the two corresponding single mutants confirming disulphide bonding between these two residues in the WT protein (Fig 5D). A disulphide-linked dimer was, however, still observed revealing the presence of one free cysteine and suggesting that the remaining Cys-255–Cys-279 bridge failed to form. The free cysteine in the C230A–C315A mutant is likely to be Cys-255, since Cys-279, even in a misfolded protein, probably resides in the core of the domain and is thus inaccessible for inter-molecular disulphide bond formation. The migration pattern of the last C255A–C279A double mutant remained as complex as that of the corresponding single mutants, indicating that the two remaining cysteines, Cys-230 and Cys-279, both of which are exposed, became available for inter-molecular disulphide bridge formation.
Altogether, these observations determine the identity of the three-disulphide bridges in the CMG2 ectodomain and show that, while the bridge that caps the vWA domain is not essential for passing the quality control leading to ER exit, the two bridges in the Ig-like domain are. The ER retention phenotype could not be rescued by mutating all Ig-like domain cysteines (Fig 5E) indicating that the disulphide bridges are essential for proper folding/stability of CMG2.
The above-mentioned conclusion that the ≍114 kDa complexes observed for CMG2 cysteine mutants correspond to covalent dimers is based on the following experiments. Cells were co-transfected with C218A harbouring an HA tag and C218A harbouring a V5 tag. Immunoprecipitation was performed using an antibody against the HA-tag followed by Western blotting, under non-reducing conditions, with an antibody against the V5 tag. A V5-positive band was observed at ≍114 kDa, while no band was observed as expected at ≍55 kDa corresponding to the monomer. The monomer was, however, detected with the HA antibody. Similar co-transfection experiments were performed for the WT protein and for the C230A mutant. A far more pronounced co-immunoprecipitation was observed with the C230A mutant, consistent with the higher abundance of complexes of different sizes, while no co-immunoprecipitation was observed for the WT proteins.
C39F and C218R HFS mutations affect folding of the ectodomain
We next investigated whether the HFS mutations involving cysteine residues had similar effects as those observed above for cysteine to alanine mutations. We included in this analysis the previously described p.C218R mutation found in a homozygous HFS patient (Table 2; Hanks et al, 2003).
Upon expression of C39F or C218R CMG2, both the mature and the ER precursor forms were observed under reducing conditions (Fig 6A), but the relative abundance of the precursor was higher than for the WT protein, as even more apparent after EndoH treatment (Fig 6B). Partial ER retention was confirmed by immunofluorescence analysis, where we could detect both plasma membrane and ER staining, as particularly indicated by the staining of the nuclear membrane (illustrated for C39F in Fig 6C).
Figure 6. C39F and C218R HFS mutations lead to inter-disulphide bond formation and ER retention.
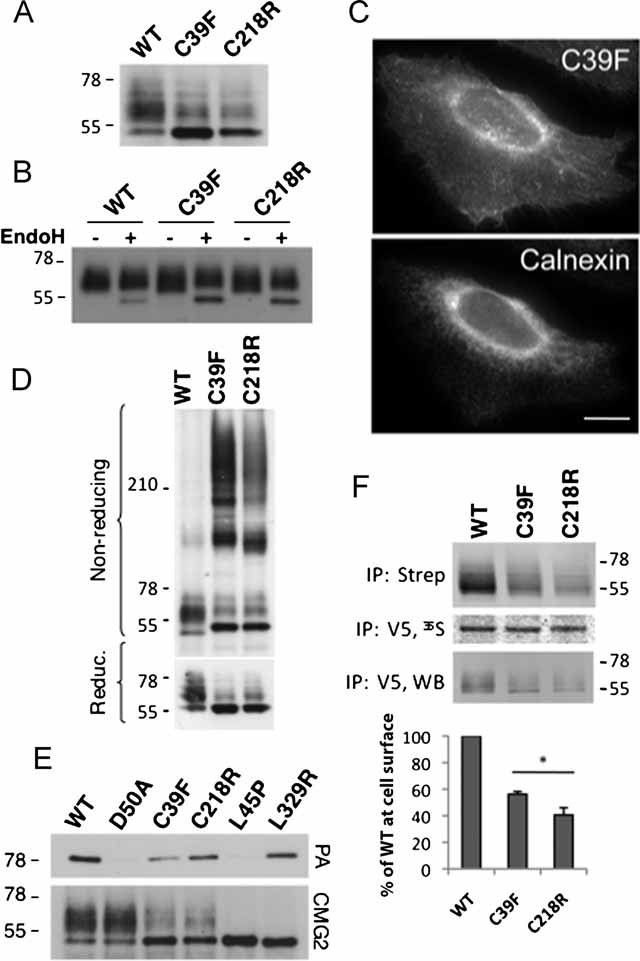
- A,D. CHO cells were transfected for 48 h with WT or mutants CMG2-V5 constructs. Cell lysates were subjected to immunoprecipitation with an anti-V5 antibody. Samples were analysed by SDS–PAGE—under reducing or non-reducing conditions—and Western blotting with an anti-V5 antibody.
- B. CHO cell lysates were subjected to immunoprecipitation with an anti-V5 antibody, treated with or without EndoH enzyme and analysed by SDS–PAGE and Western blotting with an anti-V5 antibody.
- C. HeLa cells were transfected with WT or mutant CMG2-V5 cDNA for 24 h prior to fixation and permeabilization, and labelling with anti-V5 monoclonal and anti-calnexin polyclonal antibodies. Bar: 10 µm.
- E. CHO cells were transiently transfected with WT or mutant CMG2-V5 cDNA. Cells were lysed in immunoprecipitation buffer and 1 µg/ml of PA was added in the buffer for 1 h at 4°C. After immunoprecipitation using an antibody against the V5 tag, samples were analysed by SDS–PAGE followed by Western blotting against PA or the V5 tag.
