

Abstract
Autism spectrum disorders (ASD) are important neuropsychiatric disorders, currently estimated to affect approximately 1% of children, with considerable emotional and financial costs. Significant collaborative effort has been made over the last 15 years in an attempt to unravel the genetic mechanisms underlying these conditions. This has led to important discoveries, both of the roles of specific genes, as well as larger scale chromosomal copy number changes. Here, we summarize some of the latest genetic findings in the field of ASD and attempt to link them with the results of pathophysiological studies to provide an overall picture of at least one of the mechanisms by which ASD may develop.
Keywords: autism, copy number variants, genetics, pathophysiology, underconnectivity
INTRODUCTION
Autism is characterized by impaired reciprocal social interaction and communication, as well as stereotyped behaviour and interests, with an onset typically before 3 years of age. A broader range of phenotypes is also recognized, termed autism spectrum disorders (ASDs), which include ‘strict’ autism, atypical autism, Asperger syndrome and pervasive developmental disorder—not otherwise specified (PDD-NOS; OMIM %209850). Recently, it has been recognized that relatives of those diagnosed with an ASD may demonstrate attenuated forms of these conditions, the so-called broader autism phenotype (BAP; Sousa et al, 2011).
ASDs are common, currently estimated to affect approximately 1% of children (Autism Genome Project Consortium, 2007; Baird et al, 2006), but are significantly skewed towards boys, with a sex ratio of ∼4:1 (Fombonne, 2005; Santangelo & Tsatsanis, 2005). Among siblings of children with an ASD, the prevalence increases to 2–8% (Fombonne, 2005; Muhle et al, 2004). The concordance of autism in monozygotic twins is 36–60%, versus 0% in same sex dizygotic twins (Bailey et al, 1995; Folstein & Rutter, 1977), rising when the whole ASD spectrum is taken into account (Bailey et al, 1995). The heritability of a phenotype gives an indication of the extent to which it is controlled by genetic factors and can be calculated from concordance rates. Thus, the heritability of ASDs has been estimated to be ∼90%, making ASDs the most heritable of the childhood onset neuropsychiatric disorders (Sousa et al, 2011). As such, the importance of genetic susceptibility factors has become increasingly recognized. Many genetic loci are involved in ASD susceptibility and they likely interact (Risch et al, 1999), with common variants potentially modifying the action of rare variants (Bodmer & Bonilla, 2008), either by ameliorating or enhancing the effect of the susceptibility locus. This review will discuss the most recent genetic findings for ASD and place them in context of the currently known pathophysiology.
LINKAGE STUDIES
Linkage studies identify comparatively broad genomic regions co-inherited with a phenotype. The key advantage is that they can be performed with relatively limited numbers of genetic markers. However, as the regions identified can be large, they are likely to contain multiple genes, not all of which will contribute to the phenotype under investigation.
Initial attempts to understand ASD genetics utilized linkage studies with most cohorts consisting of multiplex nuclear families. These identified many loci, including 2q (Buxbaum et al, 2001; Lamb et al, 2005; Shao et al, 2002a, 2002b), 3p (McCauley et al, 2005; Shao et al, 2002a), 3q (Auranen et al, 2002), 7q (Ashley-Koch et al, 1999; Auranen et al, 2002; Barrett et al, 1999; Lamb et al, 2005; Shao et al, 2002a), 11p (Autism Genome Project Consortium, 2007; Yonan et al, 2003), 16p (Lamb et al, 2005; McCauley et al, 2005), 17q (Cantor et al, 2005; McCauley et al, 2005; Yonan et al, 2003), 19p (McCauley et al, 2005; Philippe et al, 1999), and Xq (Auranen et al, 2002; Shao et al, 2002a). In total, 26 non-overlapping regions of the genome are implicated, pointing to the complexity of the genetics involved. In an early study, Risch et al (Risch et al, 1999) concluded that there were likely to be at least 15 loci involved.
Each of these identified regions is large, and it has therefore been important to refine the regions to identify the specific genes of importance. This has often taken the form of association studies of individual candidate genes.
