

Abstract
Improved colonoscopy is revealing precancerous lesions that were frequently missed in the past, and ∼30% of those detected today have nonpolypoid morphologies ranging from slightly raised to depressed. To characterize these lesions molecularly, we assessed transcription of 23,768 genes in 42 precancerous lesions (25 slightly elevated nonpolypoid and 17 pedunculated polypoid), each with corresponding samples of normal mucosa. Nonpolypoid versus polypoid morphology explained most gene expression variance among samples; histology, size, and degree of dysplasia were also linked to specific patterns. Expression changes in polypoid lesions frequently affected cell-cycling pathways, whereas cell-survival dysregulation predominated in nonpolypoid lesions. The latter also displayed fewer and less dramatic expression changes than polypoid lesions. Paradigmatic of this trend was progressive loss through the normal > nonpolypoid > polypoid > cancer sequence of TMIGD1 mRNA and protein. This finding, along with TMIGD1 protein expression patterns in tissues and cell lines, suggests that TMIGD1 might be associated with intestinal-cell differentiation. We conclude that molecular dysregulation in slightly elevated, nonpolypoid, precancerous colorectal lesions may be somewhat less severe than that observed in classic adenomatous polyps.
Keywords: colorectum, preinvasive tumors, TMIGD1, transcriptomics
INTRODUCTION
Two decades of in-depth investigation have shed important light on the complexity and heterogeneity of human colorectal cancer. Several distinct phenotypes have been identified at the clinical, histologic and molecular levels, and the differences can also be observed in premalignant colorectal lesions. Once referred to collectively as colorectal polyps, these precursor lesions are now classified at endoscopy as polypoid and nonpolypoid. The former category comprises lesions that protrude into the gut lumen, including pedunculated and sessile forms. At the other end of the spectrum are the nonpolypoid lesions, which are still widely referred to as ‘flat’. Most are actually slightly elevated above the mucosal surface. Mildly depressed forms are rare, but even small lesions of this type are generally farther along the road to cancer than their slightly elevated counterparts (Endoscopic Classification Review Group, 2005; Soetikno et al, 2008). O'Brien et al highlighted the impact of this new terminology in their reassessment of superficial neoplasms classified as polyps in the United States National Polyp Study. Around 30% of these tumours met the new criteria for nonpolypoid lesions, including two-thirds of those originally labelled sessile polyps (O'Brien et al, 2004). A similar picture emerged from recent studies (Kudo et al, 2008, and references herein), many of which employed high-resolution colonoscopes, magnification, and chromoendoscopy.
The more accurate classification of premalignant lesions in terms of their appearance at endoscopy has led to some important observations. For example, true polypoid lesions are more frequently detected in the distal colon (from the splenic flexure to the rectum), whereas nonpolypoid neoplasms are more common in the proximal colon (from the cecum to the splenic flexure). Furthermore, while most nonpolypoid lesions exhibit adenomatous changes with variable degrees of cellular dysplasia (like their polypoid counterparts), others display infolding of the glandular epithelium that produces a saw-toothed (or serrated) pattern in longitudinally sectioned epithelial crypts (Snover et al, 2005). These serrated lesions include morphologically distinct subsets (e.g. with or without dysplasia), and many present molecular alterations that are rare in adenomas, such as BRAF mutations or the CpG island methylator phenotype, which is characterized by silencing of several cancer-relevant gene promoters (O'Brien, 2007). The DNA mismatch repair gene MLH1 is a very common target of CpG island methylation (Herman et al, 1998). Its silencing transforms precursor lesions into mismatch repair-deficient adenocarcinomas, which are almost always located in the proximal colon and occur 5–10 years later than other sporadic colorectal cancers.
The present study was designed to test the hypothesis that morphological differences among precancerous colorectal lesions reflect distinct tumourigenic pathways. To this end, we compared the global gene expression profile of nonpolypoid preinvasive lesions (whose roles in colon tumourigenesis are still poorly understood) with those of normal colonic mucosa and polypoid lesions.
RESULTS
As shown in Table 1, the endoscopic criteria used for lesion collection (see Materials and Methods) provided us with two relatively homogeneous tissue groups. The 17 pedunculated polypoid lesions [type Ip in the Paris classification (Paris Workshop Participants, 2003)] were taken from all segments of the colorectum, and all were adenomas. On the whole, they were appreciably smaller than the 25 slightly elevated nonpolypoid lesions (type IIa; mean diameters: 23.3 mm vs. 29 mm, respectively), which were deliberately collected from the proximal colon. The aim of this restriction was to maximize the chances of obtaining lesions associated with a nonadenomatous pathway of tumourigenesis (see Introduction). In fact, 5 (20%) of the nonpolypoid lesions exhibited serrated histology, including 3 that were nondysplastic. The other 20 were adenomas with some degree of cellular dysplasia (Table 1). High-degree dysplasia was more common in the nonpolypoid group (n = 7, 28% vs. n = 3, 17.6% of the polypoid lesions), but this difference is probably related to the larger size of the type IIa lesions (see above). The highly dysplastic lesions with polypoid morphology were much smaller than their counterparts in the nonpolypoid group (mean diameters: 25 mm vs. 40 mm, respectively). Three of the 5 serrated lesions harboured a BRAF V600E mutation (as expected), but none exhibited epigenetic silencing of MLH1 (Supporting Information Table 1).
Table 1.
Characteristics of the 37 patients with nonpolypoid and polypoid precancerous lesions included in the study (42 lesions).
| Patienta | Age | Sex | Colon segment involvedb | Maximum lesion diameter (mm) | Macroscopic appearancec | Pit patternd | Microscopic appearancee | Dysplasiaf | No. lesions presentg |
|---|---|---|---|---|---|---|---|---|---|
| Nonpolypoid | |||||||||
| 1a* | 75 | F | Tra | 70 | IIa-IIb | 3S-3L | VA | high | 7 |
| 1b* | Tra | 20 | IIa | 3S-3L | VA | high | |||
| 2a* | 72 | F | Cec | 30 | IIa | 4 | TA | low | 9 |
| 2b* | Tra | 20 | IIa | 3S-3L | TA | low | |||
| 3 | 75 | F | Tra | 40 | IIa-IIc | 3S-3L | TA | high | 1 |
| 4 | 73 | F | Asc | 40 | IIa-IIc | 2-3L | SA (mixed) ≠ | high | 1 |
| 5 | 44 | M | HF | 35 | IIa | 3S-3L | TA | low | 2 |
| 6 | 79 | F | Asc | 20 | IIa | 4 | TA | low | 1 |
| 7 | 67 | F | Asc | 20 | IIa | 3L | TA | low | 1 |
| 8 | 48 | M | HF | 15 | IIa | 3L | TA | low | 1 |
| 9 | 69 | F | Asc | 40 | IIa | 3L | TVA | low | 1 |
| 10 | 83 | M | Asc | 25 | IIa | 3L | TA | low | 4 |
| 11 | 66 | M | Cec | 25 | IIa | 3L | TA | high | 2 |
| 12 | 79 | M | Asc | 35 | IIa-IIc | 5N | TVA | high | 2 |
| 13a* | 54 | F | Cec | 45 | IIa | 3L | TVA | low | 16 |
| 13b* | Asc | 12 | IIa | 3L | TA | low | |||
| 14 | 74 | F | Cec | 50 | IIa | 3L | TVA | low | 1 |
| 15 | 62 | F | Asc | 15 | IIa | 3S-3L | SA (mixed) | low | 1 |
| 16 | 52 | F | Tra | 15 | IIa | 2 | SSA ≠ | no dysplasia | 1 |
| 17a* | 54 | F | Cec | 50 | IIa | 2-3L | TA | high | 2 |
| 17b* | Asc | 20 | IIa | 2-3L | TA | low | |||
| 18 | 72 | M | Asc | 25 | IIa | 3S-3L | TA | low | 1 |
| 19a* | 79 | M | Asc | 25 | IIa | 3L | VA | low | 3 |
| 20 | 47 | M | Asc | 20-25 | IIa-IIc | 3S-3L | MVSP ≠ | no dysplasia | 3 |
| 21 | 66 | M | Asc | 12 | IIa | 3L | MVSP | no dysplasia | 1 |
| Polypoid | |||||||||
| 22 | 64 | F | Asc | 15 | Ip | nr | TA | low | 2 |
| 23 | 56 | M | Asc | 15 | Ip | nr | TA | low | 2 |
| 24 | 79 | M | Asc | 20 | Ip | nr | TVA | low | 3 |
| 25 | 27 | M | Asc | 15 | Ip | nr | TVA | high | 3 |
| 26 | 40 | F | Sig | 30 | Ip | nr | TVA | low | 51 § |
| 27 | 83 | M | Sig | 12 | Ip | nr | TVA | low | 3 |
| 28 | 50 | M | Sig | 30 | Ip | nr | TVA | high | 3 |
| 29 | 74 | F | Sig | 20 | Ip | nr | TVA | low | 1 |
| 30 | 69 | M | Rec | 40 | Ip | nr | TA | low | 3 |
| 31 | 69 | M | Sig | 30 | Ip | nr | TA | low | 7 |
| 32 | 56 | M | Sig | 30 | Ip | nr | TA | low | 3 |
| 33 | 58 | F | Des | 20 | Ip | nr | TA | low | 3 |
| 34 | 52 | F | Sig | 30 | Ip | nr | TA | low | 3 |
| 35 | 69 | F | Sig | 15 | Ip | nr | TVA | low | 3 |
| 36 | 58 | F | Asc | 15 | Ip | nr | TVA | low | 1 |
| 19b* | 79 | M | Tra | 30 | Ip | nr | VA | low | 3 |
| 37 | 73 | M | Asc | 30 | Ip | nr | TVA | high | 1 |
Two lesions were analyzed from patients marked with an asterisk.