- F. HeLa cells were transfected for 48 h with WT or mutant CMG2-V5 cDNA. Surface biotinylation was performed as in 4B. Error bars represent the standard deviation (n = 3). Asterisks represent significant difference with respect to WT (p < 0.05).
Moreover, analysis of C39F and C218R CMG2 by non-reducing SDS–PAGE showed that, in contrast to the corresponding alanine mutants, which only formed dimers, patient mutations led to the formation of higher order complexes (Fig 6D) which were not seen under reducing conditions (Supplementary Fig 4A). This reveals the availability of more than one cysteine to inter-molecular disulphide bonding. In patients, the presence of a bulky phenylalanine residue at position 39 or a long charged arginine side chain at position 218 is therefore significantly more disruptive than merely preventing formation of the 39-218 bridge. Folding of the vWA domain was, however, not significantly impaired as revealed by the ability of the mutant CMG2 proteins to bind the anthrax protective antigent (PA) in cell lysates (Fig 6E). WT and the previously analysed ER retained HFS L329R mutant (Table 2) were used as positive controls (Deuquet et al, 2009), while D50A, mutated in the vWA ligand binding site (Scobie & Young, 2006) and L45P affected in vWA domain folding (Deuquet et al, 2009), were used as a negative control.
Finally, surface biotinylation experiments showed that plasma membrane targeting of C39F and C218R was impaired (Fig 6F) as we previously reported for the C218R mutation (Deuquet et al, 2009). When comparing Figs 4B and 6F, it is again apparent that the mutations found in the patients have a more severe effect on transport than cysteine to alanine mutations.
Ig-like domain HFS mutations lead to aberrant disulphide bond formation and severe ER retention
We next analysed the effects of the mutations observed in Patients 2 and 3 as well as of a previously reported mutation in the Ig-like domain that leads to the insertion of a glutamine residue at position 293 (p.InsQ293) (Hanks et al, 2003). Upon expression in CHO cells, V310F, C315W, and InsQ293 migrated as essentially a single (Fig 7A), EndoH sensitive (Fig 7B) band. Immunofluorescence analysis showed that the mutant proteins were retained in the ER (Fig 7C). Non-reducing SDS–PAGE revealed that these mutants efficiently formed high order inter-molecular disulphide bonded complexes (Fig 7D) indicative of the availability of two or more cysteine residues. Finally, surface biotinylation analysis showed that only a small fraction reached the plasma membrane (Fig 7E). ER retention of CMG2 could not be alleviated by the mere removal of the mutation-sensitive Ig-like domain as indicated by the analysis of an isoform of CMG2 (isoform 2, Uniprot Ref P58335-2), which lacks exons 8–11 encoding the six last residues of the vWA domain—including Cys-218—as well as the entire Ig-like domain. CMG2 isoform 2 indeed failed to reach the plasma membrane (Fig 7E) and localizes to the ER as demonstrated by immunofluorescence (Bell et al, 2001).
Figure 7. Various HFS mutations in the CMG2 Ig-like domain lead to inter-disulphide bond formation and ER retention.
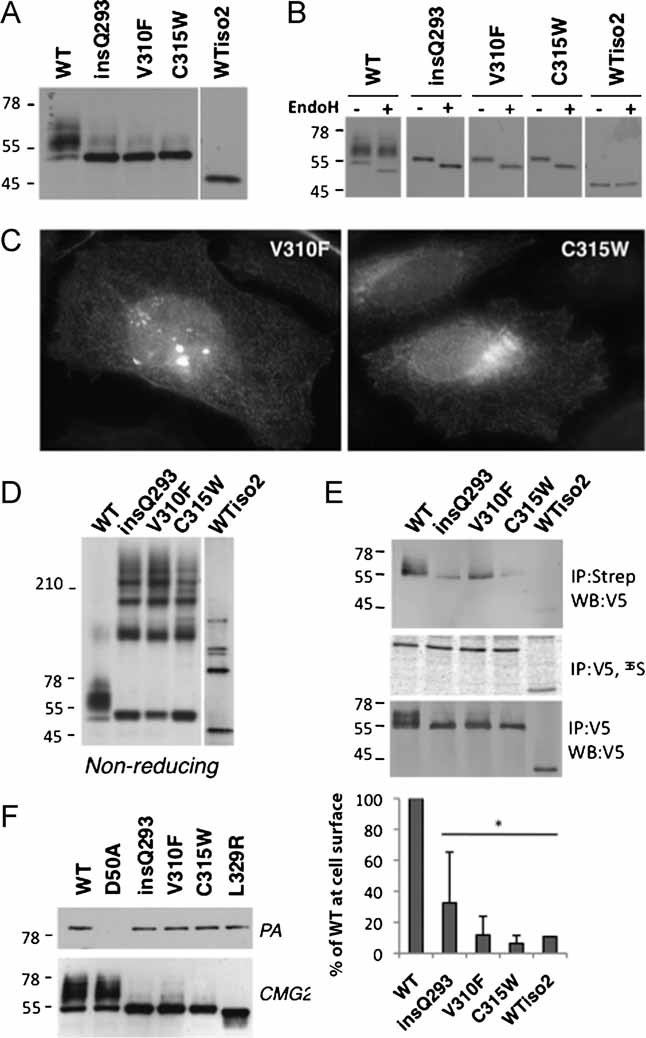
- A,D. CHO cells were transfected for 48 h with WT or mutants CMG2-V5 constructs. Cell lysates were subjected to immunoprecipitation with an anti-V5 antibody. Samples were analysed by SDS–PAGE—under reducing (A) or non-reducing conditions (D)—and Western blotting with an anti-V5 antibody.
- B. CHO cell lysates were subjected to immunoprecipitation with an anti-V5 antibody, treated with or without EndoH and analysed by SDS–PAGE and Western blotting with an anti-V5 antibody.