CANDIDATE GENE STUDIES
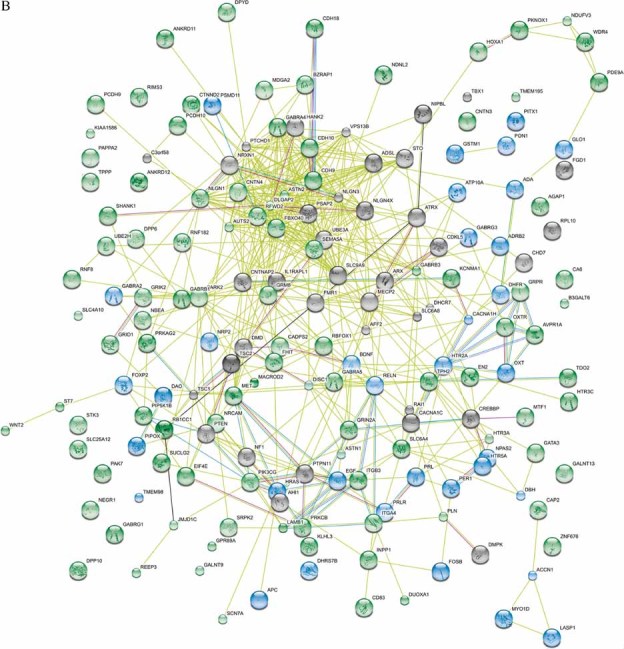
Candidate gene association analysis for ASD has been an extremely active field, with over two hundred genes examined in the last 15 years. Of these, approximately one hundred, spread across virtually every chromosome, are reported as showing at least nominal association with ASD (Sousa et al, 2011; Fig 1A), although this likely contains some false positive results. Tools such as the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) 9.0 (http://string.embl.de/) have been developed, which show the potential interactions between the proteins encoded by these genes (Szlarczyk et al, 2010; Fig 1B). While many of these interactions are predicted using methods such as text mining, and therefore are likely to contain some false positive results, they do demonstrate both the connectedness and complexity that is likely to underlie susceptibility to ASDs. Despite these caveats, the importance of such candidate gene studies is clear, particularly in light of the difficulty for genome-wide association studies to detect weak effects. Candidate genes can be identified by different means, including function, linkage, and genome-wide association. Several examples highlight the benefits and difficulties of candidate gene studies in ASDs.
Figure 1. The complexity of the genetics underlying ASD.

- Ideogram showing the relative locations of genes implicated in ASD susceptibility, adapted from the UCSC Genome Browser (http://genome.ucsc.edu/). Genes in black are listed as ‘known ASD genes’ in Pinto et al (2010) Supporting information Table 9. Genes in green are listed as ‘ASD candidates’ in Pinto et al (2010) Supporting information Table 9. Genes in blue are additional genes reported as showing association with ASD (Sousa et al, 2011, Table 2.1).
- Network of known and predicted interactions between proteins encoded by genes implicated in ASD susceptibility produced by the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) 9.0 (http://string.embl.de/) using default settings. Proteins are represented by spheres, the colours corresponding to the genes in (A) (protein names may differ from gene names, for example, SHANK3 encodes PSAP2). Lines linking proteins indicate evidence for interactions; pale green = textmining, light blue = databases, pink = experimental, pale purple = homology, black = co-expression, bright green = neighbourhood (the genes reside within 300 bp on the same strand in the genome).
Some genes are selected for association studies based solely on their known function indicating that they plausibly affect the phenotype. The serotonin transporter gene SLC6A4 is one such example as serotonin levels have been shown to be altered in autism cohorts (Anderson et al, 1987; Piven et al, 1991). Initial association to the gene was identified for an insertion/deletion variant located in the promoter region (Cook et al, 1997). Subsequently, multiple studies have also identified association to variants in this gene (e.g. Coutinho et al, 2007; Devlin et al, 2005), but several reports showing a lack of association have also been published (e.g. Betancur et al, 2002; Maestrini et al, 1999).
An important example of linkage data being utilized to guide candidate gene association analysis is CNTNAP2, found associated in two studies (Alarcón et al, 2008; Arking et al, 2008), with further evidence from a subsequent genome-wide association study (GWAS; Anney et al, 2010). CNTNAP2 is a neurexin, and there is strong evidence that this gene family is influential in ASD (Autism Genome Project Consortium, 2007; Feng et al, 2006; Gauthier et al, 2011; Kim et al, 2008). It is expressed in brain regions related to ASD (Bakkaloglu et al, 2008) and there is evidence from Copy number variations (CNVs) identified in schizophrenia, such as a patient with schizophrenia and autistic features who carried a CNV affecting this gene (Friedman et al, 2008). Recently, Zeeland et al compared functional neuroimaging of brains of autistic and healthy individuals to the CNTNAP2 genotype and found risk genotype individuals had reduced long range, but increased local, connectivity, indicative of a more immature pattern (Scott-Van Zeeland et al, 2010).
Glossary
Association study
Identification of marker alleles that is either (a) more frequent in cases than controls or (b) more frequently inherited by cases, than expected by chance. Marker alleles that are associated with the phenotype are likely to reside near susceptibility factors.
Common variant
A polymorphism such as a SNP, CNV or microsatellite where the minor (least frequent) allele has a frequency greater than 1% in the general population.