Abbreviations: Cec, cecum; Asc, ascending colon; HF, hepatic flexure; Tra, transversum; Des, descending colon; Sig, sigma; Rec, rectum.
Classified according to the Paris Endoscopic Classification of Superficial Neoplastic Lesions (Paris Workshop Participants, 2003). All nonpolypoid lesions were slightly elevated (type IIa); a few also included a small region that was completely flat (IIb) or depressed (IIc).
Kudo classification of colonic crypt morphology (Kudo et al, 2001). Abbreviations: nr, Pit pattern not reported by the endoscopist.
Abbreviations: TA, tubular adenoma; TVA, tubulovillous adenoma; VA, villous adenoma; MVSP, microvescicular serrated polyp; SA, serrated adenoma; SSA, sessile serrated adenoma. ≠, Lesions carrying BRAFV600E mutation (valine to glutamate substitution at codon 600).
Highest degree of dysplasia in the lesion based on the WHO classification of tumors of the digestive system (Editorial and consensus conference in Lyon, France, November 6-9, 1999 [IARC]).
Total number of lesions noted during the study colonoscopy, including those used in the present study. §, Patient with suspected attenuated polyposis. The mutational analysis of the APC and MYH genes is currently not available.
Expression of 23,768 transcript clusters was analysed in all 42 precancerous lesions and their corresponding samples of normal mucosa (see Materials and Methods and Supporting Information). Unsupervised principal component analysis (PCA; Fig 1A) demonstrated that much of the variance among the samples was due to interindividual variability (PC axis 1, PC1), but a considerable portion of the residual variance was accounted for by tissue type (i.e. normal mucosa, polypoid lesions and nonpolypoid lesions; PC2). This variable was therefore used as the prime grouping factor in the supervised between-group analysis (BGA) based on correspondence analysis (CoA; Fig 1B). Normal mucosa samples were clearly segregated from the precancerous lesions, and the polypoid and nonpolypoid subsets within the latter category were also clearly distinct. [These distinctions were still evident after exclusion from the BGA of the three nondysplastic serrated lesions (Supporting Information Fig 1).]
Figure 1. Principal component and between-group analyses of gene expression data for the 84 colorectal tissue samples.
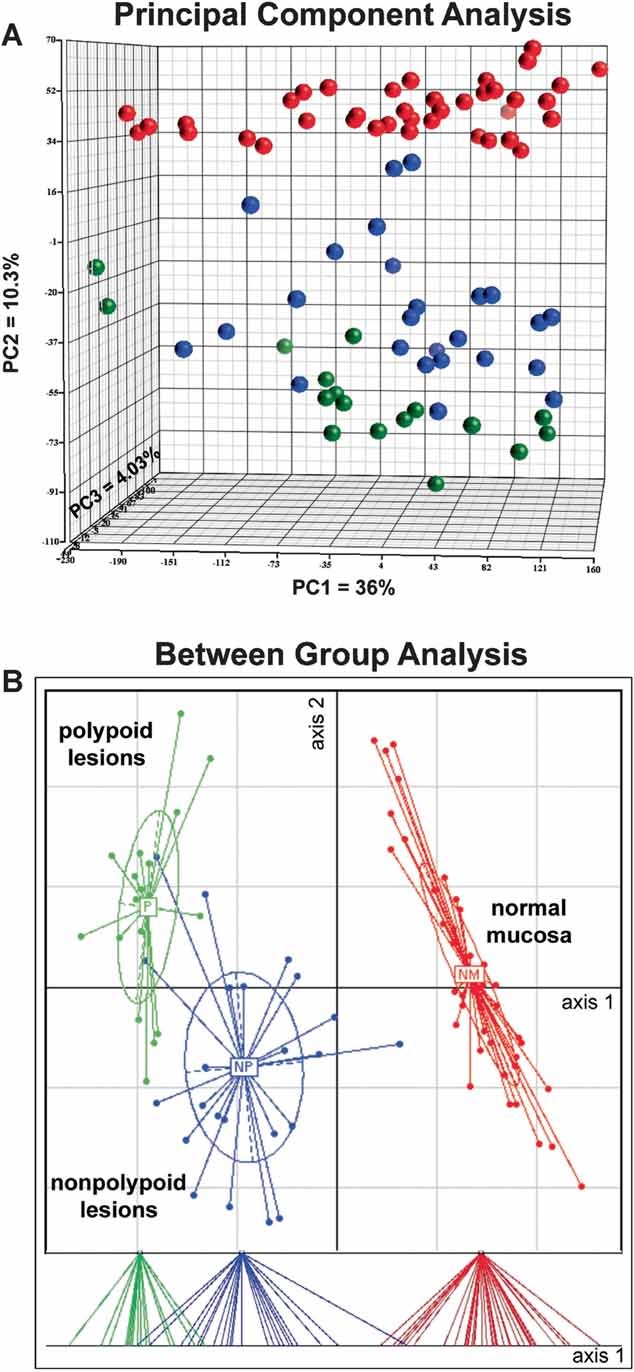
- Three-dimensional PCA score plot of log2 expression intensity values for the 42 normal mucosa samples (red spheres); 25 nonpolypoid lesions (blue); and 17 polypoid lesions (green). The first three principal components (PCs) account for 50.3% of total variance. PC axis 1 (PC1), the main direction of spread within all three tissue groups, reflects intragroup, interindividual variability. PC2 reflects intergroup variance based on tissue type, the factor responsible for segregating specimens into three groups. No specific biological variables correlated with the variance explained by PC3 (<5% of total) or the remaining variance in the data set (49.7% of the total). The data quality and model reliability are reflected in the absence of outliers and the high proportion of variance explained by PCs 1 and 2 (46.3% vs. 25% in a randomized PCA model of this set of samples and genes).
- BGA based on correspondence analysis (CoA) of log2 gene expression intensity values for samples grouped by tissue type. CoA discriminated between normal and lesional samples (axis 1) and between polypoid and nonpolypoid lesions (axis 2). The dispersion of scores along axis 2 reflects high-interindividual variability. Lower panel: Projection of scores on axis 1.
To identify other clinical variables that would explain the detected gene expression changes, we reduced the noise of the interindividual variability that had emerged from both the BGA and PCA. To this end, transcript levels were expressed as ratios of the lesional value to the basal level observed in the corresponding sample of normal mucosa. These log2 ratio expression values were then subjected to redundancy analysis (RDA), in which clinical descriptors (tissue type, sex, age, histology, diameter, and degree of dysplasia) were independent (or explanatory) variables, and the gene expression profile was the dependent (or explained) variable predicted by the clinical descriptors (Supporting Information). As shown in Fig 2A, over half (53.2%) of the total variance in this data set was explained by four variables—tissue type, histology, diameter, and degree of dysplasia. The most important was tissue type (i.e. nonpolypoid vs. polypoid lesions): it was described by the longest vectors, which pointed in opposite directions, reflecting the negative correlations between nonpolypoid and polypoid lesions for gene expression. A map of the sample scores and the 20 genes whose expression levels most effectively discriminated between polypoid and nonpolypoid lesions are shown in Fig 2B. (The top 200 discriminating genes are listed in Supporting Information Table 2.)
Figure 2. Clinical and pathologic variables that predict the gene expression changes.