- C. HeLa cells were transfected with mutant CMG2-V5 cDNAs for 24 h prior to fixation and permeabilization, and labelling with anti-V5 antibodies.
- E. CHO cells were transiently transfected with WT or mutant CMG2-V5 cDNA. Cells were lysed in immunoprecipitation buffer and 1 µg/ml of PA was added in the buffer for 1 h at 4°C. After immunoprecipitation using an antibody against the V5 tag, samples were analysed by SDS–PAGE followed by Western blotting against PA or the V5 tag.
- F. HeLa cells were transfected for 48 h with WT or mutant CMG2-V5 cDNA. Surface biotinylation was performed as described in 4B. Error bars represent the standard deviation (n = 3). Asterisks represent significant difference with respect to WT (p < 0.05).
That HFS mutations in the Ig-like domain affect folding is supported by analysis of the Ig-like domain model: Val-293 is predicted to be located in the middle of a β-strand. Inserting a residue at this position would lead to an out-of-register β-strand, leading to the exposure of initially buried hydrophobic residues, a change expected to have major impact on folding. Val-310 makes a direct hydrophobic contact with Val-293, in the core of the Ig-like domain. Introduction of a bulkier side chain—phenylalanine versus valine—should not be easily accommodated by the structure.
The CMG2 vWA and Ig-like domains are two independent folding units
The above described observations show that HSF mutations in the Ig-like domain affect folding of this domain. As readout for proper vWA domain folding, we investigated whether the insQ293, V310F, and C315W CMG2 mutants are able to bind the anthrax PA. PA binding was again performed in cell lysates as above (Fig 6E). Ig-like domain mutants were competent of PA binding (Fig 7F), indicating that even if the Ig-like domain did not fold properly, the vWA domain did.
To further evaluate the ability of the vWA and Ig-like domains to fold independently, we investigated whether previously analysed vWA domain HFS mutations (Deuquet et al, 2009) lead to the formation of inter-molecular disulphide bond formation, used as an indication of defect Ig-like domain folding. Dimeric forms were observed for the previously described L45P, G105D, and I189T mutations, but bands corresponding to trimers and tetramers were absent, suggesting that these mutations affected the formation of the Cys39–Cys218 bond (Supplementary Fig 4B), but not those in the Ig-like domain.
Altogether, these observations indicate that the vWA and Ig-like domains constitute independent folding units. This is important since it indicates that if Ig-like domain HFS CMG2 mutants were rescued from ER retention, they would be competent for ligand binding and likely functional, and thus that the ER folding/quality control and ERAD components are potential therapeutic targets for HFS.
Proteasome inhibitors can rescue CMG2 in HFS patient cells
In view of a possible therapeutic intervention, we tested the effect of BZ, also known as Velcade, the first therapeutic proteasome inhibitor tested in humans (Adams & Kauffman, 2004), on CMG2 levels in HFS patient fibroblasts. The effect of BZ (Fig 8A) was very similar to that of MG132 (Fig 2C) with an increase in the levels of CMG2 for all patients including control. The highest levels of CMG2 were reached for Patient 1. We wondered whether this was because Patient 1 harbours a mutation in the vWA domain, while the missense mutations in Patients 2 and 3 map to the Ig-like domain. To address this possibility, we analysed CMG2 in fibroblasts from a previously described patient (Patient 5) heterozygous for the p.I189T mutation in the vWA domain and a frame-shift mutation in exon 13 (Deuquet et al, 2009; Dowling et al, 2003). We have previously shown that the I189T mutation does not affect the structure of the vWA domain per se, but slows down folding of the domain as to render CMG2 detectable by the ER quality control in the cellular context (Deuquet et al, 2009). As shown in Fig 2B, the mRNA levels of CMG2 in Patient 5 were very similar to that found in Patient 2. In agreement with our hypothesis, full-length CMG2 was detectable in fibroblasts from Patient 5, albeit at lower levels than for Patient 1 (Fig 8A) and this level was increased upon treatment with either MG132 (Supplementary Fig 4C) or BZ (Fig 8A) but to a lesser extent than for Patient 1.
Figure 8. Rescue of CMG2 cell surface expression and function in HFS patient cells upon exposure to proteasome inhibitors.
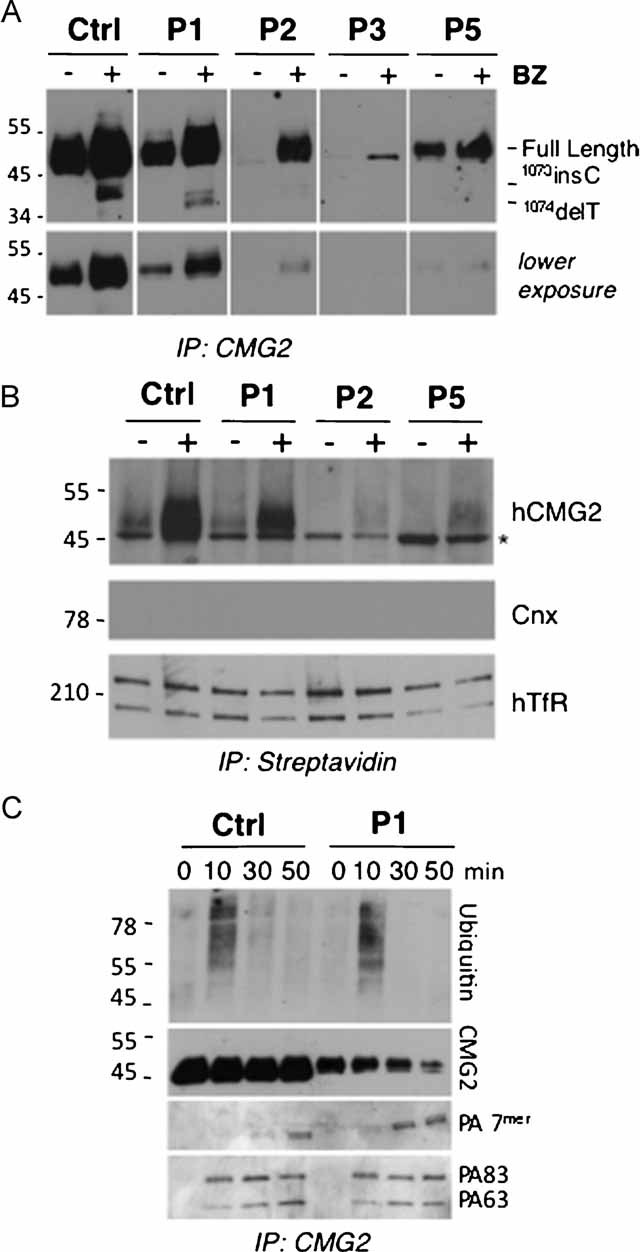
- Patient-derived fibroblasts were incubated with 10 µM of BZ or not for 16 h. CMG2 was immunoprecipitated using the goat anti-human CMG2 antibody from 300 µg of cell lysates and analysed by SDS–PAGE under non-reducing conditions and Western blotting using the 2F6 anti-hCMG2 antibody.