Copy number variation (CNV)
Changes in the amount of DNA present in a genome from the typical two copies in autosomes or one copy in sex chromosomes (in males). The region affected must be larger than 1 kb in order to be classified as a CNV. Variants present at a frequency greater than 1% in the general population are sometimes referred to as copy number polymorphisms (CNPs).
Joint attention
The ability to follow gazes and gestures of others towards objects of interest, and to direct the attention of others in a similar manner.
Linkage study
Identification of sections of DNA that are consistently co-inherited with a phenotype. Such regions are likely to harbour susceptibility genes.
Microsatellite
Region of the genome, which can have a variable number of repeats of short sequences of DNA.
Non-coding mutation
A change in a nucleotide that does not affect the protein coding sequence. Such a change may affect elements such as gene promoters, enhancers or silencers, thus altering the expression of a gene and leading to an altered phenotype. In contrast, a coding mutation is a nucleotide change in an exon, which alters an amino acid in the resulting protein.
Rare variant
A polymorphism such as a SNP, CNV or microsatellite, where the minor (least frequent) allele has a frequency less than 1% in the general population.
Synapse
The junction between a neuron and another cell. Electrical messages propagated from one neuron are transmitted to the adjoining neuronal cell across the synapse via chemical transmissions.
Whole exome sequencing
The determination of the nucleotide sequence of all exons in all known genes in the genome.
Whole genome sequencing
Determination of the sequence of all nucleotides of the genome.
A common way in which candidate genes are identified for further association analysis is by combining linkage and functional data to identify genes with a function plausibly related to the phenotype and located in a region of linkage. For example, MET is a proto-oncogene, involved in a number of biological pathways, including neuronal development (Campbell et al, 2006; Sousa et al, 2009). Due to its location in a region of linkage on chromosome 7q31, and its potential functional roles, Campbell et al investigated its association with ASD and identified a positive result to a single nucleotide polymorphism (SNP) in the promoter region. This SNP was shown to result in decreased activity of the MET promoter by altering the ability of the transcription factor SP1 to bind (Campbell et al, 2006). Three further studies have found association to MET (Campbell et al, 2008; Sousa et al, 2009; Thanseem et al, 2010). Two associated SNPs were located in intron 1 and are predicted to alter transcription factor binding (Sousa et al, 2009; Thanseem et al, 2010). These studies indicate the importance of non-coding mutations in ASD, particularly relevant in light of recent genome-wide studies (see below). MET is also associated with schizophrenia, and again, the associated SNPs appear to affect gene expression (Burdick et al, 2010).
In contrast to the linkage/functional data approach, some candidate gene studies followed from GWAS data, as in the case of the leucine rich repeat (LRR) genes. Such an approach is of particular importance given the increasing amount of GWAS data containing results of interest. There are 313 members in this class of genes (Sousa et al, 2010), some of which have been implicated by GWAS, but do not reach genome-wide significance thresholds (Anney et al, 2010; Wang et al, 2009). To better understand if such genes are likely to be involved in ASD, Sousa et al examined four LRR genes that are enriched in the brain and identified significant associations in two, LRRN3 and LRRTM3 (Sousa et al, 2010). While these results have not yet been replicated, Gauthier et al identified a truncating mutation of NRXN1 which was shown to affect the ability of the protein to bind to LRRTM2 (Gauthier et al, 2011). Therefore, there is a need to examine GWAS results, not just those that meet stringent criteria, but other less strongly associated results in order not to miss potentially relevant findings.
However, candidate gene studies are not always so clear and can give conflicting results. For example, RELN is involved in neuronal migration, similar regions of the brain are altered in individuals with ASD and reeler mutant mice, and the gene is located in the chromosome 7 region of linkage. Initial association to a triplet repeat in the 5′-untranslated region was promising (Persico et al, 2001). However, despite many studies, replication of this result has been mixed, with an approximately equal amount of work reporting positive (e.g. Ashley-Koch et al, 2007; Holt et al, 2010; Serajee et al, 2006) and negative (e.g. Bonora et al, 2003; Dutta et al, 2008) results, as well as tantalizing evidence from one GWAS (Anney et al, 2010). Such differences in results can be due to factors such as the genotyping strategy, and size and ascertainment of the cohorts used. However, on balance, RELN is likely to be involved in ASD, but its investigation highlights the need for multiple studies in independent cohorts.
GENOME-WIDE ASSOCIATION STUDIES
Rapid advances in technology have enabled hundreds of thousands of variants spread across the genome to be genotyped in large numbers of individuals. This has allowed association studies to be performed at a genome-wide level rather than just with specific candidate genes. Several such studies have been performed for ASDs. Although these studies have not consistently identified specific genes, they have provided valuable data indicating directions for further study.
