

Abstract
Germ-line mutations in the BRCA1 gene strongly predispose women to breast cancer (lifetime risk up to 80%). Furthermore, the BRCA1 protein is absent or present at very low levels in about one third of sporadic breast cancers. However, the mechanisms underlying BRCA1 somatic inactivation appear multiple and are still not fully understood. We report here the involvement of miR-146a and miR-146b-5p that bind to the same site in the 3′UTR of BRCA1 and down-regulate its expression as demonstrated using reporter assays. This was further confirmed with the endogenous BRCA1 gene by transfecting microRNA (miRNA) precursors or inhibitors in mammary cell lines. This down-regulation was accompanied by an increased proliferation and a reduced homologous recombination rate, two processes controlled by BRCA1. Furthermore, we showed that the highest levels of miR-146a and/or miR-146b-5p are found in basal-like mammary tumour epithelial cell lines and in triple negative breast tumours, which are the closest to tumours arising in carriers of BRCA1 mutations. This work provides further evidence for the involvement of miRNAs in sporadic breast cancer through down-regulation of BRCA1.
Keywords: BRCA1, breast cancer, microRNA, post-transcriptional regulation
INTRODUCTION
Women with a germ-line mutation in the ubiquitously expressed BRCA1 gene have a highly increased risk of developing breast and ovarian cancers (reviewed in Mavaddat et al, 2010). Although BRCA1 germ-line mutations are relatively frequent (1 woman out of 1500 is a carrier), it has been estimated through population-based studies that they account for no more than 1–2% of all breast cancer cases (Anglian Breast Cancer Study Group, 2000). Very few somatic mutations have been identified in sporadic cases, a situation also observed for other tumour suppressor genes encoding proteins involved, as are BRCA1 and BRCA2, in the DNA damage response (the so-called ‘caretaker tumour suppressor genes’). Despite this absence of somatic mutations, a reduced expression of the BRCA1 gene has been observed in a significant proportion—maybe as high as 30%—of sporadic breast cancer cases (Mueller & Roskelley, 2003). These tumours share several features with familial BRCA1 breast cancers including the absence of HER2 oncogene amplification, of estrogen receptor alpha (ER) and progesterone receptor (PR) gene expression (triple negative breast cancers), and the presence of TP53 mutations. This led to the definition of a BRCAness phenotype (Turner et al, 2004). In particular, basal-like breast tumours, a subgroup of breast cancer defined through gene expression profiling and characterized by an expression signature similar to that of the basal/myoepithelial cells of the breast (Rakha et al, 2008), display lower BRCA1 expression than other breast cancer subgroups (Turner et al, 2007). In a fraction of these tumours, this low BRCA1 expression could be due to aberrant methylation of the promoter, which was reported in 11–14% of breast cancer cases (Catteau et al, 1999; Esteller et al, 2000; Rice et al, 2000; Turner et al, 2007). Loss of heterozygosity (LOH) at the BRCA1 locus is a common event that occurs in 21–42% of sporadic breast tumours (Beckmann et al, 1996; Nagai et al, 1994; Niederacher et al, 1997), but only 20% of the tumours with LOH display inactivation of the remaining allele through promoter hypermethylation (Esteller et al, 2000). Finally, down-regulation of BRCA1 expression has also been shown to be linked to overexpression of two proteins playing a role in the regulation of gene transcription: HMGA1 (high-mobility group proteins type A1) (Baldassarre et al, 2003), which belongs to a family of architectural proteins controlling DNA conformation, and ID4 (inhibitor of differentiation 4) (Beger et al, 2001; Turner et al, 2007), which belongs to a family whose members function as dominant-negative regulators of basic helix–loop–helix transcription factors. However, it seems likely that in sporadic breast tumours, BRCA1 can be repressed by other mechanisms than those already described, as these cannot account, even when combined, to the extent of reduction of the expression of BRCA1 in the large proportion of tumours that share this characteristic.
As microRNAs (miRNAs) have been shown in the past years to play a major role in post-transcriptional gene regulation, we chose to investigate the role of these single-stranded, small, non-coding RNAs of approximately 22 nucleotides (nt) in BRCA1 down-regulation. In animals, miRNAs generally inhibit translation of their target genes through imperfect base-pairing interactions, mostly in the 3′ untranslated regions (UTRs) of transcripts. The seed region of miRNAs (nt 2–9) appears crucial for target recognition, conducting perfect base-pairing, while bulges often occur in the central portion of the miRNA–messenger RNA (mRNA) hybrid. This imperfect base-pairing interaction of miRNAs with their target sites hampers the identification of regulated genes. Indeed, although the computational approaches that have been designed in the past 5 years are of considerable help, most of them predict large numbers of targets, among them many false-positive hits, and experimental validation is therefore necessary.
We hypothesised that down-regulation of the BRCA1 gene could be achieved through the action of miRNAs overexpressed in mammary tumours. In this study, we have indeed identified two miRNAs, miR-146a and miR-146b-5p, which negatively regulate BRCA1 expression and whose expression is particularly high in some mammary tumour cell lines of the basal type with concomitant low levels of BRCA1 and in triple negative mammary tumours. This novel mechanism for BRCA1 down-regulation is described.
RESULTS
Identification of microRNAs targeting the BRCA1 3′UTR
The 3′UTR of the human BRCA1 gene is comprised of 1367 nt while the mean size of 3′UTRs of human genes is around 800 nt (Mignone et al, 2002). This observation suggested that BRCA1 expression could be regulated through its 3′UTR. To identify miRNAs targeting the BRCA1 3′UTR, we performed a computational search using three different algorithms: MicroInspector (Rusinov et al, 2005), miRanda (John et al, 2004), and TargetScan (Lewis et al, 2005). The BRCA1 3′UTR was then examined with a fourth algorithm, RNA22 (Miranda et al, 2006), to predict potential binding sites for all the miRNAs predicted by the three previous algorithms. This approach allowed us to identify 14 miRNAs predicted to bind the 3′UTR of BRCA1 at 38 different locations. For each of these 14 miRNAs, one binding site was predicted by at least two algorithms (Table S1 of Supporting Information). The 38 predicted binding sites are evenly scattered all over the 1367 nt, including within the Alu sequence that lies between nt 574 and 878, without special clustering within the 3′ terminal region that displays the highest degree of interspecies conservation. Among these miRNAs, some had been previously shown to be overexpressed in breast tumours through miRNA expression profiling using microarrays (Iorio et al, 2005; Volinia et al, 2006): miR-9, miR-17-5p, and miR-146. We therefore chose to focus our analysis on these.
Repression of BRCA1 expression by miR-146a and miR-146b-5p
We first tested the influence of miR-9, miR-17-5p, miR-146a, and miR-146b-5p on their predicted messenger target by using a reporter vector into which we inserted the entire 3′UTR of BRCA1 downstream of the firefly luciferase open reading frame (ORF). This reporter vector, that we named Luc-BRCA1 3′UTR, was transfected into HeLa cells with a control vector encoding no miRNA, with a miR-Vec construct encoding let-7i for which no binding site in BRCA1 3′UTR is predicted by any of the four algorithms used, or with miR-Vec constructs encoding miR-9, miR-17-5p or miR-146a (Voorhoeve et al, 2006). These latter are expressed at low levels or are not expressed in HeLa cells according to Cheng and colleagues (Cheng et al, 2005) and/or Nelson and colleagues (Nelson et al, 2004). Whereas miR-146a expression reduced luciferase activity by ∼20% compared to control vector transfection, weak or no statistical effect was observed with miR-9, miR-17-5p or with let-7i (Fig 1A). Although miR-146a and miR-146b-5p are encoded by two different genes (located on different chromosomes), their seed region is identical and their mature sequences differ by only 2 nt (Fig 1B). The unique target site on the 3′UTR of BRCA1 (nt 489–507) predicted by three algorithms (MicroInspector, TargetScan 3.1 and RNA22) is common to both miRNAs (Fig 1B). To confirm the effect of miR-146a and to explore that of miR-146b-5p, we then transfected Luc-BRCA1 3′UTR into HeLa cells with miR-146a or miR-146b-5p synthetic precursors or with a negative control precursor that does not target any known mRNA within the human transcriptome. With both miRNAs, the degree of luciferase inhibition reached 50–60% compared to the control precursor (Fig 1C). This higher level of inhibition was expected as synthetic precursors have been shown to be more effectively delivered and more active than plasmids expressing miRNAs. As expected, considering the fact that miR-146a and miR-146b-5p share the same binding site on the 3′ UTR of BRCA1, cotransfection of both synthetic precursors did not increase the extent of inhibition (Fig 1C). When the Luc-BRCA1 3′UTR vector was mutated within this target site, miR-146a- or miR-146b-5p-mediated repression was no longer observed in cotransfection experiments (Fig 1C), suggesting specificity of the repression effect.