- Fibroblasts derived from Patients 1, 2 and 5 and control cells were incubated with 10 µM BZ or not for 16 h and subsequently incubated with 0.2 mg/ml NHS-SS-biotin. Cell lysates (300 µg of total proteins) were subjected to immunoprecipitation with streptavidin–agarose beads and analysed by SDS–PAGE and Western blotting probed with anti-hCMG2, anti-calnexin and anti-hTransferin receptor antibodies. * indicates unspecific band recognized by the 2F6 antibody after surface biotinylation and immunoprecipitation. This band is also observed for Patient 4 fibroblasts that express undetectable levels of CMG2. While calnexin (Cnx) was absent in these immunoprecipitations, it was readily detected in cell extracts (Supplementary Fig 4E). Under non-reducing conditions, human transferrin receptor (hTfR) migrates as a covalent dimer.
- Fibroblasts derived from Patient 1 and control cells were incubated with 10 µM BZ for 16 h and subsequently incubated with anthrax PA at a 500 ng/mL final concentration. After different time points, cell lysates were immunoprecipitated with an anti-hCMG2 antibody. Samples were analysed by SDS–PAGE under reducing or non-reducing conditions and Western blotting with an anti-hCMG2, an anti-ubiquitin, and anti-PA antibodies.
We next analysed plasma membrane targeting of rescued CMG2 by surface biotinylation of control fibroblasts and fibroblasts from Patient 1, 2, and 5. Patients 3 and 4 were not included in this analysis because the sensitivity of surface biotinilyation would not allow the detection of the low levels of CMG2 observed in these patients even after treatment with proteasome inhibitors. Equal amounts of proteins were submitted to precipitation with streptavidin beads. Treatment with MG132 (Supplementary Fig 4D) or BZ (Fig 8B, Supplementary Fig 4E) led to an increase in the amount of CMG2 detectable at the plasma membrane both for WT and mutant proteins, albeit to different extents. This indicates that inhibition of the proteasome not only rescued mutant CMG2 from degradation but allowed it to exit the ER and reach the plasma membrane. The increase in WT protein at the cell surface in BZ treated fibroblasts is likely due to both an increase in ER exit and a reduction in surface removal as discussed above for MG132 treatment.
We have previously shown, using anthrax PA as a ligand, that CMG2 undergoes ligand-triggered endocytosis (van der Goot & Young, 2009) and that this requires ligand-induced signalling via CMG2 (Abrami et al, 2010b). More specifically, we have shown that the toxin bound to the extracellular domain of CMG2 triggers ubiquitination on the cytosolic tail and that ubiquinitation is required for efficient endocytosis of the ligand-receptor complex via clathrin-coated pits (Abrami et al, 2003, 2006, 2010a). As evidence for the ability of CMG2 to signal, we have monitored CMG2 endocytosis in BZ-treated Patient 1 fibroblasts upon exposure to anthrax PA as a proof of concept. PA led to rapid degradation of CMG2 showing that the toxin led to receptor endocytosis and targeting to lysosomes (Fig 8C). Moreover, PA triggered ubiquitination of CMG2 not only in BZ-treated control but also patient fibroblasts (Fig 8C), revealing the ability of CMG2 to signal. This ubiquination signal was weakly detectable for the WT protein in the absence of BZ treatment, as expected due to the lower levels of CMG2, and undetectable for the mutant protein (Supplementary Fig 4F) consistent with the absence of surface CMG2 in Patient 1 cells.
DISCUSSION
The severity of HFS ranges from being lethal during early childhood to chronic and highly debilitating at a later age. While the exact function of CMG2 remains to be unravelled, our studies show that the majority of the HFS mutations lead to loss of function due either to the instability of the CMG2 messenger—in the case of frame-shift mutations in exon 13—or to improper folding of the ectodomain of the protein in the ER followed by ER retention and degradation. We find that CMG2 protein harbouring mutations in the ectodomain can partially be rescued through treatment with proteasome inhibitors. This rescue, which not only leads to partial recovery in terms of protein levels but also plasma membrane targeting of a functional protein, might to be more efficient for mutations that map to the vWA domain than mutations mapping to the Ig-like domain, highlighting the importance of in-depth genotype–phenotype analysis for potential personalized therapeutic treatment.