The first GWAS for ASD was performed by Lauritsen et al on a small number of cases and controls from the relatively isolated population of the Faroe Islands using ∼600 microsatellite markers, the most significant result identified at 3p25.3 (Lauritsen et al, 2006). Subsequently, advances in genotyping technology have allowed more thorough studies, using hundreds of thousands to millions of SNPs in large cohorts (Anney et al, 2010; Arking et al, 2008; Ma et al, 2009; Wang et al, 2009; Weiss et al, 2009). While these studies have identified potential susceptibility loci including 5p14.1 (Ma et al, 2009; Wang et al, 2009), 5p15 (Weiss et al, 2009) and MACROD2 (Anney et al, 2010), there is significant overlap of samples used and little replication of specific loci between studies. While few associations meet stringent association thresholds, further analysis of the most strongly associated variants has yielded results of interest (Arking et al, 2008; Weiss et al, 2009). Of note is the identification of the role of cadherins. Wang et al found strong association to a locus between Cadherin 9 (CDH9) and Cadherin 10 (CDH10). Both genes encode neuronal cell adhesion molecules, and CDH10 is expressed in the frontal cortex, a region of the brain associated with ASDs (Wang et al, 2009). Data generated by the Autism Genome Project (Pinto et al, 2010) implicated chromosome 16q13-21 in a posterior probability of linkage analysis containing two rare deletions of CDH8, located in this region in families with ASD or learning disability (Pagnamenta et al, 2011a). Therefore, there is strong evidence for the involvement of this class of genes in ASD susceptibility.
Several associations obtained have been in regions outside genes. As stated, Wang et al found their strongest association to an intergenic region and speculate that the association is to variants affecting regulatory elements (Wang et al, 2009). Weiss et al found significant association to a SNP between SEMA5A and TAS2R1. SEMA5A has been implicated in axonal guidance and is expressed at lower levels in cell lines and brains from individuals with ASD. The associated SNPs were ∼80 kb upstream of SEMA5A, consistent with a role in gene regulation (Weiss et al, 2009). Therefore, these results show the importance of considering the role of regulatory polymorphisms in ASDs. Currently, demonstrating an actual effect on gene function of such variants is limited, but will be of importance as improving sequencing technologies provide larger amounts of variant data.
Despite their relative successes, the major GWAS have not unambiguously identified many common susceptibility variants, resulting in a move away from the common variant—common disorder hypothesis as the primary genetic mechanism underlying the ASDs. While common variants are still likely to be of importance in ASDs, increased attention focuses on the role of rare variants of large phenotypic impact. This has been facilitated by the genome-wide SNP genotyping studies, which have allowed a more comprehensive investigation of the role that CNVs play.
COPY NUMBER VARIATIONS
Genotyping data from genome-wide studies, as well as additional technologies such as array comparative genome hybridization (aCGH) have allowed the identification of CNVs on an ever-increasing scale and resolution. While it is known that such variants are present in the general population (Conrad et al, 2010), in recent years, understanding of the importance of CNVs in ASD has increased dramatically (Autism Genome Project Consortium, 2007; Jacquemont et al, 2006). These and further studies have shown both increased rates of de novo CNVs, as well as increased numbers in specific genes and gene pathways, giving new insights into the variants and genetic mechanisms underlying ASDs.
Several studies have demonstrated an increased burden of CNVs in individuals with ASDs compared to controls. Sebat et al demonstrated a significant difference in frequence of de novo CNVs between sporadic cases (10%), familial cases (3%) and controls (1%; Sebat et al, 2007). Marshall et al also found an increase in the percentage of de novo CNVs in families with one affected child and implicated post-synaptic density genes such as SHANK3, NLGN4 and NRXN1, as well as other genes encoding proteins in the synaptic complex, such as DPP10 (Marshall et al, 2008). Christian et al identified CNVs present only in cases and not controls in ∼11% of affected individuals, with ∼60% being co-inherited by affected siblings. However, the presence of a CNV in cases, but not controls, does not decisively indicate a phenotypic effect (Christian et al, 2008). More recent SNP data from GWAS has been used to examine the occurrence of CNVs in even greater detail (Bucan et al, 2009; Glessner et al, 2009; Pinto et al, 2010).
Pinto et al performed the highest resolution genome-wide comparison of CNVs in ASD, utilizing the data generated from approximately one million SNPs. The results showed a significantly increased burden of rare CNVs affecting genes in individuals with ASD compared to controls. The difference was more pronounced when the analysis was limited to CNVs previously implicated in ASD and/or intellectual disability. However, this highlights the difficulty to confidently identify specific susceptibility CNVs when their individual frequencies are low. To overcome this they examined if there was an increased incidence of CNVs in specific pathways and identified GTPase/Ras signalling, as well as cellular proliferation, projection and motility. They also noted potential novel susceptibility genes on the basis of their presence in cases, but not controls. These included PTCHD1, likely to be involved in development of the cerebellum, and SHANK2 (Noor et al, 2010; Pinto et al, 2010). SHANK2 is related to SHANK3 (Durand et al, 2007; Moessner et al, 2007), which encodes a scaffolding protein located at synapses in the brain, since implicated in other studies (Berkel et al, 2010).