Figure 1. Binding of miR-146a and miR-146b-5p to BRCA1 3′UTR.

- Relative luciferase activity after cotransfection into HeLa cells of the Luc-BRCA1 3′UTR reporter vector and of an empty miR-Vec construct (control vector), or of miR-Vec constructs expressing different miRNAs, as indicated. Error bars represent standard error of the mean (SEM) of four independent experiments. *p < 0.05; ***p < 0.001 (Student's t-test).
- Sequence alignment of miR-146a and miR-146b-5p and their complementary site in the schematically represented BRCA1 3′UTR. The seed sequence is bolded.
- Repression of luciferase activity after cotransfection into HeLa cells of the wild-type (wt) or mutated (mut146) Luc-BRCA1 3′UTR reporter vector and of control or miR-146 synthetic precursors, as indicated. Error bars represent SEM of four independent experiments.
- Western blot analysis with an antibody against IRAK1 or BRCA1 of proteins extracted from HeLa cells transfected with a control, miR-146a, miR-146b-5p or both miR-146a and miR-146b-5p precursors. The bands corresponding to BRCA1 were quantified relative to the α-tubulin loading control (BRCA1 normalized level) using the GelDoc™XR+ Imager (Bio-Rad) and the Image Lab™ software. The results shown are representative of at least three independent experiments.
To determine whether miR-146a or miR-146b-5p affected endogenous BRCA1 expression, we compared the level of the BRCA1 protein in HeLa cells after transfection with miR-146a or miR-146b-5p synthetic precursors, or with a negative control precursor. We first showed by Northern blot analysis that miR-146a and miR-146b-5p could be detected in transfected cells only (Fig S1A of Supporting Information). The expression of these miRNAs individually or in combination led to a drastic reduction in the amount of IRAK1, a known target of miR-146a and miR-146b-5p (Perry et al, 2008; Taganov et al, 2006), and in the amount of BRCA1 protein (Fig 1D), demonstrating that miR-146a and miR-146b-5p are effective on the endogenous BRCA1 gene. Taken together, these results show that miR-146a and miR-146b-5p down-regulate the expression of the BRCA1 gene in HeLa cells.
miR-146a/b-5p expression in mammary cell lines of different subtypes
We next determined the expression level of miR-146a/b-5p by quantitative Polymerase Chain Reaction (after reverse transcription) (qRT-PCR) in three normal mammary cell lines and in 15 breast cancer cell lines (Table S2 of Supporting Information). Most of these cell lines have been characterized at the molecular level [(Elstrodt et al, 2006; Neve et al, 2006); Cancer Cell Lines Project, COSMIC: http://www.sanger.ac.uk/genetics/CGP/CellLines/] and have been shown to mirror the recurrent genomic and transcriptional characteristics of primary breast tumours (Neve et al, 2006). We found that in most instances, the pattern of expression of miR-146a and miR-146b-5p is similar, suggesting that their genes, although located on different chromosomes, are coregulated (Fig 2A). Furthermore, while miR-146a/b-5p are either weakly or not expressed in normal mammary cell lines, their expression is high in some tumour cell lines, particularly in those which have been classified as basal-like (Neve et al, 2006). We then analysed the level of expression of the BRCA1 protein in non-tumourigenic and basal-like mammary cell lines by Western blot. We found much lower levels of BRCA1 in most basal-like cell lines than in the non-tumourigenic HBL-100 cell line (Fig 2B). In the three basal-like cell lines with the highest miR-146a/b-5p expression level, the amount of BRCA1 was particularly low.
Figure 2. miR-146 a/b-5p and BRCA1 expression levels in mammary cell lines.
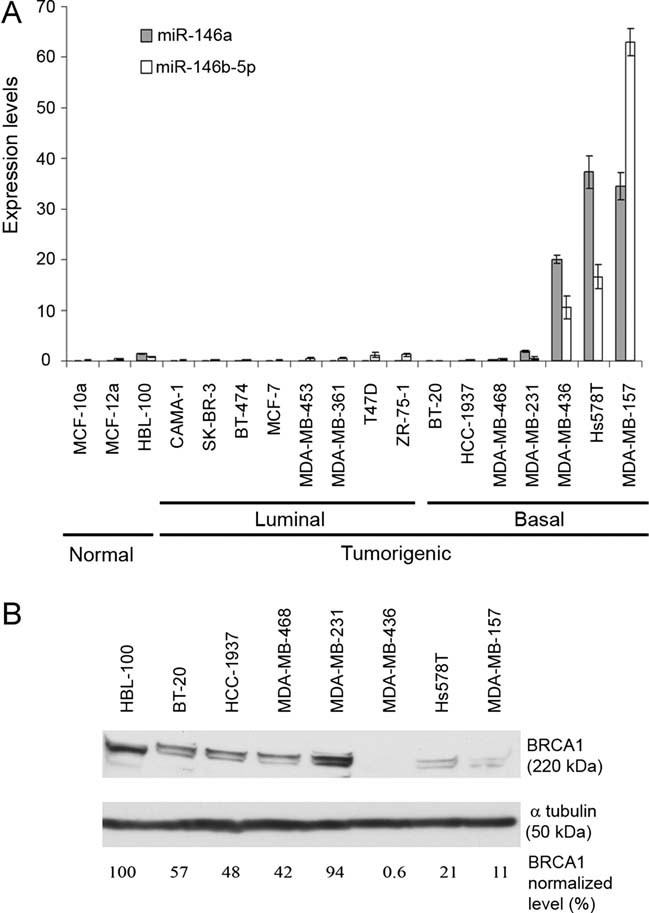
- Expression level of miR-146a and miR-146b-5p determined by quantitative RT-PCR in three mammary normal cell lines, eight tumour cell lines with a luminal transcriptional profile and seven tumour cell lines with a basal-like transcriptional profile. miR-146a and miR-146b-5p expression were normalized using RNU44 RNA expression. Error bars represent standard deviations (SD) for triplicates of one representative experiment.
- Western blot analysis with an antibody against BRCA1 of proteins extracted from a mammary normal and of seven tumour cell lines with a basal-like transcriptional profile. The bands were quantified relative to the α-tubulin loading control using the UVP BioImaging system (EC3) and the Quality One Software.