CMG2 structure, ER folding, quality control and degradation
Our work provides additional understanding of the structure of the CMG2 ectodomain. Following the vWA domain, CMG2 harbours an Ig-like domain that is well conserved in different species (47–82% identity with zebrafish and bovine, respectively) as well as in the homologous protein TEM8 (46% identity; Supplementary Fig 3A; van der Goot & Young, 2009).
We previously found that, with the exception of the p.T118K mutation (Lindvall et al, 2008), which affects the ligand binding MIDAS motif (Scobie & Young, 2006), HFS mutations that map to the vWA domain lead to slow or aberrant folding in the ER and thus recognition by the ER quality control (Deuquet et al, 2009). This, however, does not lead to complete degradation by ERAD as we show here using patient fibroblasts. The retention of CMG2 in the ER in the absence of complete degradation provides a window of opportunity for therapeutic treatments that would promote ER exit.
The N- and C-termini of the vWA domain are bridged by a disulphide bond. While formation of this bond probably occurs in the ER, its formation is not a prerequisite for ER exit. In contrast, the Ig-domain contains two disulphide bridges, formation of both of which is essential for ER exit. It has been shown that ER folding is generally under thermodynamic rather than kinetic control (Sekijima et al, 2005; Wiseman et al, 2007). Therefore, it is likely that disulphide bond formation in the Ig-like domain has a strongly stabilizing effect that drives the folding reaction forward. Improper folding of the Ig-like domain leads to the formation of inter-disulphide-linked CMG2 complexes that are presumably recognized by the ER quality control system and efficiently targeted to the ERAD pathway. As a consequence, the CMG2 protein is undetectable using our 2F6 antibody in the cells from patients harbouring mutations in the Ig-like domain.
Treatment with proteasome inhibitors
Our study shows that HFS missense mutations in the ectodomain lead to post-translational protein degradation. We hypothesize that the loss of CMG2 function is the cause of the disease. Considering that the HFS missense mutations lead to CMG2 ER retention, an alternative possibility could have been that these mutations trigger the unfolded protein response (UPR; Ron & Walter, 2007), which in turn would cause the disease. This latter explanation however seems unlikely in light of Patient 4, who shows all classical HFS symptoms. In this patient, the loss of CMG2 is due to excessive degradation of the cmg2 mRNA. The C-terminally truncated CMG2 that is produced—at very low levels—is properly targeted to the plasma membrane (Liu et al, 2007) and thus would not trigger any UPR. Similarly, the p.T118K mutation, which was reported for a homozygous HFS patient (Lindvall et al, 2008) and is defective in ligand binding (Scobie & Young, 2006), does not show any folding defect and is not expected to trigger the UPR.
Our strategy was therefore to attempt rescuing CMG2 from ERAD using proteasome inhibitors. We found that MG132—routinely used in tissue culture studies—as well as its derivative BZ—used in a number of clinical trials—allow the partial rescue of CMG2 protein in patient cells. Interestingly, the level of rescue correlates with the severity of the disease: rescue is extremely weak in patients showing typical infantile symptoms such as recurrent diarrhoea in early childhood (Patients 3 and 4), while rescue is observed for Patients 1, 2 and 5 who show a milder form of the disease (the severity of the disease cannot be compared between these patients due to the differences in medical care received during early childhood).
More in depth studies on cells from Patient 1 showed that, upon treatment with BZ, CMG2 protein levels could be restored to approximate control levels, that the protein was properly targeted to the plasma membrane, and that it was signalling competent. It is important to note that a full rescue of CMG2 protein levels is unlikely to be necessary since individuals with only one mutated cmg2 allele do not suffer from HFS indicating that an ≍50% protein level is sufficient. This is further supported by our findings that silencing cmg2 mRNA expression to ∼60% had no effect whatsoever on the gene expression profile of HUVEC (not shown). Altogether, our study shows that drugs affecting the proteasome are promising therapeutics to treat HFS patients, especially those harbouring missense mutations in exons 1–7.
MATERIALS AND METHODS
Subjects and genetic analysis for mutation detection
All patients were ascertained by physician-initiated referral. We collected blood and isolated DNA from cases diagnosed at the children's hospital of Freiburg University in accordance with the Declaration of Helsinki principles, specifically under a protocol approved by the ethics review board Baden-Württemberg, or from cases referred through the European skeletal dysplasia network (ESDN). Informed consent was obtained for all subjects, each family member was assessed by clinical criteria. Control samples and primary cells were obtained from ancestry-, sex- and age-matched healthy individuals.
For the genomic analysis and mutation detection, exons including intron–exon boundaries of ANTXR2 were amplified by PCR using standard protocols published previously (Hanks et al, 2003). Sequences of amplified PCR products were determined on an ABI3130xl capillary sequencer (Applied Biosystems). Sequence data were processed using ABI software and analysed using Sequencher (Genecodes). We compared amplicon sequences against the cDNA reference sequence for ANTXR2 (ENST00000403729) with nucleotide numbering starting from the first transcribed nucleotide of the reference sequence. Mutations and polymorphisms were confirmed in two independently amplified PCR products using bidirectional sequencing.
The paper explained
PROBLEM:
HFS is a human genetic disease caused by mutations in the anthrax toxin receptor 2 (or cmg2) gene, which encodes a membrane protein thought to be involved in the homeostasis of the extracellular matrix. The most severe form of the disease manifests in the first weeks of life by painful swelling of the skin and the large joints and oedema of the skin as well as chronic malabsorption and diarrhoea or pulmonary infection which may lead to death in the first 2 years of life. Patients suffering from the milder form usually do not present these complications but do present cutaneous tumours and debilitating loss of joint mobility. Little is known about the genotype–phenotype correlation.