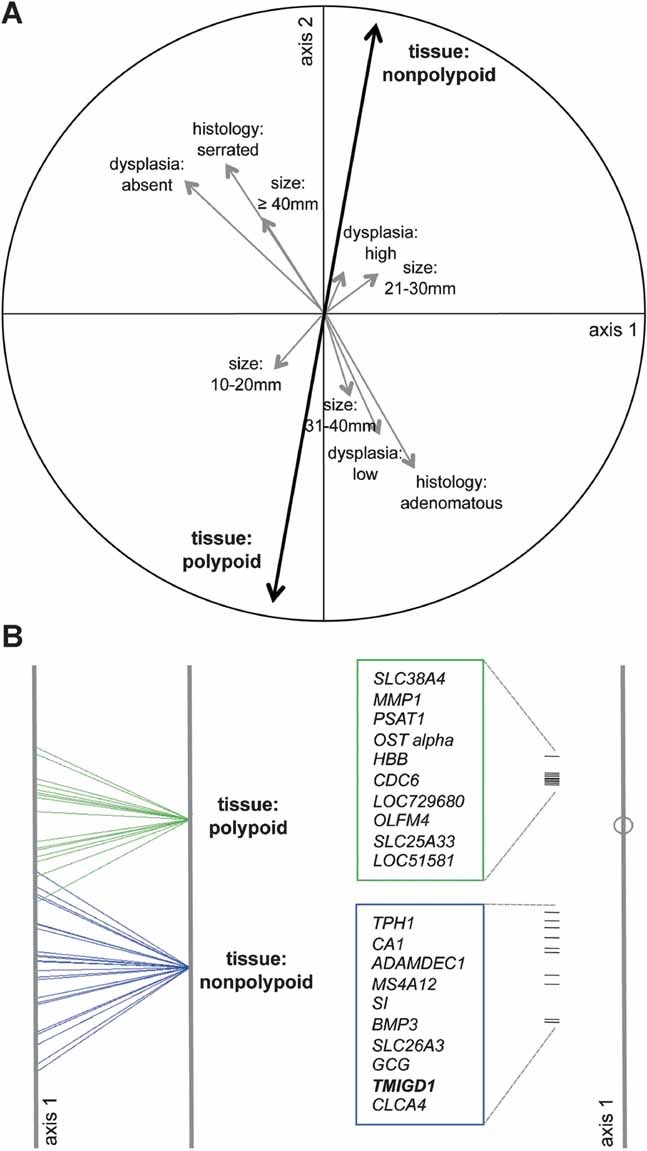
- Redundancy analysis (RDA). Correlation circle (based on log2 ratio expression intensity values) illustrates the proportion of total variance supported by different variables, each depicted as a vector. Vector length (maximum: radius of the circle) reflects the variable's relevance in the reduced coordinate system defined by RDA axes 1 and 2; vector direction reflects intervariable correlations (e.g. oppositely oriented vectors represent negatively correlated variables). Most of the variance in this data set was explained by tissue type (polypoid vs. nonpolypoid), followed by histology (adenomatous vs. serrated), degree of dysplasia (no dysplasia, low and high degree of dysplasia), and lesion diameter.
- Left: BGA of log2 ratio expression intensity values grouped by tissue type (nonpolypoid vs. polypoid). When grouping is based on a dichotomous variable like this, only the scores on BGA axis 1 are meaningful. Right: The 20 genes whose expression levels were mostly responsible for segregation of polypoid and nonpolypoid samples are plotted along the same BGA axis 1 (ranked according to score magnitude—positive or negative—on axis 1, which is proportional to the distance from the center of the axes to the correlation circle).
Polypoid and nonpolypoid lesions differed significantly in terms of the numbers of genes with expression levels different from those in normal mucosa, and this significance persisted at various false discovery rates (FDR 0.001–0.05; Supporting Information Fig 2). Genes altered in both groups of lesions represented over half of those dysregulated in nonpolypoid lesions and a lower proportion of those that were differentially expressed in polypoid lesions. In most cases, the direction of the alteration (upregulation/downregulation) was the same in both groups.
Because all our nonpolypoid lesions came from the proximal colon (i.e. not beyond the splenic flexure), some of the differences they exhibited with respect to the polypoid lesions (which came from all parts of the colon and rectum) might be related to their location instead of their morphology. In fact, gene expression profiles within our polypoid lesion group displayed obvious colon-segment-related differences (as expected; Supporting Information Fig 3). Nonetheless, the distal colon and proximal colon subsets in this group were both clearly distinguishable from the nonpolypoid lesions. Furthermore, when tissue type and location were included in a two-way ANOVA as potentially interacting factors, most of the genes whose expression levels discriminated between the nonpolypoid and polypoid lesion groups were found to be unrelated to the colon segment of origin (Supporting Information Table 2).
Specific gene expression patterns were also associated with lesion histology, size, and degree of dysplasia (Fig 3). Diameter and degree of dysplasia were significantly associated with distinct expression profile clusters, which confirms our previous observations (Sabates-Bellver et al, 2007) in 32 polypoid lesions analysed with a different microarray platform. However, as shown in Fig 3A, the present analysis also revealed that serrated and adenomatous lesions are clearly distinct at the transcriptome level.
Figure 3. Pathologic features of precancerous lesions that correlate with specific gene expression profiles.
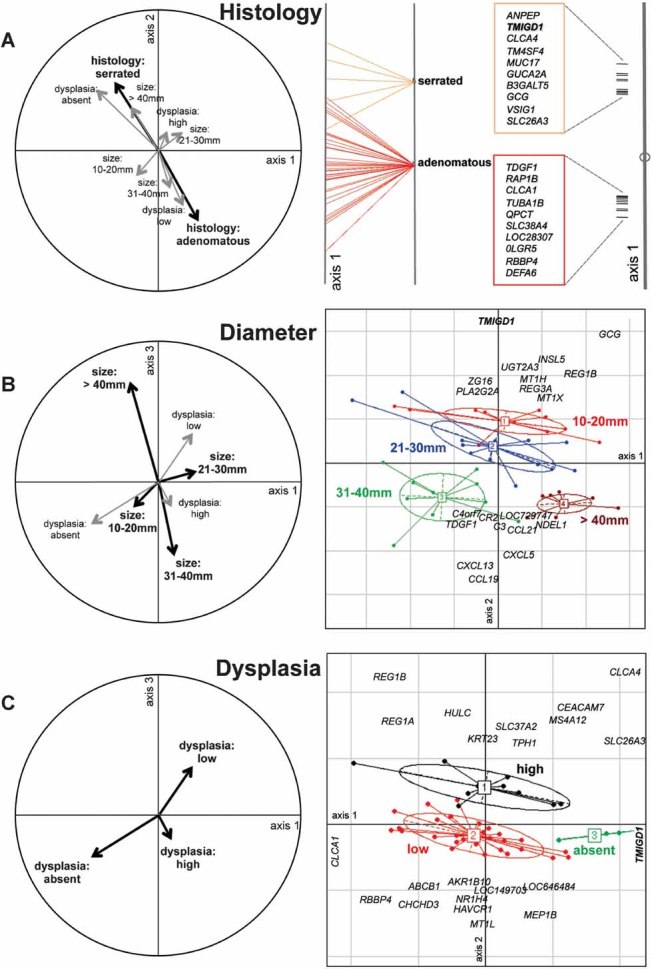
Correlation circles (left) and BGA plots (right) for variables identified by RDA as the second (A), third (B) and fourth (C) most significant predictors of gene expression patterns (after tissue type; Fig 2).
- Lesion histology (adenomatous vs. serrated). One-dimensional plot of scores on BGA axis 1 with the top 20 genes responsible for this discrimination (see Fig 2 for details).
- Lesion diameter (four size groups). For nondichotomous classifiers like this (and dysplasia, shown in panel C), scores on BGA axes 1 and 2 are meaningful, and results are visualized as biplots. Each size group is delimited by a probability ellipse (reflecting the two-dimensional 67% boundaries of a gaussian distribution, i.e. 1 standard deviation unit) with a labelled centroid. Along BGA axis 2, the red and blue ellipses representing smaller lesions (diameter ≤ 30 mm) are clearly separated from those representing larger (≥31 mm) lesions (green and brown). The top 20 genes responsible for segregating these lesions into these two categories are listed on the graph.
- Degree of dysplasia (high vs. low vs. absent). Lesions with high and low degrees of dysplasia (black and red ellipses, respectively), are completely segregated on axis 2, and the 20 genes that most responsible for this separation are reported. Lesions with and without dysplasia are segregated along axis 1. Results are visualized only for the genes with highest positive (TMIGD1) and negative (CLCA1) scores.
The transcriptomes of polypoid and nonpolypoid lesions were also analysed to identify the molecular pathways that are dysregulated in the two types of tissues. As shown in Fig 4A, the pathways altered in polypoid lesions were predominantly concerned with cell-cycle regulation, whereas alterations in oxidative phosphorylation, ubiquinone metabolism, and IGF-1 signalling were more characteristic of the nonpolypoid transformation process. Other pathways exhibited similar degrees of dysregulation in the two types of lesions, including the Wnt signalling cascade (Fig 4B). The latter finding was confirmed by the results of immunohistochemical assessment of β-catenin expression (Supporting Information Fig 4).
Figure 4. Cellular pathways that play major roles in the development of polypoid versus nonpolypoid precancerous lesions of the colon.