Glessner et al identified CNVs enriched in specific genes in cases compared to controls including some encoding proteins involved in neuronal cell adhesion, such as NLGN1 and ASTN1. They also found an enrichment of CNVs in cases in regions containing genes involved in ubiquitin degradation (Glessner et al, 2009). Bucan et al focused their analysis on those CNVs in genes, again looking for instances, which occurred only in cases. Of more than 150 CNVs identified in their initial cohorts, 27 were replicated. They identified novel loci, such as BZRAP1, as well as previously implicated ASD susceptibility genes involved in synaptic function. An additional important observation was a lack of perfect segregation of these rare variants with affection status in families (Bucan et al, 2009). Such results and others (Fernandez et al, 2010) indicate that CNVs may lack complete penetrance or are under the influence of modifying factors. Finally, CNV data obtained by Morrow et al implicated CNVs within genes such as PCDH10, CNTNAP2, NLGN3, NLGN4 and NRXN1, as well as non-coding variants, such as CNVs near CNTN3, involved in axon outgrowth, which may affect transcription control regions. Therefore, the regulation of gene expression may be of importance in ASDs in keeping with GWAS and candidate gene studies (Morrow et al, 2008).
CNV studies have also proved to be a fertile ground for schizophrenia (Levinson et al, 2011) and some CNVs implicated in schizophrenia overlap with CNVs implicated in ASD, indicating common underlying pathways (Guilmatre et al, 2009; Levinson et al, 2011; Mefford et al, 2008). This overlap extends to other neuropsychiatric conditions such as attention-deficit hyperactivity disorder (ADHD; Williams et al, 2010) and chromosomal variants more commonly associated with other syndromes (Cohen et al, 2005; Hendriksen & Vles, 2008; Young et al, 2008). The variability in phenotype associated with CNVs affecting NRXN1 has been examined. The CNVs varied in size and nature, and the associated phenotypes included ASD, mental retardation and language delays (Ching et al, 2010). Therefore, CNVs show variable expressivity and the potential for modifying genetic or environmental factors to be in effect.
Recently, there has been an increased focus on a ‘multi-hit’ model of CNVs in ASD and neuropsychiatric disorder susceptibility (Cook and Scherer, 2008). Christian et al noted that some individuals inherited two ‘autism-specific’ CNVs (Christian et al, 2008) and Marshall et al observed examples of cases with multiple potentially etiologic CNVs, both de novo and inherited (Marshall et al, 2008). Girirajan et al investigating developmental delay, found a deletion of 16p12.1 at increased incidence in cases versus controls. They also found that those who carried this deletion and displayed developmental delay were significantly more likely to harbour a second large CNV. Their conclusion was that the CNVs at 16p12.1 were capable of predisposing to developmental delay, with the phenotype being exacerbated in the presence of a secondary variant (Girirajan et al, 2010). Pagnamenta et al identified a single family in which both affected sons carried duplications of dystrophin and a rare large deletion of the 5′-part of TRPM3 (Pagnamenta et al, 2011b). Mutations of dystrophin are known to cause Duchenne muscular dystrophy (DMD) and the less severe Becker muscular dystrophy (BMD), in which there is an increased incidence of ASD (Hendriksen & Vles, 2008; Young et al, 2008). TRPM3, while not previously implicated in ASD, is an intriguing candidate. It encodes a brain-expressed calcium channel, is localized to oligodendrocytes during their differentiation and to neurons prior to myelinization (Hoffmann et al, 2010) and is most closely related to TRPM1, located in the 15q13.3 region (Pagnamenta et al, 2011b). Another member of the family, TRPM2, has been linked to bipolar affective disorder (Xu et al, 2006).
POINT MUTATIONS AND SEQUENCING
Technology has reached the stage where it is possible to sequence the whole exome in large numbers of individuals. Studies for autism and other neuropsychiatric disorders are already underway, such as the UK10K project (http://www.uk10k.org/). This will progress to the stage where it will be viable to perform whole genome sequencing on large cohorts. These studies will potentially identify every variant in an individual, from single base point mutations to large chromosomal abnormalities. Significantly, the importance of identifying rare point mutations in ASD has been demonstrated by multiple studies.