RESULTS:
Using patient cells and in vitro approaches, we show that there is a drastic decrease of the CMG2 protein in the four patients analysed in this study, which harbour novel mutations. These observations suggest that the disease is caused by a loss of CMG2 function. We find that in three of the four patients, the protein is prematurely degraded, while in the fourth patient, it is the cmg2 mRNA, which is prematurely degraded. We show that premature protein degradation is due to improper folding of CMG2 in the ER, which is, therefore, recognized by the ER quality control and targeted for degradation by the proteasome. Finally, we show that proteasome inhibitors can partly rescue the CMG2 protein, which then properly folds, is released from the ER, and targeted to the cell surface where it is competent for signalling.
IMPACT:
This work illustrates that the proteasome, and presumably other components of the ER degradation system, are potential therapeutic targets for HFS.
For the allele frequency analysis, in 120 normal controls of European descent, exons 1 and 11 of ANTXR2 were amplified and sequenced unidirectionally to screen for HFS-associated mutations. An additional 105 controls of Turkish decent was screened for the ANTXR2 c.945T>G mutation, detected in family 3. No mutations were found in the non-affected patients.
Sequence alignments and modelling
For the multiple sequence alignment the following genes were used: P58335, Q6DFX2, Q00IM8, Q08DG9, A4QP34 for CMG2 and Q9H6X2, Q9CZ52, Q0PMD2, XP_873063.2, XP_692362.2 for TEM8. The computer program MUSCLE (Edgar, 2004) was used to generate the alignment, and Jalview (Waterhouse et al, 2009) to visualize it.
We used HHpred (Biegert et al, 2006; Soding, 2005) to predict the fold of the 107 residues of the CMG2 protein, located between the vWA domain and the transmembrane domain. The two best hits identified were 2CXK (pval 1.3E-06) and 2UZX (pval = 2.1E-03), clearly identifying an Ig-like fold. Phyre (Bennett-Lovsey et al, 2008)] also predicted an Ig-like fold. We then used CDD (Marchler-Bauer et al, 2009) to gather a representative set of structures containing an Ig-like fold domain (2cxk, 2as5, 1vkx, 1imh, 1ttu, 1cyg, 1clu, 7cgt, 1qhp, 1uad, 2uzx, 1gji, 2v2t, 1a3q, 2yrp, 1kcl and 1nfk), which were structurally superposed using Swiss-PdbViewer (Guex & Peitsch, 1997). We then manually aligned CMG2 onto this framework using a combination of the predictions returned by HHpred and Phyre (Supplementary Fig 3B). Finally, the predicted Ig-like model has been manually grafted onto the crystal structure of the vWA domain (pdb entry 1tzn, chain ‘a’).
Reagents, proteasome inhibition and EndoH treatment
The anti-human CMG2 monoclonal antibody 2F6 was obtained by genetic immunization of rats with an hCMG2 construct (Genovac, Germany); polyclonal goat anti human CMG2 antibodies were from R&D (Ref. AF2940), PA of anthrax toxin, anti-PA and anti-CMG2 antibodies were provided from the Leppla laboratory; goat anti-human CMG2 antibody was from R&D Systems; anti-HA-HRP antibody, anti-HA beads were from Roche (Basel, Switzerland); anti human-transferin receptor from Invitrogen (Carlsbad, CA); protein G-agarose-conjugated beads were from GE Healthcare; anti-V5 antibody was from Invitrogen; streptavidin–agarose conjugated beads from Sigma–Aldrich (St. Louis, MO); anti-ubiquitin antibody from Santa Cruz; HRP secondary antibodies from Pierce Chemical Co. (Rockford, IL); Alexa-conjugated secondary antibodies from Jackson Immunoresearch. The proteasome inhibitors—MG132 was from Sigma, BZ—were used at a final concentration of 10 µM for 16 h in complete medium. Treatments with EndoH were performed as previously described (Abrami et al, 2006).
Cells, plasmids, transfection and real-time PCR
Primary fibroblasts derived from patient biopsies were grown in DMEM medium complemented with fetal bovine serum and penicillin/streptavidin, counting at 1.5 × 106 cells in a 10 cm diameter dish, for 24 h. HeLa and anthrax toxin receptor-deficient CHO (here designated as CHO) cells were grown as described (Abrami et al, 2006, 2004). The human CMG2 (isoform 4, Swiss-Prot P58335-4, GenBank AK091721.1) gene, tagged with a V5 epitope at the C-terminus, was cloned in the pcDNA3.1/V5-HIS-TOPO expression vector (Invitrogen). Mutations were generated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Transfections were performed with Fugene (Roche Diagnostics Corporation).
For real-time PCR, RNA was extracted from a confluent six-well dish of patient fibroblasts using commercial RNA easy mini extraction kits (Quiagen). RNA was quantified by spectrometry and 1 mg was used for reverse transcription using hexanucleotides (Roche). A 1:40 dilution of the cDNA was used to perform the real-time PCR using the Sybr Green reagent (Roche). mRNA levels were normalized using three house-keeping genes: TATA-binding protein (TBP), β-microglobulin and β-glucoronidase (GUS).
Immunofluorescence
HeLa cells were fixed with paraformaldehyde and permeabilized with 1% Triton and labelled with an anti-V5 monoclonal and anti-calnexin polyclonal antibodies followed by the appropriate secondary antibodies. Images were acquired using a Plan-Apochromat 100×/1.4 oil emersion objective on an Axiovert 200M (Carl Zeiss Microimaging, Thornwood, NY), equipped with Axiocam camera using the Axiovision acquisition software. The Adobe Photoshop software was used to prepare the figures.
Surface biotinylation, metabolic labelling and immunoprecipitation
For immunoprecipitation, confluent cells were washed three times with PBS. Cells were lysed by incubation for 30 min at 4°C with 0.5% NP-40, 500 mM Tris–HCl, pH 7.4, 20 mM EDTA, 10 mM NaF, 30 mM sodium pyrophosphate decahydrate, 2 mM benzamidine, 1 mM PMSF, 1 mM NEM and a cocktail of protease inhibitors (Roche). Cells lysates were incubated over-night at 4°C with anti-V5 or goat anti-human CMG2 antibodies and protein G sepharose beads. For CMG2 detection, western blots were revealed using the rat monoclonal 2F6 anti human CMG2 antibody.