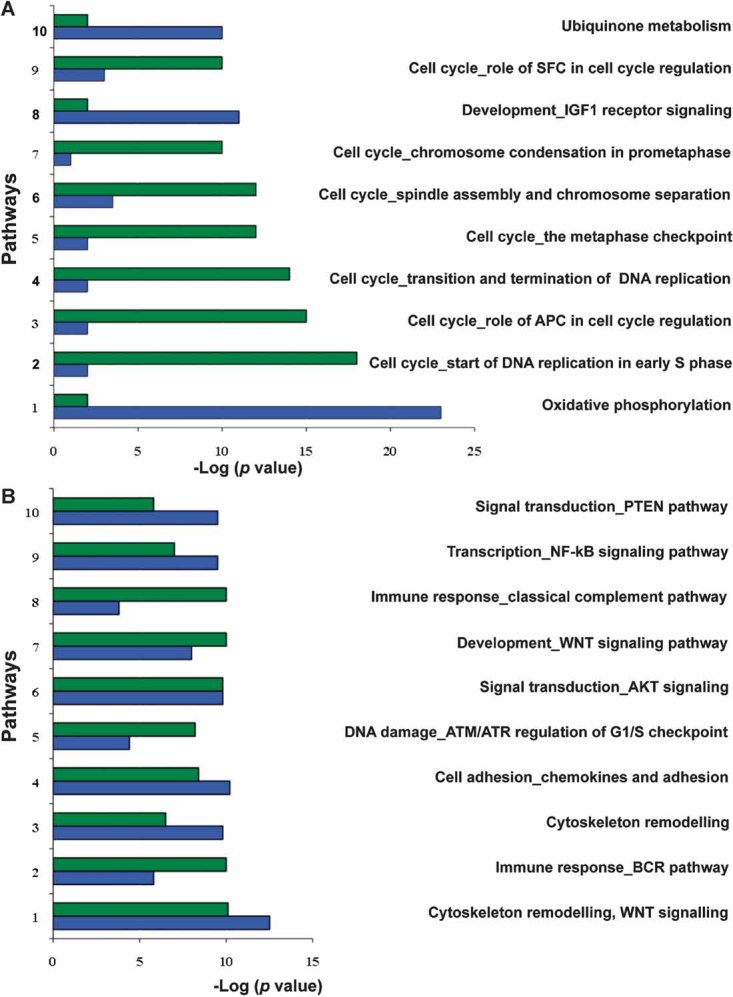
Two lists of genes whose expression was significantly altered (p < 0.05 vs. normal mucosa) in polypoid and nonpolypoid lesions were analysed with the GeneGo MetaCore software (www.genego.com; St. Joseph, MI, USA), which assigned each gene to a specific cellular pathway on the basis of data available in the Gene Ontology catalogue (Ashburner et al, 2000). We calculated the proportion of total genes in a given pathway that were differentially expressed. The statistical significance of the involvement of a given pathway is positively related to this proportion (p-value on the x-axis). Green and blue bars refer to polypoid and nonpolypoid lesions, respectively (compared with the corresponding normal mucosa).
- The 10 pathways whose dysregulation in polypoid differed most markedly from that observed in nonpolypoid lesions. For example, in nonpolypoid lesions 50% (49/98) of the genes involved in oxidative phosphorylation were differentially expressed (vs. normal mucosa) compared with only 15% (15/98) in polypoid lesions.
- The 10 pathways whose dysregulation in polypoid lesions was most similar to that observed in nonpolypoid lesions.
TMIGD1 ranked very high on the list of genes whose expression levels varied markedly with tissue type (Fig 2B), lesion size, and histology (Fig. .3; Supporting Information Table 2). The constant representation of this gene in the multivariate analyses reported thus far reflects its progressive downregulation as transformation advances. As shown in Fig 5A, its expression was clearly decreased in nonpolypoid lesions (fold-change; FC vs. normal mucosa: −19), but the downregulation was much more evident in polypoid lesions (FC −66) and even more dramatic in a small series of advanced cancers (FC −125) we recently examined with exon arrays.
Figure 5. TMIGD1 mRNA expression in colorectal tissues and cancer cell lines.
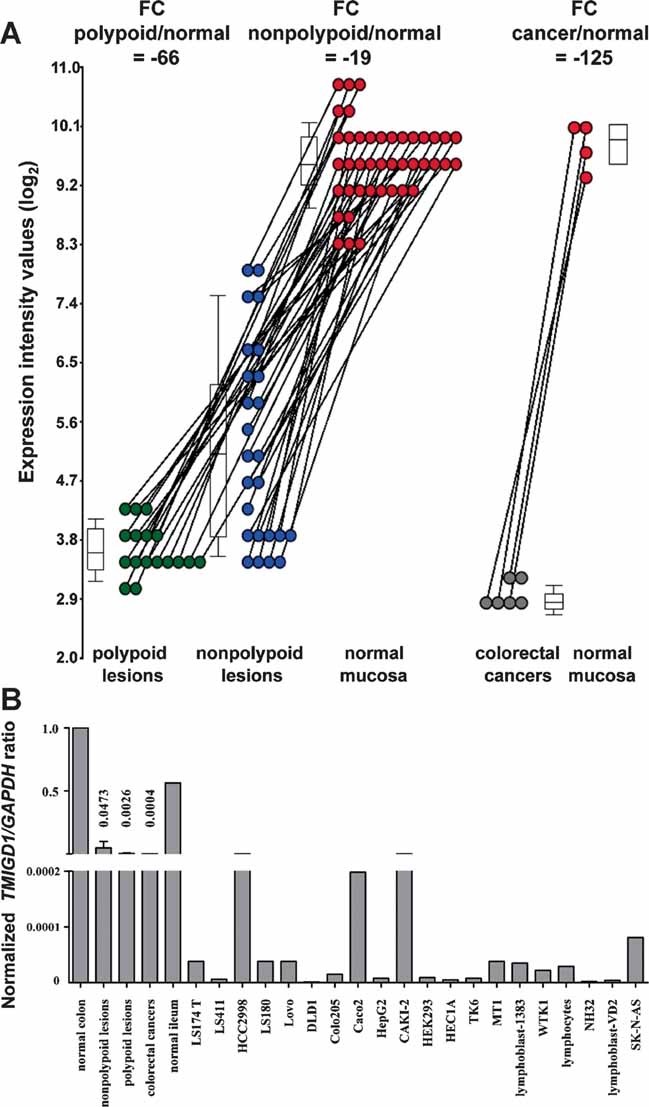
- Normalized log2 expression intensity values of TMIGD1 mRNA detected by exon array analysis in polypoid and nonpolypoid precancerous lesions (green and blue dots, respectively). Grey dots: Corresponding values for six colorectal cancers recently investigated with the same arrays. Results are expressed as fold changes (FCs) versus values observed in corresponding samples of normal mucosa (red dots). Box plots show 25th, 50th (median), and 75th percentiles of lesional expression values.
- Real time quantitative RT-PCR confirmed TMIGD1 mRNA expression levels in the colorectal tissues investigated in the microarray study. Transcript was also detected in the normal terminal ileum and in several cell lines. Other cell lines (SW480, Colo741, HT29, HT29M6, Co115, SW48, CX1, SW837, HCT116, SW620, Vaco481, SW403, LnCaP, MCF7, Hela, BEAS2B, U2OS, HEK293T, A2780, LAN-1, and Sk-N-SH) did not express TMIGD1. TMIGD1 (target) and GAPDH (reference) coding regions were amplified. y-axis: [target concentration/reference concentration] for sample/[target concentration/reference concentration] for normal mucosa.
This trend has never been described (probes for this gene were not present in previous-generation microarray platforms), and the function of TMIGD1 is unknown, so we attempted to better characterize the expression pattern of this gene in colorectal and other tissues. Real time quantitative polymerase chain reaction (after reverse transcription) (RT-PCR) analysis (Fig 5B) confirmed that TMIGD1 transcription was significantly decreased in the precancerous and cancerous lesions we examined with microarrays [compared with normal mucosal expression in different segments of the colon, the rectum (data not shown), and the terminal ileum]. TMIGD1 transcript was also found in normal epithelial cells from the kidney and trachea (Supporting Information Fig 5).
Promoter hypermethylation does not seem to be the cause of the downregulated TMIGD1 expression. The TMIGD1 promoter lacks canonical CpG islands whose epigenetic alteration can repress transcription. Furthermore, treatment of the TMIGD1-negative colon cancer cell line Co115 with a DNA methyltransferase inhibitor (5-aza-2′ deoxycytidine) and a histone deacetylase inhibitor (trichostatin A) had no effect on TMIGD1 expression (data not shown). Using the TRANSFAC database (Matys et al, 2003), we identified several putative transcription factor binding sites (TFBSs) around the transcription start site of TMIGD1. Three of these sequences were predicted to bind factors encoded by the MEIS1, HNF4A, and NFE2L1 genes, which were transcriptionally underexpressed in the precancerous lesions (Supporting Information Fig 6). HNF4A has been shown to upregulate the expression of murine Tmigd1 (Ishikawa et al, 2008), and a recent genome-wide ChIP-chip study in human hepatoma cells (HepG2) identified an HNF4A binding site distal to the TMIGD1 start site (Wallerman et al, 2009). These findings are interesting because the HNF4 transcription factor family seems to regulate the expression of numerous cell differentiation-induced genes in the enterocytes of small intestinal villi in mice (Stegmann et al, 2006). Our data therefore raise the possibility that TMIGD1 expression is somehow associated with differentiation of intestinal epithelial cells.
