

See related article in EMBO Mol Med (Zhou et al (2011) EMBO Mol Med http://dx.doi.org/10.1002/emmm.201100138)
Amyloid precursor protein (APP) is a transmembrane protein, which is sequentially cleaved at several sites by different enzymes. Cleavage leads to the production of different Aβ (β amyloid) species, which—when overproduced and aggregated—are a major cause of Alzheimer's disease.
The main cleavage sites in APP are the β- and γ/ε-sites mediated by the respective secretases (Fig 1). Importantly, mutations in APP and the presenilins have been shown to cause Alzheimer's disease by a remarkable number of mechanisms. Now, it seems there is one at a previously underestimated site.
Figure 1. Pathogenic mutations in APP.
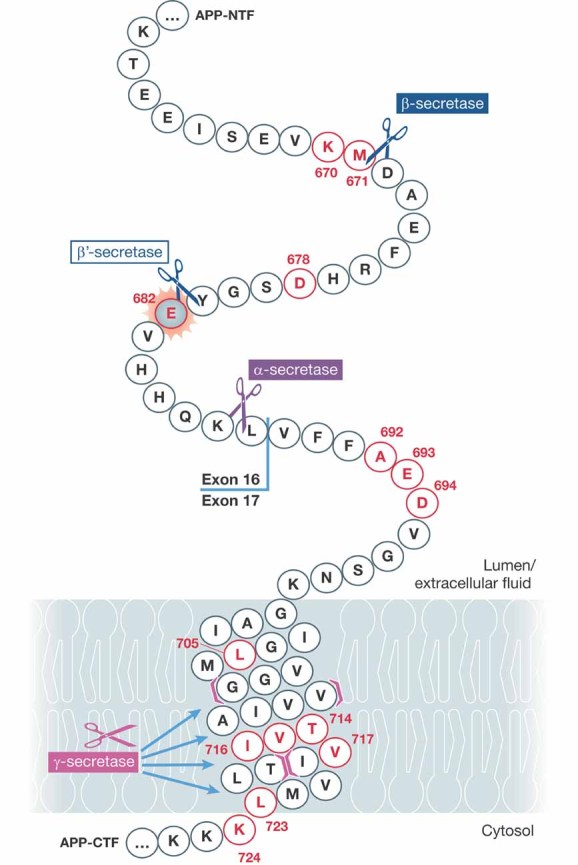
Amino acids highlighted in red are known to be pathogenic. The newly discovered mutation affecting cleavage at the β′-site is highlighted in grey. Cleavage sites for different secretases are indicated. Note that the γ-secretase cleaves APP at multiple sites starting between amino acid 718 and 719 and then proceeding towards 709 and 710. Modified from http://www.alzforum.org/res/com/mut/app/diagram1.asp, courtesy of Richard Crook and the Alzheimer Research Forum.
APP gene duplications and Down syndrome seem to simply cause more flux through the processing pathway by APP gene dose (Rovelet-Lecrux et al, 2006), the London series of mutations and presenilin mutations increase the proportion of long Aβ species by marginally altering the production from the γ-secretase cleavage (Scheuner et al, 1996), and the Flemish and similar Alzheimer causing mutations appear to reduce flux through the competing α-secretase pathway (De Jonghe et al, 1998). Perhaps most famously, the Swedish mutation increased the flux through the β pathway and produced more Aβ beginning at residue 1 of Aβ (APP residue 672). In one of the most elegant experiments in the Alzheimer literature, Citron and colleagues (Citron et al, 1995) showed by exhaustive mutagenesis that only the Swedish mutation (M671L) was a better substrate for β-secretase cutting at this site: this specificity is one of the most powerful arguments for the amyloid hypothesis of the disease (Hardy & Selkoe, 2002). However, the β-secretase cleavage of APP is more complicated than simply cutting between residues 671 and 672 yielding peptides beginning with D672 since there is an alternative β cleavage (named β′) between residue 681 and 682 (positions 10 and 11 of the Aβ sequence) (Yang et al, 2004). This cleavage generates an alternative series of Aβ molecules beginning at E682 (position 12 of Aβ).
Remarkably, in this issue of EMBO Molecular Medicine Zhou et al (2010) report a mutation (APP E682K) occurring in a case of early onset Alzheimer's disease, which disrupts this β′ cleavage and which, like the Swedish mutation, leads to more cleavage between residues 671 and 672 and thus more full-length Aβ. This is potentially important because it shows that these two cleavage sites are genuine alternatives to each other and implies that the peptides starting at position 11 are significantly less amyloidogenic than those starting at position 1. Clearly, it also implies that if we could modulate β-secretase cleavage towards the β′-site, it would have potential therapeutic benefit (although it is not obvious how this might be achieved).
»… if we could modulate β-secretase cleavage towards the β′-site, it would have potential therapeutic benefit…«
These interesting results and interpretations are subject to a caveat however, that we cannot be sure that the mutation was genuinely pathogenic. Genetic causation can only be proven by either linkage (segregation within a family) or association (more frequent occurrence in affected individuals than in controls). In this case, the mutation has only been found in a single individual and so neither the criteria for linkage nor association have been met. However, clearly, we now know something of the biology of autosomal dominant Alzheimer's disease initiation and it is therefore tempting, and to some extent, appropriate, to assign pathogenicity based on the relevant biology of the mutation. We have suggested a formal process for this assessment in the regard to APP and presenilin mutations (Guerreiro et al, 2010), and, under this system, this variant (APP E682K) would be assigned ‘possible’ pathogenic mutation status. As the authors briefly discuss, the problems in assigning pathogenic status even to APP variants in Alzheimer's disease are real: assigning pathogenic status to variants in a genome wide fashion will clearly be extremely difficult.
Acknowledgments
The authors declare that they have no conflict of interest.
References
- Citron M, et al. Neuron. 1995;14:661–670. doi: 10.1016/0896-6273(95)90323-2. [DOI] [PubMed] [Google Scholar]
- De Jonghe C, et al. Neurobiol Dis. 1998;5:281–286. doi: 10.1006/nbdi.1998.0202. [DOI] [PubMed] [Google Scholar]
- Guerreiro RJ, et al. Neurobiol Aging. 2010;31:725–731. doi: 10.1016/j.neurobiolaging.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, et al. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Scheuner D, et al. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Yang HC, et al. J Neurochem. 2004;91:1249–1259. doi: 10.1111/j.1471-4159.2004.02764.x. [DOI] [PubMed] [Google Scholar]
- Zhou L, et al. EMBO Mol Med. 2011 DOI: 10.1002/emmm.201100138. [Google Scholar]