We next focused our analysis on three breast cancer basal-like cell lines: MDA-MB-468 express low levels of miR-146a/b-5p and intermediate levels of BRCA1, while MDA-MB-157 and MDA-MB-436 express high levels of miR-146a/b-5p and low levels of BRCA1 (Fig 2). Northern blotting confirmed the absence of miR-146a/b-5p in MDA-MB-468 non-transfected cells and showed that transfection of synthetic precursors in these cells produced the appropriately processed miRNAs (Fig S1B of Supporting Information). Transient overexpression of these miRNAs either individually or in combination resulted in a ∼6- to 50-fold reduction in the amount of BRCA1 as compared with cells transfected with a control precursor, while transfecting these cells with a BRCA1 siRNA led to a ∼20-fold reduction (Fig 3A). Conversely, transfection of Locked Nucleic Acid (LNA)-modified anti-miR-146a and anti-miR-146b-5p in MDA-MB-436 cells was accompanied by a decrease in the amount of miR-146a/b-5p (Fig S1C of Supporting Information) and a 1.3- to 6-fold increase in the amount of BRCA1, as compared with cells transfected with a control LNA (Fig 3B). The increase in the amount of BRCA1 was higher when both anti-miR-146a and anti-miR-146b-5p were cotransfected in MDA-MB-436 cells. In MDA-MB-157 cells, with likewise high levels of miR-146a/b-5p, cotransfection of anti-miR-146a and anti-miR-146b-5p produced a similar effect on BRCA1 protein expression (3- to 8.6-fold increase; Fig 3C). It should be noted that the MDA-MB-436 cell line contains a BRCA1 splicing mutation on one allele and shows loss of the other allele (Elstrodt et al, 2006). The mutation (c.5396+1G>A) leads to in frame-skipping of exon 20 and to a mutant BRCA1 protein with a deletion of 28 amino acids indistinguishable from wild-type (wt) BRCA1 (3390 vs. 3418 amino acids). These results demonstrate that miR-146a and miR-146b-5p down-regulate the expression of wt and mutant alleles of the BRCA1 gene in mammary cell lines.
Figure 3. Modulation of miR-146a/b-5p and BRCA1 expression in three tumour mammary cell lines.
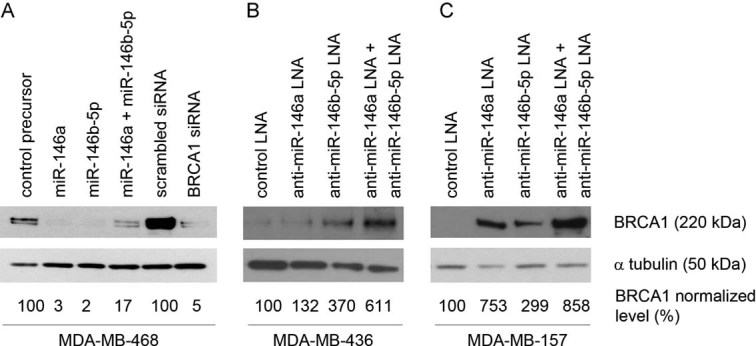
Western blot analysis with an antibody against BRCA1 of proteins extracted from MDA-MB-468 (A), MDA-MB-436 (B) or MDA-MB-157 (C) cells.
- A. MDA-MB-468 cells were transfected with a control, miR-146a, miR-146b-5p or both miR-146a and miR-146b-5p precursors, a scrambled siRNA or a siRNA targeting the BRCA1 gene.
- B, C. MDA-MB-436 or MDA-MB-157 cellswere transfected with a control, anti-miR-146a, anti-miR-146b-5p or both anti-miR-146a and anti-miR-146b-5p LNA. The bands were quantified relative to the α-tubulin loading control as in Fig 1D. The results shown are representative of at least three independent experiments.
miR-146a/b-5p control of BRCA1-mediated proliferation and homologous recombination
BRCA1 has been repeatedly shown to inhibit cellular proliferation when overexpressed in different cell types (Abbott et al, 1999; Aprelikova et al, 1999; Holt et al, 1996). Conversely, BRCA1 depletion through RNA interference has been shown to stimulate proliferation. Therefore, we studied the consequences of miR-146a and miR-146b-5p expression in HeLa (Fig 4A) or MDA-MB-468 (Fig 4B) cells on proliferation. As expected, miR-146a and miR-146b-5p precursor transfection increased cell proliferation in HeLa and in MDA-MB-468 cells, as did BRCA1 siRNA (Fig S2 of Supporting Information). In HeLa cells, the increase of proliferation seen with miRNAs was equivalent to that obtained with siRNAs. Furthermore, in these latter cells, cotransfection with a BRCA1-expressing vector lacking the BRCA1 3′UTR [pBRCA1 (1–24)] and thus insensitive to miR-146a/b-5p did not produce any change in cell proliferation, indicating that the increase seen previously was linked to down-expression of BRCA1 (Fig 4A).
Figure 4. Proliferation and homologous recombination rate of cells transfected with miR-146a/b-5p precursors.
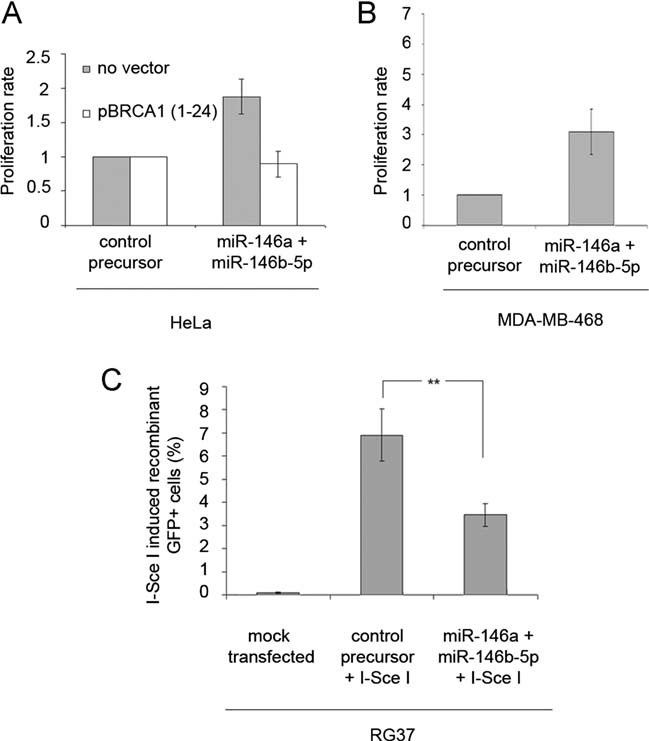
- Proliferation rate of HeLa cells transfected with a control or miR-146a and miR-146b-5p precursors. Proliferation rate was also measured after cotransfection with a BRCA1 expressing vector lacking the BRCA1 3′UTR [pBRCA1 (1–24)]. Error bars represent SEM for four independent experiments.
- Proliferation rate of MDA-MB-468 cells transfected with a control or miR-146a and miR-146b-5p precursors. Error bars represent SEM for four independent experiments.
- Rate of induced recombinant GFP positive cells (GFP+) either mock transfected or cotransfected with a control ormiR-146a and miR-146b-5p precursors and an I-SceI expressing plasmid. Error bars represent SEM for three independent experiments.
We next tested the effects of miR-146a and miR-146b-5p binding to the BRCA1 3′UTR on a well-documented biological function of BRCA1, i.e. homologous recombination. For doing so, we took advantage of an established recombination assay that allows the monitoring of gene conversion (Dumay et al, 2006; Pierce et al, 1999; Plo et al, 2008). RG37, a human SV40-immortalized fibroblast cell line, contains a single chromosomally integrated copy of a tandem repeat of two inactive GFP genes. This locus can be converted to a functional entity by homologous recombination following double strand breakage by meganuclease digestion at a targeted I-SceI unique site. Silencing of the BRCA1 gene by RNA interference leads to a significant reduction in gene conversion with this recombination assay (Plo et al, 2008). We showed that transfection of miR-146a and miR-146b-5p synthetic precursors in the RG37 cells led to a marked reduction in the amount of BRCA1 (Fig S3 of Supporting Information) and a concomitant two-fold decrease in the frequency of I-SceI-induced GFP+ cells (Fig 4C). These results show that down-regulation of BRCA1 by miR-146a and miR-146b-5p impairs two cellular processes controlled by BRCA1.
miR-146a/b-5p expression in breast tumours
To further assess the involvement of miR-146a/b-5p in breast tumourigenesis, their level of expression was determined in 76 primary breast tumours by qRT-PCR (Table S3 of Supporting Information). The expression of miR-146a and miR-146b-5p, relative to the expression of the RNU44 small nucleolar RNA used as an internal control, is coregulated in most cases, as seen in tumour mammary cell lines: in 65 tumours (86%) miR-146a and miR-146b-5p expression levels are both inferior to their respective median value, or both superior. The median miR-146a expression is 0.21 (range: 0–2.54; mean: 0.37). Twenty-six tumours (34%) show a miR-146a level 1.5-fold greater than the median value, and for 7 of them (9%), the increase is >fivefold. Concerning miR-146b-5p, the median expression is 0.25 (range: 0.05–3.36; mean: 0.41); 27 (35%) and 3 (4%) tumours have a miR-146b-5p level 1.5- or 5-fold greater than the median value, respectively. We next determined the levels of BRCA1 in these tumours by Western blotting (Fig S5 of Supporting Information). We were able to analyse protein expression in 35 samples out of 76; 22 of them presented a lower extra band in the α-tubulin loading control, likely to correspond to detyrosinated tubulin, consistent with the fact that this post-translational modification has been reported to be a frequent occurrence in breast cancer (Mialhe et al, 2001). Among these 35 exploitable samples, BRCA1 was found to be absent in 15 tumours (43%) and present in 20 (57%) (Table 1). We observe a statistically significant inverse correlation between BRCA1 and miR-146 (p = 0.05), but not between IRAK1 and miR-146, which suggests that IRAK1 gene regulation is probably complex and exerted by multiple factors upon different layers.