Immunohistochemistry studies revealed TMIGD1 protein levels that paralleled those of TMIGD1 transcript. In normal colorectal mucosa, the protein is confined mainly to the upper crypt compartments, which contain differentiated cells (Fig 6); no expression was noted in the proliferative crypt compartment. Similar patterns were observed in the small intestine, where TMIGD1 expression was limited to the villi (and was maximal at the brush border), and in the kidney (Supporting Information Fig 7). In line with our gene expression data, TMIGD1 expression in colorectal tissues diminished markedly in precancerous and malignant lesions, paralleling losses of differentiation in these tissues (Fig 6).
Figure 6. Immunohistochemical staining of normal and neoplastic colonic tissues with antibodies against TMIGD1.
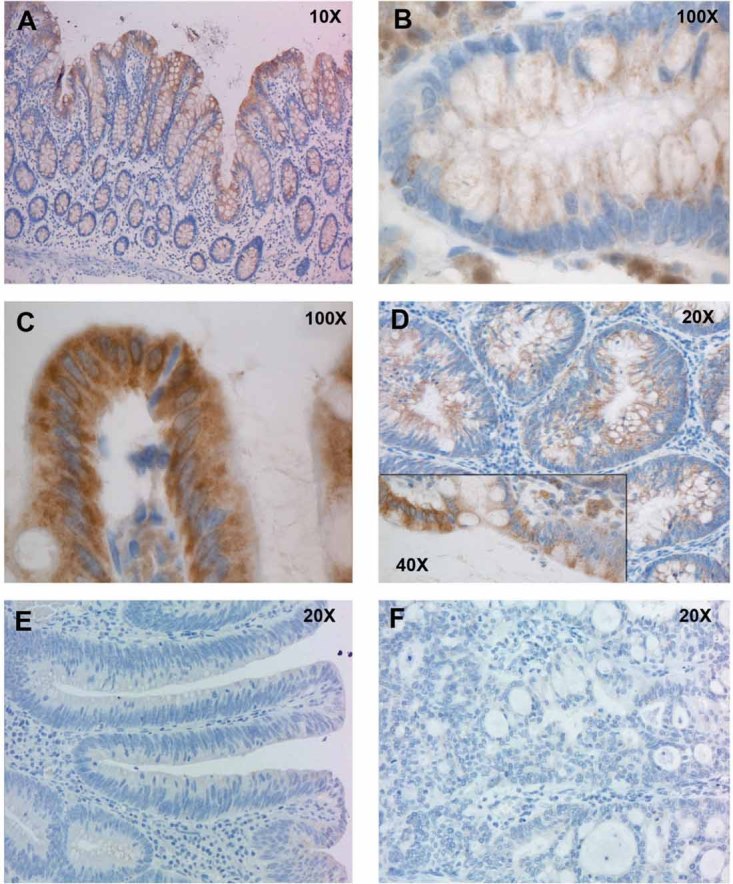
- In normal mucosa, TMIGD1 expression is limited to the upper portion of the epithelial crypts, where differentiated cells are located.
- Higher magnification views of TMIGD1 staining at base of a colonic crypt.
- Higher magnification views of TMIGD1 staining at mouth of a colonic crypt. TMIGD1 is located in the cytoplasm and probably in the cell membrane.
- Its expression was markedly reduced in nonpolypoid lesions. The inset shows different levels of expression at the interface between normal (left) and dysplastic (right) epithelium.
- More marked reduction was observed in polypoid lesions, and
- expression was lost in colorectal cancers.
The specificity of the antibody used in these studies was ascertained with Western blot and immunocytochemistry experiments performed in SW480 colon cancer cells transfected with TMIGD1 cDNA (Fig 7A and B). The results indicate that TMIGD1 is a nonsecreted, glycosylated protein located in the cytoplasm and cell membrane. Its possible relation to cell differentiation was explored in Caco2 colon cancer cells, where persistent contact-mediated inhibition of growth triggers differentiation towards the absorptive intestinal cell lineage (Peterson & Mooseker, 1992) reflected by increased expression of the differentiation marker VIL1. As shown in Fig 7C, confluence-induced differentiation in these cells was accompanied by increasingly marked upregulation of TMIGD1 transcription and mildly increased expression of the corresponding protein, which was undetectable in extracts from log-phase Caco2 cells (Fig 7D).
Figure 7. TMIGD1 mRNA and protein expression studies in colon cancer cells.
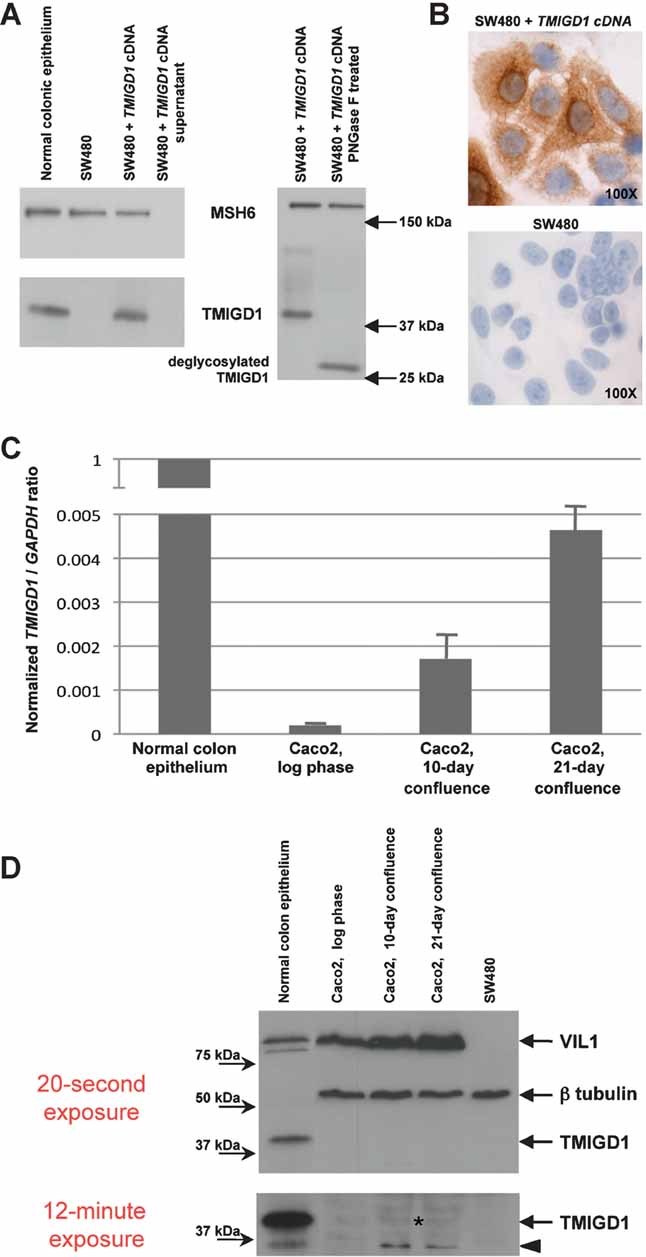
- Western blot analysis of TMIGD1 protein expression in total cell extracts from epithelial colonic crypts; SW480 colon cancer cells (which do not express TMIGD1 mRNA—see legend to Fig 5) and SW480 cells transiently transfected with the TMIGD1 cDNA (positive control). Proteins were separated on a 12% polyacrylamide gel. TMIGD1 (apparent molecular weight: ∼39 kDa) was not found in cell supernatants, and migration accelerated (right-hand blot) when protein extracts were treated with the deglycosylating enzyme PNGase F, behaviour suggestive of a nonsecreted glycosylated protein. The nuclear protein MSH6 (nonsecreted and nonglycosylated) was used as a control.
- Immunocytochemistry. TMIGD1 expression in SW480 cells transiently transfected with full-length TMIGD1 cDNA. The protein is highly expressed in the perinuclear compartment (probably associated with the rough endoplasmic reticulum) and cell membrane. Negative control: SW480 cells transfected with the empty vector.
- Real time quantitative RT-PCR assay of TMIGD1 mRNA levels in Caco2 colon cancer cells: Compared with proliferating cells (log phase), those undergoing confluence-induced differentiation exhibited progressive increases in TMIGD1 mRNA levels (8.7-fold and 23.6-fold increases on post-confluence days 10 and 21, respectively). TMIGD1 (target) and GAPDH (reference) coding regions were amplified. y-axis: [target concentration/reference concentration] for sample/[target concentration/reference concentration] for normal mucosa. Columns and bars represent mean values and SD, respectively, of duplicate experiments. (Reference expression was the mean observed in the normal mucosa sample, indicated as 1).
- Western blot assay of TMIGD1 protein expression in proliferating and differentiating Caco2 cells. Upper panel (20-s exposure): Confluence-induced differentiation is reflected by increasing expression of the intestinal differentiation marker VIL1 on post-confluence days 10 and 21. SW480 colon cancer cells: negative control for VIL1. Lower panel (12-min exposure): In Caco2 cells undergoing confluence-induced differentiation (post-confluence days 10 and 21), there was a slight increase in TMIGD1 protein expression (asterisk) compared with the undetectable expression that characterized proliferating cells (log-phase). In the differentiating colon-cancer cells (and in the normal mucosa positive control as well), there was also a second lower molecular weight band (arrowhead) possibly representing a TMIGD1 degradation product or isoform.