In SHANK3, Durand et al identified mutations in individuals with ASD including a single base insertion resulting in a frameshift, rare non-coding mutations not present in controls, as well as CNVs of potential significance (Durand et al, 2007). Moessner et al also investigated SHANK3 variation in ASD and identified several point mutations, as well as CNVs, concluding that one was likely of significance (Moessner et al, 2007). Mice with homozygous SHANK3 deletions demonstrate repetitive behaviour, increased anxiety and abnormal social interaction. They also have altered composition of the post-synapse, potential disruption of glutamatergic signalling and changes in the development of medium spiny neurons and striatal glutamatergic synapses. This confirms the importance of post-synaptic impairment and neuronal connectivity in ASD and the role of SHANK3 in these processes (Peça et al, 2011). Similarly, a recent study identified a series of rare point mutations in SHANK2 with a putative effect on ASD susceptibility. While these variants did not always co-segregate with affection status in the families, it was suggested that they potentially contribute to the phenotype in combination with other unidentified factors (Berkel et al, 2010).
Detailed sequencing of other genes in individuals with ASD has also been performed. Jamain et al screened the X linked genes NLGN3, NLGN4 and NLGN4Y and found a frameshift mutation resulting in a premature stop codon and a missense mutation in NLGN4 in separate families, which co-segregate with affected status and are absent in controls (Jamain et al, 2003). Subsequently, several groups have attempted to identify variants in neuroligin genes in additional cohorts. While some studies have been successful (Zhang et al, 2009), most of these studies have either failed to identify variants likely to be pathogenic (Vincent et al, 2004; Wermter et al, 2008), or have also found them present in controls (Blasi et al, 2006).
Other genes, including those involved in the synapse, have also been screened for mutations in autism cohorts. Gauthier et al identified truncating mutations of NRXN1 and NRXN2 in patients with schizophrenia and/or ASD. A truncating mutation in NRXN1 is of particular interest as it affects the interaction of NRXN1 with LRRTM2 and NLGN2 (Gauthier et al, 2011). Hamdan et al identified both a CNV and point mutations in FOXP1 in individuals with autistic features (Hamdan et al, 2010). FOXP1 is related to FOXP2, which is involved in a severe speech and language disorder (Lai et al, 2001). In individuals with epilepsy and autistic features, Marini et al found point mutations of PCDH19, a gene believed to be involved in synapse signalling and neuronal connectivity (Marini et al, 2010). Traditionally, the emphasis on mutations in ASD is on loss of function. However, some research indicates the importance of gain of function in ASD. Mejias et al found association to the glutamate receptor gene GRIP1, involved in synaptic function, in an autistic cohort. Screening for variants identified two present only in the cases, which significantly increased the amount of surface Glu2A in hippocampal neurons (Mejias et al, 2011).
However, the presence of an apparently pathogenic variant in the sequence of a gene does not immediately confer genuine causal status. For instance, Kolevzon et al studied what was thought to be a pathogenic single base pair insertion in exon 11 of SHANK3 in a child with ASD. They showed that it was in fact likely to be non-pathogenic, due to few spliceforms containing this exon (Kolevzon et al, 2011). Despite this, there is strong evidence for the role of point mutations in various genes in ASD susceptibility.
UNDERCONNECTIVITY AND GENETICS
Despite the limited number of samples available and a lack of perfect concordance, studies comparing brains from autistic and control individuals have identified several key differences. Comparison of these changes with the genetic factors, which have been implicated, shows correlations, which help provided greater insight into ASDs as a whole. On a gross scale, localized failure of cerebellar development, cerebral cortical abnormalities, altered amygdala development and decreased corpus callosum size have been reported (Amaral et al, 2008; Bauman & Kemper, 2005; Hughes, 2007; Wegiel et al, 2010). Excessive growth of white matter in the first 2 years of life, followed by undergrowth, leading to macrocephaly in ∼20% of children with ASD has also been noted (Amaral et al, 2008; Geschwind & Levitt, 2007; Hughes, 2007). While macrocephaly may not persist into adulthood, there is evidence of abnormal brain growth continuing in adolescence (Amaral et al, 2008).