Table 1.
Relation between BRCA1 or IRAK1 status and expression levels of miR-146a/b-5p determined by quantitative RT-PCR
| No of tumours with miR-146 level < median | No of tumours with miR-146 level > median | Total no of tumours | |
|---|---|---|---|
| BRCA1 protein status | |||
| Negative | 4 (27%) | 11 (73%) | 15 |
| Positive | 12 (60%) | 8 (40%) | 20 |
| p = 0.05 | |||
| IRAK1 protein status | |||
| Negative | 3 (37%) | 5 (63%) | 8 |
| Positive | 13 (48%) | 14 (52%) | 27 |
| p > 0.05 | |||
A tumour was defined as ‘< median’ when the expression levels of both miR-146a and miR-146b-5p were less than their respective median level of expression in the analysed tumours. It was defined as ‘> median’ when the expression level of either miR-146a or miR-146b-5p was increased compared to their respective median level of expression in the analysed tumours. Total number of tumors analysed was 35.
We next determined the level of expression of miR-146a and miR-146b-5p in another series of 167 breast tumours (Table S3 of Supporting Information) for which clinical variables were known. We found highly similar results as those found in the previous series concerning miR-146a expression level: 59 (35%) and 11 (7%) tumours show levels 1.5- and 5-fold greater than the median, respectively (0.66; range: 0.11–15.33; mean: 1.24). For miR-146b-5p, the figures are higher or similar as those observed in the previous series: 83 (50%) and 10 (6%), respectively (median: 0.40; range: 0.10–3.08; mean: 0.63). We found that miR-146a/miR-146b-5p expression levels are significantly higher in triple negative versus non-triple negative, in ER-PR- versus ER+ and/or PR+, and in SBR grade III versus grade II breast tumours (Fig. 5). miR-146a/miR-146b-5p expression levels were not found to be associated with ERBB2 amplification, pTNM stage and metastasis or menopausal status (data not shown). These results are consistent with the fact that breast tumours developed by BRCA1 mutation carriers commonly lack ER and PR expression, do not overexpress ERBB2 and are of a higher grade than those found in controls. Taken together, the data strongly suggest that miR-146a/b-5p overexpression in triple-negative tumours results in BRCA1 inactivation.
Figure 5. Relationship between miR-146a/b-5p expression and hormonal status in 167 mammary tumours.
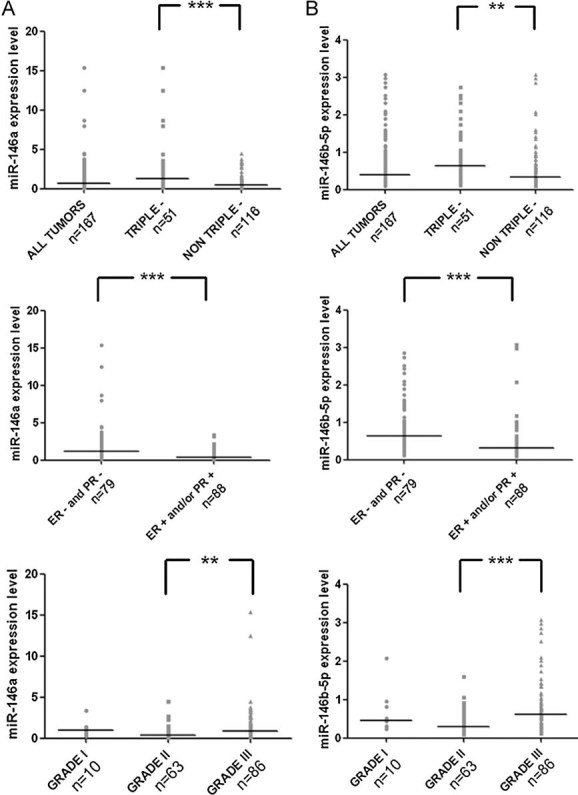
Expression level of miR-146a or miR-146b-5b in 167 mammary tumours and in different subgroups classified according to hormonal status (triple negative, i.e. ER negative/PR negative/no ERBB2 expression), or to SBR grades (I–III). Each point represents the expression level in one tumour while the line represents the median expression. p < 0.01; p < 0.001 (one-way analysis of variance with Tukey's multiple comparison test (ANOVA)).
- Expression level of miR-146a.
- Expression level of miR-146b-5p.
DISCUSSION
miR-146a and miR-146b-5p share the same seed region and have closely related mature sequences (20/22 nt identity) encoded by two genes located on chromosomes 5 and 10, respectively. Bioinformatics tools identified a binding site for miR-146a and miR-146b-5p in the 3′UTR of BRCA1. These predictions were validated as we have shown that the BRCA1 gene is down-regulated by miR-146a and miR-146b-5p in breast cancer cell lines of the basal-like subtype and in triple negative breast tumours.
While we were conducting this study, Shen and colleagues reported that miR-146a could bind to the 3′UTR of BRCA1 and BRCA2 mRNAs using luciferase reporter assays, but without studying the involvement of this miRNA in breast cancer any further (Shen et al, 2008). While our work confirms the binding of miR-146a to BRCA1 3′UTR, we did not find any evidence for its binding to BRCA2 3′UTR (Fig S4 of Supporting Information).
Given the involvement of BRCA1 in ovarian cancer susceptibility and possibly in sporadic ovarian cancer (Weberpals et al, 2008), it is interesting to note that miR-146b-5p has been found to be up-regulated in ovarian cancer tissues and cell lines (Dahiya et al, 2008), especially in stage III ovarian cancers (Eitan et al, 2009) and in the ovarian serous carcinoma subtype (Wyman et al, 2009). As ovarian tumours developed by BRCA1 mutation carriers are mostly stage III serous carcinoma (Lakhani et al, 2004), these results raise the possibility that miR-146b-5p and possibly miR-146a could also be involved in BRCA1 down-regulation in sporadic ovarian cancer.
The role of miR-146a in the regulation of inflammation induced via the innate immune response is largely documented (Williams et al, 2008). This role is likely to be exerted, at least partly, through its two validated targets, TNF receptor-associated factor 6 (TRAF6) and IL-1 receptor-associated kinase 1 (IRAK1), whose involvement in Toll-like receptor and proinflammatory cytokine signalling is well established. miR-146a has also been shown to modulate IL-2 expression and activation-induced cell death in T lymphocytes, suggesting a role of this miRNA in the adaptive immune response as well (Curtale et al, 2010). Elevated basal expression of miR-146a has been reported in tissues associated with psoriasis (Sonkoly et al, 2007), low-grade osteoarthritis cartilage (Yamasaki et al, 2009), and rheumatoid arthritis (Nakasa et al, 2008; Pauley et al, 2008), suggesting that miR-146a dysregulation could play an important role in the pathophysiology of these chronic inflammatory diseases (Williams et al, 2008).
Another identified miR-146a target is the CXCR4 (chemokine receptor 4) mRNA: CXCR4 and its ligand SDF-1 are key molecules in the process of homing/mobilization of haematopoietic cells and miR-146a has been shown to be a major constituent of a pathway that controls megakaryopoiesis (Labbaye et al, 2008).