DISCUSSION
Not all precancerous colorectal lesions develop into carcinomas. This fate is more commonly seen in depressed (type IIc) lesions (Endoscopic Classification Review Group, 2005; Soetikno et al, 2008), but this subset includes only ∼1% of all precancerous lesions. Type-IIa nonpolypoid lesions, which are slightly elevated above the mucosal surface, are much more common. For several reasons, they, too, are commonly believed to be more likely to undergo malignant transformation than polypoid lesions. For one thing, they are easier to miss during standard colonoscopy (especially those located in the proximal colon; Lambert et al, 2009, and references herein). In addition, epigenetic silencing of the DNA mismatch repair gene MLH1 seems to be particularly frequent in nonpolypoid lesions in the proximal colon (O'Brien, 2007, and references herein). (Mismatch repair deficiency dramatically increases mutation rates, and this is believed to markedly accelerate the transformation process.) And finally, Soetikno et al have reported a frequency of in situ or submucosal carcinoma in nondepressed, nonpolypoid lesions that was four times higher than that observed in polypoid lesions (Soetikno et al, 2008).
Reliable characterization of the malignant potential of nonpolypoid lesions requires high-throughput molecular analysis of large numbers of lesions. Our study represents the first attempt to fill this gap. We investigated the entire complement of RNA transcripts in 25 nonpolypoid precancerous lesions and compared the results with those obtained in corresponding specimens of normal colonic mucosa and in 17 polypoid lesions (A total of 84 samples were investigated, a relatively large series for this type of study.) The exon array platform we used explores the expression of all exons, annotated and putative, in the human genome. We limited the present data analysis to a subset of 23,768 well-annotated genes, because our main interest lies in expression changes involving proteins or regulatory RNAs with potential roles in the transformation process. When we extended our analysis to the 235,234 non-annotated transcript clusters (many of which probably represent noncoding genomic regions), the discriminatory power of the expression changes dropped sharply (data not shown). We also excluded data on the expression of alternatively spliced transcript isoforms. This information can be obtained with the exon platform, but we have found serious limitations in the software tools available for its analysis.
The clinical variable that explained the largest proportion of the variation in our data set was tissue type. This means that the transcriptomes of nonpolypoid and polypoid lesions are not only different from that of the normal mucosa: they can also be readily distinguished from one another. At the molecular level, nonpolypoid lesions differed from their polypoid counterparts in two respects.
The first was quantitative. As shown in Supporting Information Fig 2, compared with the polypoid lesions, those in the nonpolypoid group had fewer genes displaying transcript levels significantly different from those found in the normal mucosa. Consequently, the nonpolypoid transcriptome was somewhat closer to that of the normal mucosa (Fig 1B). Many genes, however, displayed altered expression in both types of lesions, and in these cases the alterations were usually concordant in terms of direction, but those observed in the nonpolypoid lesions were less dramatic. In-depth evaluation of expression intensity values for single genes across all the samples (Supporting Information Fig 8) revealed that type IIa lesions occupy an intermediate position between normal mucosa and polypoid lesions. In other words, many genes exhibit progressive up- or downregulation of transcription across the normal mucosa → nonpolypoid → polypoid sequence.
These findings suggest that—in general—nonpolypoid lesions might be less advanced on the road to cancer than polypoid lesions. For example, loss of cell differentiation, a hallmark of tumourigenesis, might be less pronounced in these slightly elevated precursor lesions, as reflected by our findings for many genes known to be involved in colorectal epithelial cell differentiation (e.g. CA1, GCG, CLCA4, AQP8, and GUCA2A; Fig 2B and Supporting Information Table 2) and also those for TMIGD1. The latter genes's association with cell differentiation is suggested by our preliminary experiments, which showed that the protein it encodes is expressed exclusively in the cell-differentiation compartment of normal colorectal crypts. Furthermore, its expression in Caco2 cells is restored by confluence-induced differentiation, an effect that was evident mainly at the transcriptional level. Indeed, the increase in TMIGD1 protein expression was far more modest. It is important to recall, however, that Caco2 cells are a colon cancer cell line, and although they can be forced to undergo some form of in vitro differentiation mediated by transcriptional regulation resembling that seen during normal-cell differentiation, they may also retain feedback mechanisms that truncate all or part of this process, e.g. by post-transcriptional suppression of the expression of one or more proteins associated with normal-cell differentiation. Therefore, the absence of substantially upregulated TMIGD1 protein expression in differentiating Caco2 cells does not exclude the possibility that this protein is associated with normal colonocyte differentiation. Like the ascertained differentiation genes listed above, TMIGD1 exhibited downregulation that was less marked in nonpolypoid precursors than in polypoid lesions and cancers. Our findings contradict those of Soetikno et al (Soetikno et al, 2008), but they are more consistent with three recent studies of large series of superficial colorectal neoplasms, which revealed frequencies of submucosal carcinoma in nondepressed, nonpolypolid lesions that were similar to (Bianco et al, 2010; Park et al, 2008) or substantially lower than (Kudo et al, 2008) those observed in polypoid lesions.
It is important to note that endoscopic type IIa lesions are more heterogeneous than type Ip lesions. Indeed, log2 expression intensity values for most genes (e.g. Fig 5A) displayed somewhat higher standard deviations (average, 0.43 vs. 0.39 for polypoid lesions) in the nonpolypoid group. This variability is in part a reflection of the transcriptome differences between the serrated and adenomatous subsets in this group (Fig 3A). However, the subsets in this group are fairly small, which raises the possibility of artefacts related to low statistical power and data overfitting in the BGA. Conclusions regarding these subtypes must, therefore, be regarded with caution. Neverthless, the list of genes identified by BGA is highly enriched for those whose dysregulated expression might be relevant to colorectal tumourigenesis, and this list is, therefore, highly useful for planning future studies.
The second difference between the two types of lesions was qualitative. In polypoid lesions, GeneGO Metacore analysis revealed a striking preponderance of dysregulated genes related to cell-cycle regulation (Fig 4A). Important controllers of DNA replication initiation (e.g. CDC6 and MCM7, Supporting Information Fig 9) or S phase and mitotic checkpoints (e.g. CCNA2, CDC2, CDC14A, DSCC1 and BUB1) were generally upregulated in these lesions. In contrast, nonpolypoid lesions typically displayed alterations in cell-survival pathways, including the IGF-1 receptor signalling (known to confer survival advantages; Samani et al, 2007) and those involved in oxidative phosphorylation (including ubiquinone metabolism). As shown in Supporting Information Fig 10, these findings suggest that oxidative phosphorylation may be compromised to some extent in nonpolypoid lesions. Partial or complete defects of this type have been shown to induce resistance to apoptosis in yeast (Harris et al, 2000) and mammalian cells (Tomiyama et al, 2006), and Jass has suggested that evasion of apoptosis may be the pathogenic mechanism that gives rise to the glandular serrated phenotype (Higuchi & Jass, 2004). It is tempting to speculate that cell proliferation in polypoid and nonpolypoid lesions is enhanced by different means: profound dysregulation of the cell cycle in the former, hyperactivation of growth factor signalling pathways and suppression of apoptosis in the latter. It is important to recall, however, that polypoid and nonpolypoid lesions also share important features, including the dysregulation of pathways believed to play fundamental roles in the early stages of colorectal transformation, such as those of the WNT signalling, cytoskeleton remodelling and immune responses (Fig 4B).
Our current data must be used with caution in formulating conclusions on the malignant potentials of these preinvasive lesions. Although polypoid and nonpolypoid lesions are clearly different at the molecular level, tumourigenesis in each group (the latter in particular) may well be a heterogeneous process. This possibility needs to be explored in much larger series of lesions that differ in stage (size) and segment of origin. We intend to investigate this complicated process of transformation with a two-pronged approach. The first involves integration of our transcriptomic findings with data from epigenomic, metabolomic, and proteomic studies (currently ongoing in our laboratory). The second entails the functional characterization of single molecules whose expression is dramatically altered in tumours. Our preliminary experiments point to TMIGD1 as a possible cell-differentiation marker in the lower intestine (and probably in the kidney and trachea as well), whose expression is lost during cellular transformation. In silico analysis indicates that TMIGD1 might play a role in cell adhesion since it contains an immunoglobulin-like domain similar to that found in adhesion molecules of the Ig-CAM family. Our next step is to explore its functional roles and determine whether its underexpression does indeed promote progression of tumourigenesis in the intestinal epithelium.