Similarly, differences are also manifested at a cellular level. Abnormal genesis, migration, shape, arrangement and maturation of neurons are reported, leading to general disorganization of the grey and white matter (Wegiel et al, 2010). Neurons are reported as being smaller in size, but of increased density (Amaral et al, 2008; Bauman & Kemper, 2005; Casanova et al, 2006; Geschwind & Levitt, 2007), and their organization within minicolumns being altered (Amaral et al, 2008; Casanova et al, 2006; Geschwind & Levitt, 2007). Minicolumns are of decreased width and increased number, resulting in the increase in density of neurons reported (Casanova et al, 2006). Changes in minicolumns and neuron size may promote shorter connecting fibres, increasing local connectivity and processing at the expense of connections between different cortical regions causing slower transmission of signals (Casanova et al, 2006; Courchesne & Pierce, 2005). This led to the postulation of the underconnectivity theory, which postulates that fewer long range connections between different regions are present, leading to decreased synchronization between regions of the brain and decreased global information processing (Casanova et al, 2006). Alterations in the growth of white matter and its decreased density in ASD as well as changes in axon number, pathfinding and synaptogenesis (Geschwind & Levitt, 2007) are also thought to contribute (Hughes, 2007). Although underconnectivity appears to be a general feature in ASD brains, it is particularly likely to affect connections between frontal and temporal lobe regions, and what connectivity there is, is likely to be unorganized (Courchesne & Pierce, 2005; Geschwind & Levitt, 2007; Hughes, 2007; Just et al, 2007). The theory of underconnectivity has been born out of neuroimaging studies, which have demonstrated a lack of synchronization in the activation of brain regions in autistic individuals, indicating decreased communication and connectivity between them, resulting in a lack of integration of information (Just et al, 2007, 2004; Koshino et al, 2008). Lastly, it is interesting to note that underconnectivity has also been implicated in schizophrenia (Just et al, 2007, 2004).
The underconnectivity theory appears to provide a good explanation for the development of ASD. For example, individuals with ASD show reduced capacity for joint attention. Joint attention utilizes the prefrontal and anterior regions of the brain. If there is reduced connectivity between these regions, it would explain the resultant deficits observed (Casanova et al, 2006) as the resulting decrease in joint attention could lead to decreased language ability and social behaviour (Geschwind & Levitt, 2007). In addition, social encounters require significant integration of information. If the ability to perform this is reduced due to lack of connectivity, it would help explain the social deficit of autism and the reliance on stereotyped patterns of behaviour (Just et al, 2004).
The genetic data are consistent with such a theory. Genes such as FLRT3 (Anney et al, 2010), SEMA5A (Weiss et al, 2009) and MET (Campbell et al, 2006; Geschwind & Levitt, 2007; Levitt et al, 2004; Powell et al, 2001; Sousa et al, 2009) that are involved in neuronal development, migration, growth and maturation have been implicated. This is also true for RELN (Persico et al, 2001; Wegiel et al, 2010), which is important in neuronal migration (Wegiel et al, 2010) and the proper formation of brain structures (Skaar et al, 2005).
Genetics has also pointed to the role of genes involved in axonal growth, such as PLD2, PLD5, PCDH10, CDH8, MET and CNTN3 (Anney et al, 2010; Bekirov et al, 2008; Hussman et al, 2011; Levitt et al, 2004; Morrow et al, 2008; Pagnamenta et al, 2011a; Sousa et al, 2009). In particular, many genes involved in the synapse and synaptic complexity have been identified in ASD, for example, SHANK3, CNTNAP2, NLGN3, NLGN4, NRXN1, PCDH10, CDH8, CDH9 and CDH10 (Morrow et al, 2008; Pagnamenta et al, 2011a; Pinto et al, 2010; Wang et al, 2009). Pathway analyses have pointed to groups of genes involved in cell adhesion (Bucan et al, 2009), cellular projection and GTPase/Ras signalling (Pinto et al, 2010)—the latter known to partially govern neurite differentiation (Hussman et al, 2011)—and the ubiquitin pathway, which can alter dendritic spines, the post-synaptic density and turnover of neuronal cell adhesion molecules (Glessner et al, 2009; Yaspan et al, 2011). Glessner et al also found the neuronal cell adhesion pathway to be implicated, which affects axon guidance and the formation of the synapse (Glessner et al, 2009).
Therefore, there appears to be strong concordance between observed structural alterations in autistic brains and the underlying genetic susceptibility loci identified. However, this does not mean that this is the only mechanism involved in ASD susceptibility (Fig 2). For example, in a recent pathway analysis Yaspan et al identified 17 pathways, including the uridine diphosphate glycosyltransferase 2 protein family and genes involved in synthesis and degradation of ketone bodies. The latter could affect γ-aminobutyric acid (GABA) levels, a neurotransmitter system implicated in ASD (Yaspan et al, 2011).
Figure 2. General overview of the potential route of the underlying mechanisms of ASD.