Interestingly, miR-146a and miR-146b-5p have also been implicated in several cancer types by microarray analyses. Indeed, miRNA expression profiles of six solid tumour types revealed an overexpression of miR-146 in breast carcinomas (79 samples), endocrine pancreatic tumours (39 samples), and prostate cancers (56 samples) (Volinia et al, 2006); increased miR-146a and miR-146b-5p expression was also reported, respectively, in 4 cervical cancers (Wang et al, 2008) and 15 papillary thyroid carcinomas (He et al, 2005), whereas reduced expression of miR-146a and miR-146b-5p were found in two androgen-independent prostate cell lines (Lin et al, 2008). These latter finding was confirmed by FISH analysis in prostate cancer tissue arrays containing 60 patient samples (Lin et al, 2008).
Rapid induction of miR-146a gene transcription in response to a variety of microbial components and proinflammatory cytokines is mediated through NF-κB (Pacifico et al, 2010; Taganov et al, 2006), as confirmed by promoter analysis of the miR-146a gene that contains three NF-κB binding sites (Taganov et al, 2006). The NF-κB transcription factor is a key molecular link between inflammation and cancer (Karin & Greten, 2005). Given this data, it was tempting to hypothesize that increased expression of miR-146a/b-5p might be linked to cancer through inappropriate regulation of the inflammatory response. The identification of KIT, a proto-oncogene which encodes the c-Kit receptor, as a target for miR-146b-5p (He et al, 2005) rather suggested a direct association between changes in miR-146a/b-5p expression and the development of cancer. In this work, we provide further evidence in favour of a direct involvement of miR-146a/b-5p in tumourigenesis.
A substantial amount of new information has been recently published concerning miR-146a/b-5p expression. Of particular interest, comparative analysis of purified, highly tumourigenic CD44+CD24−/lowlineage− cancer stem cell populations with non-tumourigenic cancer cells from three different breast cancers revealed 37 miRNAs showing differential expression, including both miR-146a and miR-146b-5p that were found overexpressed. These two miRs were upregulated in an additional six and nine breast cancer samples, respectively, suggesting a fundamental role in promoting breast tumourigenesis (Shimono et al, 2009). Our demonstration that miR-146a and miR-146b-5p silence BRCA1 provides a highly appealing explanation for their fundamental role in breast tumourigenesis.
It has also been shown, somehow in contradiction with the previous observation, that Breast cancer metastasis suppressor 1 (BMRS1) up-regulates miR-146a/b-5p, which in turn suppress breast cancer metastasis (Hurst et al, 2009). Furthermore, miR-146a was found to be the most up-regulated miRNA in MCF-7 cells resistant to cisplatin in a miRNA microarray expression analysis (Pogribny et al, 2010) and to suppress invasion of pancreatic cancer cells (Li et al, 2010). Further work is needed to better understand the complex connections that exist between the pleiotropic activities of miRNAs in general, and miR-146a and miR-146b-5p in particular.
This novel mechanism of inactivation of the BRCA1 gene in breast and possibly in ovarian sporadic tumours adds up to those already described, i.e. somatic mutation, promoter methylation, haploinsufficiency and transcriptional inhibition. Recently, another molecular mechanism resulting in a BRCA1-deficient-like phenotype, acting this time at the protein level, has been described. Indeed, the BRCA1 protein can be sequestrated in the cytoplasm by the AKT1 serine/threonine protein kinase, which impairs homologous recombination and leads to genetic instability (Plo et al, 2008). It is possible that in some tumours, these mechanisms may act individually, but it is likely that in most cases, a few of them cooperate in order to silence BRCA1, an indeed important breast tumourigenesis actor.
MATERIALS AND METHODS
microRNA target prediction algorithms
Four publicly available algorithms were used in order to predict the miRNAs targeting the 3′ UTR of the BRCA1 gene: Microinspector (http://mirna.imbb.forth.gr/microinspector/), MiRanda (http://www.microrna.org/), RNA22 (http://cbcsrv.watson.ibm.com/rna22.html) and TargetScan (http://www.targetscan.org/archi-ves.html). We used for all algorithms the default parameters. To limit the number of predictions, only the miRNAs predicted to bind their target with a folding energy below −25 kcal/mol were considered for analyses. Furthermore, to reduce the number of false positives, only the miRNAs predicted by at least two algorithms were further considered.
Breast primary tumours
Tissue specimens were obtained from Eric Tabone (Biological Resources Department, Centre Léon Bérard, French agreement number DC-2008-99) and were collected before any therapy from 76 patients suffering from breast cancer diagnosed between 1992 and 1999 who underwent surgery at the Centre Léon Bérard (Lyon, France). A second series of 167 mammary tumours was collected at the Centre René Huguenin, Saint-Cloud, France in accordance with French regulations.
Constructs
The Luc-BRCA1 3′UTR wt vector was constructed by cloning the 3′UTR of BRCA1, amplified by PCR using forward primer 5′-TCGCGACGTCCTGCAGCCAGCCACAGG-3′ (containing the sequence of the Aat II restriction site upstream of the first seventeen nt of the 3′UTR) and reverse primer 5′-GGAATTCCATATGGTTTGCTACCAAAGTTTATTTGCAGTG-3′ (containing the 27th last nt of the 3′UTR upstream the sequence of the Nde I restriction site). The PCR fragment was cloned between the Aat II and Nde I restriction sites in the pGL3-spacer vector, provided by R. Agami, directly downstream the firefly luciferase coding sequence. The integrity of the 3′UTR of BRCA1 was checked by sequencing. To mutate the potential miR-146a/b-5p binding site in the 3′ UTR of BRCA1, site-directed mutagenesis was performed with the two following primers: forward primer 5′-CAGAATAGTCCTTGGGCTGTACTCAAATGTTGGAGTGG-3′ and reverse primer 5′-CCACTCCAACATTTGAGTACAGCCCAAGGACTATTCTG-3′ using the QuickChange XL Site-Directed Mutagenesis kit (Stratagene, Amsterdam, The Netherlands) according to the manufacturer's instructions. The sequence complementary to the miR-146a/b-5p seed sequence, ‘5′-CAGTTCTC-3′’ is mutated to ‘5′-CTGTACTC-3′’ in the resulting vector named Luc-BRCA1 3′UTR mut146.
The pRL-SV40 Renilla luciferase vector (Promega, Charbonnières-les-Bains, France) was used as a transfection control. Vectors expressing the predicted miRNAs (pMSCV-Blast-miR vectors) were kindly provided by R. Agami and are described elsewhere (Voorhoeve et al, 2006). The I-Sce I expression plasmid expresses HA-tagged I-Sce I recombinant proteins (Plo et al, 2008), and the pBRCA1 (1–24) plasmid expresses full length BRCA1 (Anczukow et al, 2008).
All the plasmids used for transfections were prepared with the Nucleobond Xtra Midi Plus kit (Macherey-Nagel, Hoerdt, France) following the manufacturer's instruction.
Cell culture
RG37 and HeLa cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% foetal calf serum and 1% penicillin–streptomycin (Gibco, Cergy Pontoise, France). The human breast cancer cell lines were grown in different cell culture media as described (Neve et al, 2006). All cell lines were grown in a 5% CO2 incubator at 37°C.
miR-146 expression or inhibition in mammary cell lines
In order to express miR-146a and miR-146b-5p, 2 ng of has-miR-146a (ref #PM10722) and has-miR-146b-5p (ref #PM10105) synthetic pre-miR precursors (Applied Biosystem, Warrington, UK) were transfected in MDA-MB-468 mammary cell line. The Pre-miR™ miRNA Precursor Negative Control #1 (ref #AM17110; Applied Biosystem) was used.