MATERIALS AND METHODS
Endoscopic samples and RNA processing
The study protocol was approved by the local Ethics Committee, and each participating tissue donor provided written informed consent. We prospectively collected 42 precancerous colorectal lesions (each accompanied by three biopsies of normal mucosa from the same colon segment, but >2 cm from the lesion) during colonoscopy. Twenty-five were slightly elevated (<2.5 mm above the mucosal surface) nonpolypoid lesions (type IIa or mixed lesions in the Paris classification; Paris Workshop Participants, 2003); the other 17 were pedunculated (type Ip) polypoid lesions. All but one (collected from a patient with suspected attenuated familial polyposis) were sporadic lesions.
Immediately after removal, a small sample (20–30 mg) of epithelial tissue was cut from each lesion, leaving the underlying muscularis mucosae intact. The rest of the specimen was submitted for pathologic analysis. (We used only lesions measuring ≥1 cm to ensure that the sampling procedure would not interfere with the histologic diagnosis.) This approach provided specimens with a high percentage of epithelial cells without microdissection, which can diminish the quantity and quality of the extracted RNA.
The tissue fragments (lesional epithelium and normal mucosa) were stored in RNAlater (Ambion, Huntington, UK) and later homogenized. Total RNA was extracted with the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and its integrity verified by capillary gel electrophoresis (Experion, BioRad, Hercules, CA, USA). Only RNAs with a 28S:18S ribosomal RNA ratio between 1.5 and 2.2 were processed for microarray analysis. After ribosomal RNA reduction, the RNA sample was reverse-transcribed to cDNA with random hexamers tagged with a T7 promoter. The cDNA was amplified with T7 RNA polymerase and subjected to a second cDNA synthesis. The sense-oriented, single-stranded DNA produced with this step was then fragmented, biotin-labelled and hybridized to the GeneChip Human Exon 1.0 ST array (Affymetrix, Santa Clara, CA, USA). Arrays were analysed in the Affymetrix GeneChip Scanner 3000 7G. Cell intensities (CEL files) were measured with Affymetrix GeneChip Operating Software (GCOS) and Affymetrix Expression Console Software was used for quality assessment.
The paper explained
PROBLEM
Colorectal cancers arise from precancerous lesions with different endoscopic and microscopic features. Thanks to improvements in colonoscopy technique and technology, precancerous lesions are being detected that were often missed in the past. This is particularly true for nonpolypoid neoplasms, which currently account for ∼30% of precancerous colorectal lesions. They are often described as ‘flat’ since they tend to spread laterally (instead of protruding into the gut lumen like the more familiar colorectal polyp). A greater understanding of the molecular features that distinguish polypoid and nonpolypoid precursors of colorectal cancer is essential for their effective clinical management and for cancer prevention.
RESULTS
Using endoscopy, we collected biopsies from 42 precancerous colorectal lesions representing the two most frequent endoscopic types: stalked colorectal polyps and slightly elevated, nonpolypoid lesions—each with corresponding samples of normal colorectal mucosa. Each sample was analysed to identify its transcriptome—that is, messenger RNA expression levels for each of the over 20,000 genes making up the human genome. As expected, the transcriptomes of the precancerous lesions as a whole differed markedly from that of the normal colorectal mucosa, but the two types of precancerous lesions were also clearly different from one another. Compared with the classical polyps, the nonpolypoid lesions displayed fewer genes with significantly altered expression, and when the same gene was altered in both lesion types, the dysregulation in the polypoid group was usually less dramatic. In addition, the expression changes observed in the two groups often affected different molecular pathways.
IMPACT
Our findings indicate that slightly elevated nonpolypoid colorectal lesions progress towards cancer along a relatively distinct route, exploiting certain specific molecular pathways along with others shared with their polypoid counterparts (although the dysregulation at this level is less severe). These data confirm recently published clinical evidence suggesting that slightly elevated nonpolypoid lesions may have a lower malignant potential than classic colorectal polyps. With time (and growth), however, these lesions, too, can transform into cancer, so high-quality routine colonoscopy is encouraged to ensure their early detection and removal.
Raw expression data generated by the GCOS were preprocessed with the Partek Genomics Suite (Partek, St. Louis, MO, USA) and analysed in the R statistics environment with BioConductor packages (www.bioconductor.org). Probe expression intensity in each tissue sample was subjected to background adjustment and normalization with the robust multiarray analysis (RMA) algorithm (Irizarry et al, 2003). Raw transcriptomic data have been deposited in GEO (accession number GSE21962).
Microarray analysis
Our data (not shown) and others' (Robinson & Speed, 2007) confirm that the human exon array offers improved sensitivity and specificity and more reliable detection of biological variability compared with previous-generation arrays. The platform includes ∼5.4 million probes, which target all the exons—annotated and predicted—in the genome. Earlier platforms interrogated only the 3′ end of mRNA sequences, but exon arrays contain a set of four probes for each putative exonic region. These probesets (1.4 million) can be virtually reassembled into ∼260,000 transcript clusters, one tenth of which are likely to encode proteins. The so-called core probesets target exons with RefSeq mRNA; those that target exons with EST evidence only are referred to as extended probesets. The present analysis was limited to data obtained with the 228,871 core probesets (17,881 transcript clusters) plus the most completely annotated sets of the extended category (74,732, corresponding to 5887 transcript clusters). All 23,768 of these well-annotated transcript clusters displayed above-background expression levels in the 84 tissue samples we examined.
RMA-preprocessed expression data were subjected to unsupervised analysis PCA and supervised multivariate analyses (BGA-based on CoA, and RDA; Supporting Information; Baty et al, 2008; Culhane et al, 2002; Quinn and Kenough, 2006; Ringner, 2008). PCA was used to obtain an overview of the data structure (most obvious sample clusters, sample-to-sample variance and homogeneity within sample groups) and to identify outliers. BGA and RDA were used to cluster the samples and enrich the data set for tumourigenesis-related genes, with respect to single (BGA) or multiple clinical variables (RDA; details in Supporting Information). As for RDA, it was used to prioritize clinical variables on the basis of their associations with a specific sample cluster defined by similar gene expression profiles, while BGA served to identify the genes in each cluster whose expression was unique to that cluster, i.e. differentially expressed. Although BGA and RDA are also used as classification tools or predictive models, we used them exclusively with the aims specified above, mainly because of our limited sample number.
GeneGo MetaCore software (www.genego.com) was then used to identify the molecular processes most likely to be determinants of the polypoid/nonpolypoid status of the lesions. This analysis was based on the frequency of significant expression changes involving genes related to the various processes (according to the Gene Ontology database; Ashburner et al, 2000).
TMIGD1 expression studies
TMIGD1 TFBSs were predicted with TFBS track, which was downloaded from the UCSC Genome Browser (http://genome.ucsc.edu/). The TFBS locations and scores were generated with the TRANSFAC Matrix Database (v7.0), which contains position-weight matrices for 398 TFBSs. This database was created by Biobase (http://www.biobase-international.com/) and is available at TFBS track. The TMIGD1 gene region analysed included 20 kb upstream and 10 kb downstream from the transcription start site.
First-strand cDNA synthesis and quantitative real-time RT-PCR were performed as previously described (Menigatti et al, 2009) with the Roche LightCycler 480 real-time PCR system and a LightCycler 480 SYBR Green I Master kit (amplification conditions and primers available on request).
Tissue sections were immunostained (24 h, 4°C) for TMIGD1 expression, as previously described (Truninger et al, 2005). The primary antibody (rabbit polyclonal anti-TMIGD1—HPA021946, Sigma–Aldrich, St. Louis, MO, USA) was used at a 1:500 dilution. It was also used to evaluate TMIGD1 expression in SW480 colon cancer cells. Briefly, cells were seeded onto glass cover-slips, grown for an appropriate period and fixed in 50% ethanol/50% methanol (15 min at RT). Fixed cells were permeabilized with 0.25% Triton X-100, blocked with goat serum, and incubated 24 h with anti-TMIGD1 antibodies. The cells were incubated with a secondary antibody (labelled polymer-HRP anti-rabbit, DakoCytomation EnVision+ System-HRP; K4010) and the cytochemical detection performed by using DAB (3,3-diaminobenzidine tetrahydrochloride).
Western blot analysis was performed as previously described (Menigatti et al, 2009) on total cell extracts from epithelial colonic crypts (isolated with the procedure reported by Fujimoto et al, 2002) and two colon cancer cell lines (SW480 and Caco 2) obtained from the Zurich Cancer Network cell-line repository. Rabbit polyclonal anti-TMIGD1 antibodies (1:300; HPA021946) were purchased from Sigma–Aldrich; mouse monoclonal anti-MSH6 antibodies (1:2000; BD610918) were from BD Transduction Laboratories (San Jose, CA, USA); mouse monoclonal anti-VIL1 antibodies (1:1000; MAB1639) were from Chemicon International. Caco2 cells were cultured to confluence (day 0) and harvested 10 and 21 days later to induce differentiation. Medium was changed every 2 days (Papetti & Augenlicht, 2011).
Acknowledgments
We thank the patients who participated in this study. F. Verrey, M. Risio, S. Pedersen and L. C. LaPointe for productive discussion. S. Camargo, S. V. Straface, E. Valtorta, A. Patrignani, M. Okoniewski, I. Toller, J. Eloranta and M. Papetti for technical assistance and Marian Everett Kent for manuscript editing. The research was supported by a grant from the Swiss National Science Foundation.