For metabolic labelling, HeLa cells were transfected for 48 h with CMG2 cDNAs, starved with methionine/cysteine-free medium, incubated for a 20 min pulse at 37°C with 50 µCi/mL [35S] methionine (Hartman Analytics) and then incubated for 10 min at 37°C in complete medium with a 10-fold excess of non-radioactive methionine and cysteine.
For surface biotinylation, CMG2 transfected Hela cells or patient-derived fibroblasts were incubated with 0.2 mg/ml NHS-SS-biotin (Pierce) in PBS for 30 min at 4°C and washed with sterile PBS containing 100 mM NH4Cl. Lysates were immunoprecipitated with anti-streptavidin coated agarose-beads, or with anti-V5 or anti-CMG2 and protein G sepharose beads. After SDS–PAGE and Western blotting against the V5 tag or endogenous CMG2 protein, biotinylated CMG2 proteins and expression level were quantified with Image Quant TL 2005/Typhoon software (GE Healthcare). The signal of biotinylated CMG2 was normalized to the level of CMG2 synthesis as determined by metabolic labelling. Expression of CMG2 mutants at the cell surface was normalized to that of the WT protein.
Acknowledgments
We are very grateful to the patients and their parents for accepting to be part of this study and for their collaborative attitude. We thank the Associazione I.S.I (http://www.associazioneisi.it) for linking patients as families suffering for HFS. We thank Béatrice Kunz for performing the qPCR analysis and Jérôme Bürgi for generating the CMG2/TEM8 alignments. This work was supported by the Swiss National Science Foundation (G.v.d.G.), the Fondation Telethon Action Suisse, the Fondation S.A.N.T.E.-Vaduz/Aide au soutien des nouvelles therapies and the Gerbert Rüf Stiftung. The computations were performed at the Vital-IT (http://www.vital-it.ch) Center for high-performance computing of the Swiss Institute of Bioinformatics. J. D. is recipient of a fellowship within the Marie Curie ENDOCYTE training network. G. v. d. G. is an international scholar of the Howard Hughes Medical Institute.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
JD designed and performed the majority of the experiments and wrote the paper, EL did most of the genetic analysis, NG performed the modelling of the Ig-like domain, LA did the experiments on the anthrax toxin, SS did all the maintenance of patient cell cultures, AL performed qPCR analysis and intellectual input for the NMD pathway, MCMR did some of the genetic analysis, JAM edited the paper and provided medical background on the disease, DR is the physician of Patient 4, LB is the physician of Patient 1, AS-F edited the paper and provided medical background on the disease and FGvdG designed the study, the experiments and wrote the paper.
For more information
Portal for rare diseases and orphan drugs
Infantile Systemic Hyalinosis
http://www.ncbi.nlm.nih.gov/omim/236490
Juvenile Hyaline Fibromatosis
http://www.ncbi.nlm.nih.gov/omim/228600
Association ISI
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160:321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Lindsay M, Parton RG, Leppla SH, van der Goot FG. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J Cell Biol. 2004;166:645–651. doi: 10.1083/jcb.200312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Leppla SH, van der Goot FG. Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J Cell Biol. 2006;172:309–320. doi: 10.1083/jcb.200507067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Bischofberger M, Kunz B, Groux R, van der Goot FG. Endocytosis of the anthrax toxin is mediated by clathrin, actin and unconventional adaptors. PLoS Pathog. 2010a;6:e1000792. doi: 10.1371/journal.ppat.1000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Kunz B, van der Goot FG. Anthrax toxin triggers the activation of src-like kinases to mediate its own uptake. Proc Natl Acad Sci USA. 2010b;107:1420–1424. doi: 10.1073/pnas.0910782107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Invest. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- Bell SE, Mavila A, Salazar R, Bayless KJ, Kanagala S, Maxwell SA, Davis GE. Differential gene expression during capillary morphogenesis in 3D collagen matrices: regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J Cell Sci. 2001;114:2755–2773. doi: 10.1242/jcs.114.15.2755. [DOI] [PubMed] [Google Scholar]
- Bennett-Lovsey RM, Herbert AD, Sternberg MJ, Kelley LA. Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins. 2008;70:611–625. doi: 10.1002/prot.21688. [DOI] [PubMed] [Google Scholar]
- Biegert A, Mayer C, Remmert M, Soding J, Lupas AN. The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res. 2006;34:W335–W339. doi: 10.1093/nar/gkl217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuquet J, Abrami L, Difeo A, Ramirez MC, Martignetti JA, van der Goot FG. Systemic hyalinosis mutations in the CMG2 ectodomain leading to loss of function through retention in the endoplasmic reticulum. Hum Mutat. 2009;30:583–589. doi: 10.1002/humu.20872. [DOI] [PubMed] [Google Scholar]
- Dowling O, Difeo A, Ramirez MC, Tukel T, Narla G, Bonafe L, Kayserili H, Yuksel-Apak M, Paller AS, Norton K, et al. Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet. 2003;73:957–966. doi: 10.1086/378781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Kamah GY, Fong K, El-Ruby M, Affifi HH, Clements SE, Lai-Cheong JE, Amr K, El-Darouti M, McGrath JA. Spectrum of mutations in the ANTXR2 (CMG2) gene in infantile systemic hyalinosis and juvenile hyaline fibromatosis. Br J Dermatol. 2010;163:213–215. doi: 10.1111/j.1365-2133.2010.09769.x. [DOI] [PubMed] [Google Scholar]