In order to inhibit miR-146a and miR-146b-5p, 2 ng of miRCURY LNA knockdown probe (Exiqon, Vedbaek, Denmark) for has-miR146a (ref #138210-00) and has-miR-146b-5p (ref #138604-00) were transfected in MDA-MB-436 or MDA-MB-157 mammary cells. The Control miRCURY knockdown probe (ref #199002-00) commercialized by Exiqon, referred to as the ‘control LNA’, was used as a negative control.
In both cases, 300,000 cells were seeded per well in a six-well plate and transfected 24 h later with the Lipofectamin 2000 transfection reagent (Invitrogen, Cergy Pontoise, France) in the OptiMEM medium (Gibco) following the manufacturer's instructions. After 5 h, the OptiMEM transfection medium was removed. Cells were washed by DMEM during 15 min and recovered in 2 ml of DMEM medium with foetal calf serum and penicillin–streptomycin. Forty-eight hours or 72 h post-transfection, the cells were washed with PBS 1X and recovered.
The paper explained
PROBLEM
The cloning of BRCA1 in 1994 was a major breakthrough in breast cancer research, but the failure to identify somatic mutations in sporadic tumours was highly deceiving as part of the excitement about the identification of BRCA1 came from the expectation that mutations in this gene would trigger breast cancer not only in familial cases but also in the much more common sporadic forms of the disease. Nevertheless, evidence is now accumulating that BRCA1 silencing is of critical importance in the pathogenesis of a significant proportion of sporadic, non-familial cancers and has provided new therapeutic options such as poly(ADP-ribose) polymerase inhibitors. However, the mechanisms underlying BRCA1 somatic inactivation are still not fully understood.
RESULTS
This study reports the identification of two microRNAs negatively regulating BRCA1 expression. In breast tumours, levels of these microRNAs are inversely correlated with that of the BRCA1 protein. Furthermore, we found that they are overexpressed in basal-like mammary tumour epithelial cell lines and in triple negative (ER-, PR-, HER2-) breast cancers. These subtypes share many similarities with BRCA1-associated breast cancers, either transcriptionally or histologically.
IMPACT
This work provides further evidence for the involvement of microRNAs in sporadic breast cancer. It also strengthens the importance for mammary tumorigenesis to silence BRCA1, as several BRCA1 inactivating mechanisms have been described in sporadic breast tumours that are very likely to cooperate in most cases.
RNA interference
The sequences of the small interfering RNA (siRNA) used for inhibiting the BRCA1 gene and of the non-specific siRNA used as a negative control were 5′-GGAACCUGUCUCCACAAAGdTdT-3′ and 5′-CACGAUGUGACAGUGAUAUdTdT-3′, respectively. Both primers were synthesized by Proligo (Sigma–Aldrich, St. Quentin Fallavier, France). Mammary cells were seeded at 300,000 cells per well in six-well plates. Twenty-four hours later, 0.7 µg of siRNA were transfected with the Lipofectamin 2000 transfection reagent (Invitrogen). Forty-eight hours after transfection, the cells were washed with PBS 1X and recovered.
Quantitative RT-PCR
Total RNA was isolated by using the TRI-reagent (Sigma–Aldrich), chloroform extraction and isopropanol precipitation. Ten nanograms of total RNA were reverse-transcribed using the TaqMan® microRNA Reverse Transcription Kit (Applied Biosystem) with a miRNA-specific primer, under the following conditions: 16°C for 30 min; 42°C for 30 min; 85°C for 5 min; and then hold at 4°C. 1.33 µl of each reverse transcription reaction was used in triplicate for the quantitative PCR. The TaqMan® miRNA Assays (Applied Biosystems) that we used to quantitate miRNAs target only mature ones. Reactions were performed with primers specific for hsa-miR-146a (ref #001097) or hsa-miR-146b-5p (ref #00468) in the ABI PRISM® apparatus in 96-well plates at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Target gene expression was normalized based on the values of RNU44 RNA expression (ref #001094). The comparative Ct method was employed for quantification of transcripts according to the manufacturer's protocol.
Northern blot analysis
Total RNA was extracted 48 h after transfection as previously described. Twenty micrograms of total RNA were loaded on a 15% denaturing TBE-urea polyacrylamide gel. After migration, RNA was transferred during 15 min by electroblotting to Hybond N+ membranes (GE Healthcare Amersham Biosciences, Saclay, France). RNA was fixed onto the membrane with UV Stratalinker 1800 (Stratagene). Membranes were probed with γ-32P-ATP end-labelled anti-miR oligonucleotide: anti-parallel miR-146 5′-AACCCATGGAATTCAGTTCTCAGGACAGAG-3′ and PolyT 5′-TTTTTTTTTCTCTGTCC-3′. Hybridization was carried out overnight at 35°C in Church solution containing at least 106 cpm/ml of either probe. After washing four times for 5 min with 2X SSC/0.1% SDS at 35°C, the membrane was subjected to autoradiography for 5 h. 5S RNA expression was used for normalization, and was monitored with the following probe: 5′-TTAGCTTCCGAGATCAGACGA-3′.
Western blot analysis
Cell lines were washed with PBS 1X and were collected by scrapping. After centrifugation at 2000 round per minute, cells were lysed in RIPA buffer (Tris 1.5 M pH 8, NaCl 5 M, NP40 10%, DOC 10%, SDS 10%) with 0.5 mM DTT and 1/50 of complete protease inhibitor cocktail tablets (Roche, Neuilly sur Seine, France). Mammary tumour tissues were homogenized with a cryogenic grinder and total proteins were extracted in the same manner. Equal amounts of total proteins (30 µg for cells and 50 µg for tumours), measured by Bradford assay (Bio-Rad, Marnes-la-Coquette, France), was separated on precast NuPAGE® Novex 3–8% tris-acetate gels at 110 V in NuPAGE® buffer following the manufacturer's instructions (Invitrogen). They were then transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA) and preincubated with methanol during 2 h at 100 V in NuPAGE® Transfert buffer with 10% ethanol and 0.1% antioxidant (sodium bisulfite and N,N-dimethylformamide). Membranes were blocked in 5% milk-TBS Tween 20% and incubated overnight with the #4359 rabbit polyclonal anti-IRAK1 antibody (Cell Signaling Technology Inc., Danvers, MA), the OP107 mouse monoclonal anti-BRCA1 antibody (Calbiochem, Darmstadt, Germany) or the T5168 mouse monoclonal anti-α-tubulin antibody (Sigma–Aldrich). The peroxydase-conjugated affiniPure goat anti-mouse immunoglobin G secondary antibody (Jackson ImmunoResearch Laboratories, Suffolk, England) was used for detection with the Lumi-Light Western blotting substrate (Roche). Signals were either quantified with the EC3 photometer (Scientec, Les Ulis, France) and with the Quantity One software, or with the GelDoc™ XR+ Imager (Bio-Rad) and Image Lab™ software.
Luciferase assay
HeLa cells were seeded at 20,000 cells per well in 96-well plates 17 h before transfection. Synthetic pre-miR precursor or negative control molecules (Applied Biosystem) were transfected as for MDA-MB-468 mammary cell line, with Lipofectamin 2000 transfection reagent (Invitrogen). In the case of the miR-Vec constructs, cells were seeded at 300,000 cells per well in a six-well plate and 100 ng of the plasmid were transfected per well with the Lipofectamin 2000 reagent (Invitrogen). After 5 h, the OptiMEM transfection medium was removed. Cells were washed by DMEM during 15 min and recovered in 2 ml of DMEM medium with foetal calf serum and penicillin–streptomycin. Cells were then immediately transfected with the plasmids encoding the firefly and Renilla luciferase proteins, pGL3-3′UTR-BRCA1 wt or mut146 and the pRL-SV40 vector, respectively (100 ng of each supplemented with DNA carrier up to 3 µg), using the jetPEI reagent (Polyplus Transfection, Illkirch, France) according to the manufacturer's instructions. Cells were washed as for the Lipofectamin 2000 transfection but 24 h after transfection. Forty-eight hours post-transfection with Lipofectamin 2000, cells were washed with 1X PBS and lysed. Firefly and Renilla luciferase activities were measured using the Dual-Glo® Luciferase Assay (Promega) according to the manufacturer's instructions. Firefly luciferase expression was adjusted to Renilla luciferase expression to normalize for transfection efficiency.