Supporting Information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
EC performed microarray analyses and other experiments as part of her PhD thesis; EL performed BGA and RDA and most of the statistical analyses; FB and MAB performed endoscopies and tissue sampling; FZ and BH histologically classified tumour samples; RH performed immunohistochemistry; MM extracted nuclei acids and performed real-time RT-PCR experiments; ZB performed KRAS and BRAF mutational analysis; JS-B and AT performed studies with cell lines and Western blotting; JJ conceived important experiments during the study; and GM conceived the project, obtained funding, prepared the manuscript and served as supervising mentor for EC during her PhD.
For more information
Transcription factor binding sites:
http://www.biobase-international.com/
GeneGo MetaCore software:
Gene Expression Omnibus (GEO):
Statistics:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baty F, Jaeger D, Preiswerk F, Schumacher MM, Brutsche MH. Stability of gene contributions and identification of outliers in multivariate analysis of microarray data. BMC Bioinformatics. 2008;9:289. doi: 10.1186/1471-2105-9-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco MA, Cipolletta L, Rotondano G, Buffoli F, Gizzi G, Tessari F. Prevalence of nonpolypoid colorectal neoplasia: an Italian multicenter observational study. Endoscopy. 2010;42:279–285. doi: 10.1055/s-0029-1244020. [DOI] [PubMed] [Google Scholar]
- Culhane AC, Perriere G, Considine EC, Cotter TG, Higgins DG. Between-group analysis of microarray data. Bioinformatics. 2002;18:1600–1608. doi: 10.1093/bioinformatics/18.12.1600. [DOI] [PubMed] [Google Scholar]
- Endoscopic Classification Review Group. Update on the Paris classification of superficial neoplastic lesions in the digestive tract. Endoscopy. 2005;37:570–578. doi: 10.1055/s-2005-861352. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Beauchamp RD, Whitehead RH. Identification and isolation of candidate human colonic clonogenic cells based on cell surface integrin expression. Gastroenterology. 2002;123:1941–1948. doi: 10.1053/gast.2002.37065. [DOI] [PubMed] [Google Scholar]
- Harris MH, Vander Heiden MG, Kron SJ, Thompson CB. Role of oxidative phosphorylation in Bax toxicity. Mol Cell Biol. 2000;20:3590–3596. doi: 10.1128/mcb.20.10.3590-3596.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi T, Jass JR. My approach to serrated polyps of the colorectum. J Clin Pathol. 2004;57:682–686. doi: 10.1136/jcp.2003.015230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Ishikawa F, Nose K, Shibanuma M. Downregulation of hepatocyte nuclear factor-4alpha and its role in regulation of gene expression by TGF-beta in mammary epithelial cells. Exp Cell Res. 2008;314:2131–2140. doi: 10.1016/j.yexcr.2008.03.013. [DOI] [PubMed] [Google Scholar]
- Kudo S, Lambert R, Allen JI, Fujii H, Fujii T, Kashida H, Matsuda T, Mori M, Saito H, Shimoda T, et al. Nonpolypoid neoplastic lesions of the colorectal mucosa. Gastrointest Endosc. 2008;68:S3–S47. doi: 10.1016/j.gie.2008.07.052. [DOI] [PubMed] [Google Scholar]
- Kudo S, Rubio CA, Teixeira CR, Kashida H, Kogure E. Pit pattern in colorectal neoplasia: endoscopic magnifying view. Endoscopy. 2001;33:367–373. doi: 10.1055/s-2004-826104. [DOI] [PubMed] [Google Scholar]
- Lambert R, Kudo SE, Vieth M, Allen JI, Fujii H, Fujii T, Kashida H, Matsuda T, Mori M, Saito H, et al. Pragmatic classification of superficial neoplastic colorectal lesions. Gastrointest Endosc. 2009;70:1182–1199. doi: 10.1016/j.gie.2009.09.015. [DOI] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menigatti M, Cattaneo E, Sabates-Bellver J, Ilinsky VV, Went P, Buffoli F, Marquez VE, Jiricny J, Marra G. The protein tyrosine phosphatase receptor type R gene is an early and frequent target of silencing in human colorectal tumorigenesis. Mol Cancer. 2009;8:124. doi: 10.1186/1476-4598-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien MJ. Hyperplastic and serrated polyps of the colorectum. Gastroenterol Clin North Am. 2007;36:947–968. doi: 10.1016/j.gtc.2007.08.007. [DOI] [PubMed] [Google Scholar]
- O'Brien MJ, Winawer SJ, Zauber AG, Bushey MT, Sternberg SS, Gottlieb LS, Bond JH, Waye JD, Schapiro M. Flat adenomas in the National Polyp Study: Is there increased risk for high-grade dysplasia initially or during surveillance. Clin Gastroenterol Hepatol. 2004;2:905–911. doi: 10.1016/s1542-3565(04)00392-1. [DOI] [PubMed] [Google Scholar]
- Papetti M, Augenlicht LH. MYBL2, a link between proliferation and differentiation in maturing colon epithelial cells. J Cell Physiol. 2011;226:785–791. doi: 10.1002/jcp.22399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris Workshop Participants. The Paris endoscopic classification of superficial neoplastic lesions: esophagus, stomach, and colon: November 30 to December 1, 2002. Gastrointest Endosc. 2003;58:S3–S43. doi: 10.1016/s0016-5107(03)02159-x. [DOI] [PubMed] [Google Scholar]
- Park DH, Kim HS, Kim WH, Kim TI, Kim YH, Park DI, Kim HJ, Yang SK, Byeon JS, Lee MS, et al. Clinicopathologic characteristics and malignant potential of colorectal flat neoplasia compared with that of polypoid neoplasia. Dis Colon Rectum. 2008;51:43–49;. doi: 10.1007/s10350-007-9091-5. discussion 49. [DOI] [PubMed] [Google Scholar]
- Peterson MD, Mooseker MS. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci. 1992;102:581–600. doi: 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- Quinn GP, Kenough MJ. Experimental Design and Data Analysis for Biologists. Cambridge, UK: Cambridge University Press; 2006. [Google Scholar]
- Ringner M. What is principal component analysis. Nat Biotechnol. 2008;26:303–304. doi: 10.1038/nbt0308-303. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Speed TP. A comparison of Affymetrix gene expression arrays. BMC Bioinformatics. 2007;8:449. doi: 10.1186/1471-2105-8-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabates-Bellver J, Van der Flier LG, de Palo M, Cattaneo E, Maake C, Rehrauer H, Laczko E, Kurowski MA, Bujnicki JM, Menigatti M, et al. Transcriptome profile of human colorectal adenomas. Mol Cancer Res. 2007;5:1263–1275. doi: 10.1158/1541-7786.MCR-07-0267. [DOI] [PubMed] [Google Scholar]
- Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- Snover DC, Jass JR, Fenoglio-Preiser C, Batts KP. Serrated polyps of the large intestine: a morphologic and molecular review of an evolving concept. Am J Clin Pathol. 2005;124:380–391. doi: 10.1309/V2EP-TPLJ-RB3F-GHJL. [DOI] [PubMed] [Google Scholar]
- Soetikno RM, Kaltenbach T, Rouse RV, Park W, Maheshwari A, Sato T, Matsui S, Friedland S. Prevalence of nonpolypoid (flat and depressed) colorectal neoplasms in asymptomatic and symptomatic adults. JAMA. 2008;299:1027–1035. doi: 10.1001/jama.299.9.1027. [DOI] [PubMed] [Google Scholar]
- Stegmann A, Hansen M, Wang Y, Larsen JB, Lund LR, Ritie L, Nicholson JK, Quistorff B, Simon-Assmann P, Troelsen JT, et al. Metabolome, transcriptome, and bioinformatic cis-element analyses point to HNF-4 as a central regulator of gene expression during enterocyte differentiation. Physiol Genomics. 2006;27:141–155. doi: 10.1152/physiolgenomics.00314.2005. [DOI] [PubMed] [Google Scholar]
- Tomiyama A, Serizawa S, Tachibana K, Sakurada K, Samejima H, Kuchino Y, Kitanaka C. Critical role for mitochondrial oxidative phosphorylation in the activation of tumor suppressors Bax and Bak. J Natl Cancer Inst. 2006;98:1462–1473. doi: 10.1093/jnci/djj395. [DOI] [PubMed] [Google Scholar]
- Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, Bannwart F, Yurtsever H, Neuweiler J, Riehle HM, et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology. 2005;128:1160–1171. doi: 10.1053/j.gastro.2005.01.056. [DOI] [PubMed] [Google Scholar]
- Wallerman O, Motallebipour M, Enroth S, Patra K, Bysani MS, Komorowski J, Wadelius C. Molecular interactions between HNF4a, FOXA2 and GABP identified at regulatory DNA elements through ChIP-sequencing. Nucleic Acids Res. 2009;37:7498–7508. doi: 10.1093/nar/gkp823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.