- Fayad MN, Yacoub A, Salman S, Khudr A, Der Kaloustian VM. Juvenile hyaline fibromatosis: two new patients and review of the literature. Am J Med Genet. 1987;26:123–131. doi: 10.1002/ajmg.1320260119. [DOI] [PubMed] [Google Scholar]
- Glover MT, Lake BD, Atherton DJ. Infantile systemic hyalinosis: newly recognized disorder of collagen. Pediatrics. 1991;87:228–234. [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Hanks S, Adams S, Douglas J, Arbour L, Atherton DJ, Balci S, Bode H, Campbell ME, Feingold M, Keser G, et al. Mutations in the gene encoding capillary morphogenesis protein 2 cause juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet. 2003;73:791–800. doi: 10.1086/378418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri K, Takasaki S, Fujiwara S, Kayashima K, Ono T, Shinkai H. Purification and structural analysis of extracellular matrix of a skin tumor from a patient with juvenile hyaline fibromatosis. J Dermatol Sci. 1996;13:37–48. doi: 10.1016/0923-1811(95)00492-0. [DOI] [PubMed] [Google Scholar]
- Keser G, Karabulut B, Oksel F, Calli C, Ustun EE, Akalin T, Kocanaogullari H, Gumudis G, Doganavsargil E. Two siblings with juvenile hyaline fibromatosis: case reports and review of the literature. Clin Rheumatol. 1999;18:248–252. doi: 10.1007/s100670050094. [DOI] [PubMed] [Google Scholar]
- Lacy DB, Wigelsworth DJ, Scobie HM, Young JA, Collier RJ. Crystal structure of the von Willebrand factor A domain of human capillary morphogenesis protein 2: an anthrax toxin receptor. Proc Natl Acad Sci USA. 2004;101:6367–6372. doi: 10.1073/pnas.0401506101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landing BH, Nadorra R. Infantile systemic hyalinosis: report of four cases of a disease, fatal in infancy, apparently different from juvenile systemic hyalinosis. Pediatr Pathol. 1986;6:55–79. doi: 10.3109/15513818609025925. [DOI] [PubMed] [Google Scholar]
- Lee JY, Tsai YM, Chao SC, Tu YF. Capillary morphogenesis gene-2 mutation in infantile systemic hyalinosis: ultrastructural study and mutation analysis in a Taiwanese infant. Clin Exp Dermatol. 2005;30:176–179. doi: 10.1111/j.1365-2230.2004.01698.x. [DOI] [PubMed] [Google Scholar]
- Lindvall LE, Kormeili T, Chen E, Ramirez MC, Grum-Tokars V, Glucksman MJ, Martignetti JA, Zaragoza MV, Dyson SW. Infantile systemic hyalinosis: case report and review of the literature. J Am Acad Dermatol. 2008;58:303–307. doi: 10.1016/j.jaad.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Liu S, Leung HJ, Leppla SH. Characterization of the interaction between anthrax toxin and its cellular receptors. Cell Microbiol. 2007;9:977–987. doi: 10.1111/j.1462-5822.2006.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Crown D, Miller-Randolph S, Moayeri M, Wang H, Hu H, Morley T, Leppla SH. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc Natl Acad Sci USA. 2009;106:12424–12429. doi: 10.1073/pnas.0905409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, et al. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuphanich S, Supko JG, Carson KA, Grossman SA, Burt Nabors, Mikkelsen L, Lesser T, Rosenfeld G, Desideri S, Olson S. Phase 1 clinical trial of bortezomib in adults with recurrent malignant glioma. J Neurooncol. 2010;100:95–103. doi: 10.1007/s11060-010-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbapragada I, Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate. Curr Opin Cell Biol. 2009;21:394–402. doi: 10.1016/j.ceb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Reeves CV, Dufraine J, Young JA, Kitajewski J. Anthrax toxin receptor 2 is expressed in murine and tumor vasculature and functions in endothelial proliferation and morphogenesis. Oncogene. 2009;29:789–801. doi: 10.1038/onc.2009.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Scobie HM, Young JA. Interactions between anthrax toxin receptors and protective antigen. Curr Opin Microbiol. 2005;8:106–112. doi: 10.1016/j.mib.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Scobie HM, Young JA. Divalent metal ion coordination by residue t118 of anthrax toxin receptor 2 is not essential for protective antigen binding. PLoS ONE. 2006;1:e99. doi: 10.1371/journal.pone.0000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- Stucki U, Spycher MA, Eich G, Rossi A, Sacher P, Steinmann B, Superti-Furga A. Infantile systemic hyalinosis in siblings: clinical report, biochemical and ultrastructural findings, and review of the literature. Am J Med Genet. 2001;100:122–129. doi: 10.1002/1096-8628(20010422)100:2<122::aid-ajmg1236>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Sun J, Collier RJ. Disulfide bonds in the ectodomain of anthrax toxin receptor 2 are required for the receptor-bound protective-antigen pore to function. PLoS One. 2010;5:e10553. doi: 10.1371/journal.pone.0010553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Ebihara T, Kusubata M, Adachi E, Arai M, Kawaguchi N, Utsunomiya J, Miki Y, Hiramoto M, Hattori S, et al. Abnormal collagen deposition in fibromas from patient with juvenile hyaline fibromatosis. J Dermatol Sci. 2009;55:197–200. doi: 10.1016/j.jdermsci.2009.06.005. [DOI] [PubMed] [Google Scholar]
- van der Goot FG, Young JA. Receptors of anthrax toxin and cell entry. Mol Aspects Med. 2009;6:406–412. doi: 10.1016/j.mam.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman RL, Koulov A, Powers E, Kelly JW, Balch WE. Protein energetics in maturation of the early secretory pathway. Curr Opin Cell Biol. 2007;19:359–367. doi: 10.1016/j.ceb.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.