Proliferation assay
Cell proliferation was measured 48 h post-transfection using CellTiter-Glo® luminescent cell viability assay (Promega), based on quantification of ATP for determining the number of viable cells in culture following the manufacturer's instructions. Cells were transferred in opaque-walled 96-well plates to record luminescence with a Luminoskan Ascent luminometer (Thermo Fisher Scientific, Illkirch, France).
Homologous recombination assay
RG37 cells were plated at 2 × 105 per well in six-well plates and transfected 24 h later with 100 ng of miR-precursors using INTERFERin™ (Polyplus Transfection, Saint Quentin, France) following the manufacturer's instructions. After 48 h, cells were washed with culture medium and were directly transfected with 0.5 µg of an I-Sce I expressing vector using JetPEI reagent (Polyplus Transfection) following the manufacturer's instructions. Cells were trypsinized 72 h later, washed with PBS 1X at 4°C. GFP+ cells were detected by flow cytometry using a FACScan (Becton Dickson, Le Pont de Claix, France). The expression of I-Sce I was systematically checked with an anti-HA monoclonal antibody (Covance, California, USA) by Western blot analysis.
Statistical analysis
All statistical analyses were performed using the GraphPad Prism Software package (version 5.0). Univariate analysis was performed by using Chi-squared test to compare categorical variables and ANOVA or student test to compare quantitative variables. p-value ≤ 5% was considered as statistically significant.
Acknowledgments
We are indebted to R. Agami (The Netherlands Cancer Institute, Amsterdam, The Netherlands) for generously providing the expression plasmids for miRNAs (miR-Vec constructs), the corresponding empty vector (miR-Vec) and the pGL3-spacer vector. We thank D. G. Cox (CRCL, Lyon, France) and D. Lutringer (UMR5558 CNRS, Lyon, France) for help with the statistical analysis, O. M. Sinilnikova and M. Billaud (CRI Inserm/UJF U823, Grenoble, France) for helpful discussions and S. Kara (U735 INSERM, St-Cloud, France) for skilled technical assistance. This work was supported by the Ligue Nationale contre le Cancer, in the frame of the ‘Equipes labellisées 2008’ program (to SM) and by a grant from the IFR62 to SM and RR.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that there is no conflict of interest.
Author contributions
AG, MB, PB, BSL, IM and SM conceived and designed the experiments. AG, MB and PB performed the experiments. AG, PB, BSL, RL, IM and SM analyzed the data. RR, PB, BSL, ER and RL provided material. AG, IM and SM wrote the paper. All authors discussed the results and commented on the manuscript.
For more information
Online Mendelian Inheritance in Man (OMIM):
BRCA1 http://www.ncbi.nlm.nih.gov/omim/113705
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Abbott DW, Thompson ME, Robinson-Benion C, Tomlinson G, Jensen RA, Holt JT. BRCA1 expression restores radiation resistance in BRCA1-defective cancer cells through enhancement of transcription-coupled DNA repair. J Biol Chem. 1999;274:18808–18812. doi: 10.1074/jbc.274.26.18808. [DOI] [PubMed] [Google Scholar]
- Anczukow O, Ware MD, Buisson M, Zetoune AB, Stoppa-Lyonnet D, Sinilnikova OM, Mazoyer S. Does the nonsense-mediated mRNA decay mechanism prevent the synthesis of truncated BRCA1, CHK2, and p53 proteins. Hum Mutat. 2008;29:65–73. doi: 10.1002/humu.20590. [DOI] [PubMed] [Google Scholar]
- Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br J Cancer. 2000;83:1301–1308. doi: 10.1054/bjoc.2000.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprelikova ON, Fang BS, Meissner EG, Cotter S, Campbell M, Kuthiala A, Bessho M, Jensen RA, Liu ET. BRCA1-associated growth arrest is RB-dependent. Proc Natl Acad Sci USA. 1999;96:11866–11871. doi: 10.1073/pnas.96.21.11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldassarre G, Battista S, Belletti B, Thakur S, Pentimalli F, Trapasso F, Fedele M, Pierantoni G, Croce CM, Fusco A. Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma. Mol Cell Biol. 2003;23:2225–2238. doi: 10.1128/MCB.23.7.2225-2238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann MW, Picard F, An HX, van Roeyen CR, Dominik SI, Mosny DS, Schnurch HG, Bender HG, Niederacher D. Clinical impact of detection of loss of heterozygosity of BRCA1 and BRCA2 markers in sporadic breast cancer. Br J Cancer. 1996;73:1220–1226. doi: 10.1038/bjc.1996.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beger C, Pierce LN, Kruger M, Marcusson EG, Robbins JM, Welcsh P, Welch PJ, Welte K, King MC, Barber JR, et al. Identification of Id4 as a regulator of BRCA1 expression by using a ribozyme-library-based inverse genomics approach. Proc Natl Acad Sci USA. 2001;98:130–135. doi: 10.1073/pnas.98.1.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catteau A, Harris WH, Xu CF, Solomon E. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene. 1999;18:1957–1965. doi: 10.1038/sj.onc.1202509. [DOI] [PubMed] [Google Scholar]
- Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33:1290–1297. doi: 10.1093/nar/gki200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtale G, Citarella F, Carissimi C, Goldoni M, Carucci N, Fulci V, Franceschini D, Meloni F, Barnaba V, Macino G. An emerging player in the adaptive immune response: microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood. 2010;115:265–273. doi: 10.1182/blood-2009-06-225987. [DOI] [PubMed] [Google Scholar]
- Dahiya N, Sherman-Baust CA, Wang TL, Davidson B, Shih Ie M, Zhang Y, Wood W, III, Becker KG, Morin PJ. MicroRNA expression and identification of putative miRNA targets in ovarian cancer. PLoS One. 2008;3:e2436. doi: 10.1371/journal.pone.0002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumay A, Laulier C, Bertrand P, Saintigny Y, Lebrun F, Vayssiere JL, Lopez BS. Bax and Bid, two proapoptotic Bcl-2 family members, inhibit homologous recombination, independently of apoptosis regulation. Oncogene. 2006;25:3196–3205. doi: 10.1038/sj.onc.1209344. [DOI] [PubMed] [Google Scholar]
- Eitan R, Kushnir M, Lithwick-Yanai G, David MB, Hoshen M, Glezerman M, Hod M, Sabah G, Rosenwald S, Levavi H. Tumor microRNA expression patterns associated with resistance to platinum based chemotherapy and survival in ovarian cancer patients. Gynecol Oncol. 2009;114:253–259. doi: 10.1016/j.ygyno.2009.04.024. [DOI] [PubMed] [Google Scholar]
- Elstrodt F, Hollestelle A, Nagel JH, Gorin M, Wasielewski M, van den Ouweland A, Merajver SD, Ethier SP, Schutte M. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Res. 2006;66:41–45. doi: 10.1158/0008-5472.CAN-05-2853. [DOI] [PubMed] [Google Scholar]
- Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- He H, Jazdzewski K, Li W, Liyanarachchi S, Nagy R, Volinia S, Calin GA, Liu CG, Franssila K, Suster S, et al. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci USA. 2005;102:19075–19080. doi: 10.1073/pnas.0509603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JT, Thompson ME, Szabo C, Robinson-Benion C, Arteaga CL, King MC, Jensen RA. Growth retardation and tumour inhibition by BRCA1. Nat Genet. 1996;12:298–302. doi: 10.1038/ng0396-298. [DOI] [PubMed] [Google Scholar]
- Hurst DR, Edmonds MD, Scott GK, Benz CC, Vaidya KS, Welch DR. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer Res. 2009;69:1279–1283. doi: 10.1158/0008-5472.CAN-08-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human microRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Labbaye C, Spinello I, Quaranta MT, Pelosi E, Pasquini L, Petrucci E, Biffoni M, Nuzzolo ER, Billi M, Foa R, et al. A three-step pathway comprising PLZF/miR-146a/CXCR4 controls megakaryopoiesis. Nat Cell Biol. 2008;10:788–801. doi: 10.1038/ncb1741. [DOI] [PubMed] [Google Scholar]
- Lakhani SR, Manek S, Penault-Llorca F, Flanagan A, Arnout L, Merrett S, McGuffog L, Steele D, Devilee P, Klijn JG, et al. Pathology of ovarian cancers in BRCA1 and BRCA2 carriers. Clin Cancer Res. 2004;10:2473–2481. doi: 10.1158/1078-0432.ccr-1029-3. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Li Y, Vandenboom TG, II, Wang Z, Kong D, Ali S, Philip PA, Sarkar FH. miR-146a Suppresses invasion of pancreatic cancer cells. Cancer Res. 2010;70:1486–1495. doi: 10.1158/0008-5472.CAN-09-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SL, Chiang A, Chang D, Ying SY. Loss of mir-146a function in hormone-refractory prostate cancer. RNA. 2008;14:417–424. doi: 10.1261/rna.874808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavaddat N, Antoniou AC, Easton DF, Garcia-Closas M. Genetic susceptibility to breast cancer. Mol Oncol. 2010;4:174–191. doi: 10.1016/j.molonc.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mialhe A, Lafanechere L, Treilleux I, Peloux N, Dumontet C, Bremond A, Panh MH, Payan R, Wehland J, Margolis RL, et al. Tubulin detyrosination is a frequent occurrence in breast cancers of poor prognosis. Cancer Res. 2001;61:5024–5027. [PubMed] [Google Scholar]
- Mignone F, Gissi C, Liuni S, Pesole G. Untranslated regions of mRNAs. Genome Biol. 2002;3:S0004. doi: 10.1186/gb-2002-3-3-reviews0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- Mueller CR, Roskelley CD. Regulation of BRCA1 expression and its relationship to sporadic breast cancer. Breast Cancer Res. 2003;5:45–52. doi: 10.1186/bcr557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai MA, Yamamoto L, Salaorni S, Pacheco MM, Brentani MM, Barbosa EM, Brentani RR, Mazoyer S, Smith SA, Ponder BA, et al. Detailed deletion mapping of chromosome segment 17q12-21 in sporadic breast tumours. Genes Chromosomes Cancer. 1994;11:58–62. doi: 10.1002/gcc.2870110109. [DOI] [PubMed] [Google Scholar]
- Nakasa T, Miyaki S, Okubo A, Hashimoto M, Nishida K, Ochi M, Asahara H. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum. 2008;58:1284–1292. doi: 10.1002/art.23429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Baldwin DA, Scearce LM, Oberholtzer JC, Tobias JW, Mourelatos Z. Microarray-based, high-throughput gene expression profiling of microRNAs. Nat Methods. 2004;1:155–161. doi: 10.1038/nmeth717. [DOI] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederacher D, Picard F, van Roeyen C, An HX, Bender HG, Beckmann MW. Patterns of allelic loss on chromosome 17 in sporadic breast carcinomas detected by fluorescent-labeled microsatellite analysis. Genes Chromosomes Cancer. 1997;18:181–192. doi: 10.1002/(sici)1098-2264(199703)18:3<181::aid-gcc5>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Pacifico F, Crescenzi E, Mellone S, Iannetti A, Porrino N, Liguoro D, Moscato F, Grieco M, Formisano S, Leonardi A. Nuclear factor-{kappa}B contributes to anaplastic thyroid carcinomas through up-regulation of miR-146a. J Clin Endocrinol Metab. 2010;95:1421–1430. doi: 10.1210/jc.2009-1128. [DOI] [PubMed] [Google Scholar]
- Pauley KM, Satoh M, Chan AL, Bubb MR, Reeves WH, Chan EK. Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Res Ther. 2008;10:R101. doi: 10.1186/ar2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner-Svensson HM, Lindsay MA. Rapid changes in microRNA-146a expression negatively regulate the IL-1beta-induced inflammatory response in human lung alveolar epithelial cells. J Immunol. 2008;180:5689–5698. doi: 10.4049/jimmunol.180.8.5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plo I, Laulier C, Gauthier L, Lebrun F, Calvo F, Lopez BS. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res. 2008;68:9404–9412. doi: 10.1158/0008-5472.CAN-08-0861. [DOI] [PubMed] [Google Scholar]
- Pogribny IP, Filkowski JN, Tryndyak VP, Golubov A, Shpyleva SI, Kovalchuk O. Alterations of microRNAs and their targets are associated with acquired resistance of MCF-7 breast cancer cells to cisplatin. Int J Cancer. 2010;127:1785–1794. doi: 10.1002/ijc.25191. [DOI] [PubMed] [Google Scholar]
- Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol. 2008;26:2568–2581. doi: 10.1200/JCO.2007.13.1748. [DOI] [PubMed] [Google Scholar]
- Rice JC, Ozcelik H, Maxeiner P, Andrulis I, Futscher BW. Methylation of the BRCA1 promoter is associated with decreased BRCA1 mRNA levels in clinical breast cancer specimens. Carcinogenesis. 2000;21:1761–1765. doi: 10.1093/carcin/21.9.1761. [DOI] [PubMed] [Google Scholar]
- Rusinov V, Baev V, Minkov IN, Tabler M. MicroInspector: a web tool for detection of miRNA binding sites in an RNA sequence. Nucleic Acids Res. 2005;33:W696–W700. doi: 10.1093/nar/gki364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Ambrosone CB, DiCioccio RA, Odunsi K, Lele SB, Zhao H. A functional polymorphism in the miR-146a gene and age of familial breast/ovarian cancer diagnosis. Carcinogenesis. 2008;29:1963–1966. doi: 10.1093/carcin/bgn172. [DOI] [PubMed] [Google Scholar]
- Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkoly E, Wei T, Janson PC, Saaf A, Lundeberg L, Tengvall-Linder M, Norstedt G, Alenius H, Homey B, Scheynius A, et al. MicroRNAs: novel regulators involved in the pathogenesis of psoriasis. PLoS One. 2007;2:e610. doi: 10.1371/journal.pone.0000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- Turner NC, Reis-Filho JS, Russell AM, Springall RJ, Ryder K, Steele D, Savage K, Gillett CE, Schmitt FC, Ashworth A, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 2007;26:2126–2132. doi: 10.1038/sj.onc.1210014. [DOI] [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhoeve PM, le Sage, Schrier C, Gillis M, Stoop AJ, Nagel H, Liu R, van Duijse YP, Drost J, Griekspoor J, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–1181. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- Wang X, Tang S, Le SY, Lu R, Rader JS, Meyers C, Zheng ZM. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS One. 2008;3:e2557. doi: 10.1371/journal.pone.0002557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weberpals JI, Clark-Knowles KV, Vanderhyden BC. Sporadic epithelial ovarian cancer: clinical relevance of BRCA1 inhibition in the DNA damage and repair pathway. J Clin Oncol. 2008;26:3259–3267. doi: 10.1200/JCO.2007.11.3902. [DOI] [PubMed] [Google Scholar]
- Williams AE, Perry MM, Moschos SA, Larner-Svensson HM, Lindsay MA. Role of miRNA-146a in the regulation of the innate immune response and cancer. Biochem Soc Trans. 2008;36:1211–1215. doi: 10.1042/BST0361211. [DOI] [PubMed] [Google Scholar]
- Wyman SK, Parkin RK, Mitchell PS, Fritz BR, O'Briant K, Godwin AK, Urban N, Drescher CW, Knudsen BS, Tewari M. Repertoire of microRNAs in epithelial ovarian cancer as determined by next generation sequencing of small RNA cDNA libraries. PLoS One. 2009;4:e5311. doi: 10.1371/journal.pone.0005311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki K, Nakasa T, Miyaki S, Ishikawa M, Deie M, Adachi N, Yasunaga Y, Asahara H, Ochi M. Expression of microRNA-146a in osteoarthritis cartilage. Arthritis Rheum. 2009;60:1035–1041. doi: 10.1002/art.24404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.