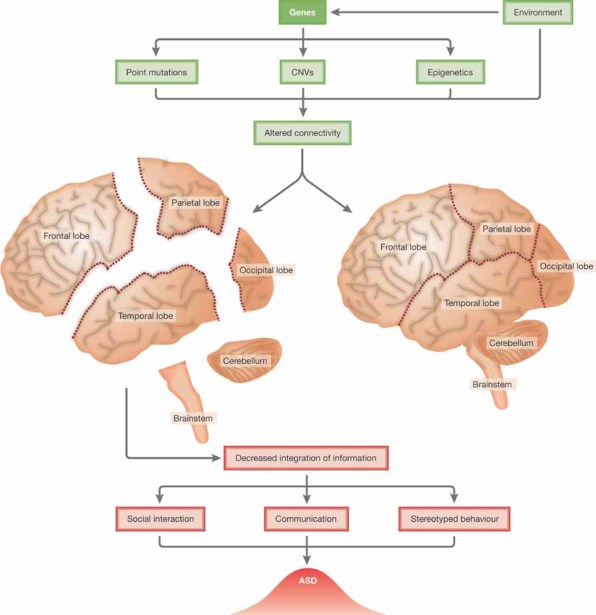
Interactions between the environment and genetic factors, including point mutations, CNVs and epigenetics, determine the development of the brain. Combinations of factors resulting in decreased connectivity between regions of the brain cause a decreased ability to integrate information. This expresses itself at the level of behaviour in the individual as alterations in the areas of social interaction, communication and behaviour, the extent of the variation in each area depending on the environmental and genetic variables, leading to the range of ASD phenotypes observed.
CONCLUDING REMARKS
Development in our understanding of the genetics of ASDs has flourished over the past 20 years due to rapid advances in technology. This will continue as studies such as the UK10K project and those funded by the Simons Foundation (http://sfari.org/) are in the process of performing whole exome sequencing for both ASD and other neuropsychiatric cohorts, with similar studies being planned. The findings from these projects, and the inevitable whole genome sequencing projects which will follow, will provide a wealth of data on variants in individuals with ASD. Therefore, there will be a need to determine which variants are likely to be pathogenically relevant, and also to determine their function experimentally. In addition, there is a need to better understand the interlinking pathways in ASD genetics. These are essential tasks if we are to translate the findings of the research community into tangible benefits for the ASD community as a whole. The occurrence of many rare, potentially unique, CNVs in ASD cases points to the complexity of the genetics underlying these disorders. In this way, genetic research is validating the view that ASDs should not be considered a set of discrete disorders, but a continuous range of individually rare conditions. The rarity of specific variants, combined with the sample sizes employed to date, mean that caution must be adopted in assigning a pathogenic role to a specific variant based solely on it's uniqueness within an ASD sample.
Therefore, there is a need to recruit additional cohorts of patients and controls, in order to better understand which are rare in the general population and which are likely to be ASD-specific. Larger cohorts will also help to identify common variants which could influence the outcome of other variants such as CNVs. In the past, the emphasis was on recruiting strictly phenotyped cases to minimize heterogeneity. With the increasing revelation of the vast spectrum of ASD there is a move towards collecting larger, more ‘lightly’ phenotyped cohorts, which better reflect the clinical reality faced by medical practitioners. Clinically recruited cohorts are already being utilized in studies of ASDs (Shen et al, 2010). This will help increase recruitment, overcoming the frequent overlap of samples, such as the AGRE cohort, increasing the power of studies, as well as give greater information on the frequency of variants in clinical scenarios to help improve translation.
Finally, there is a strong imperative to utilize current findings to benefit individuals with ASDs and their families, including making genetic testing for variants of known importance available. Various studies have examined the potential to translate current genetic and biological knowledge about autism into a clinical diagnostic setting (e.g. Schaefer et al, 2008; Shen et al, 2010). While the predicted yields vary depending on the number and variety of tests suggested, there seems to be a clear case for expecting a significant yield. As such, Miller et al have published a consensus statement in favour of clinical CNV testing for children with autism (Miller et al, 2010). The potential benefits of such testing do not necessarily lie in the ability to provide intervention to the proband, but rather to provide information and counselling to the individual and family, which are likely to have significant emotional benefits (Lenhard et al, 2005). There are issues to overcome, not least due to the overlap of variants in causing susceptibility to multiple conditions, thus limiting the predictive power of any such test in siblings, raising issues of balancing the potential stress that could be caused if younger asymptomatic siblings are tested against the ability to instigate increased surveillance and early intervention and the benefits this provides (Lord et al, 2005). However, if used correctly, such testing could significantly benefit families, and is already in effect in some countries, albeit to varying degrees.
Pending issues
Collection of new, large cohorts of cases and controls to allow better determination of the relative frequencies of rare variants and CNVs to help identify those that are likely to be of true significance in susceptibility. This will also allow improved identification of common variants of small effect.
Better characterization of CNVs in order to determine the minimal affected region and which genes or DNA regions within them are likely to be key in contributing to the phenotype.
Collection of prospective clinical cohorts screened for CNVs in order to identify genetic factors reflecting the clinical outcome of ASD. Longitudinal studies will help determine the probabilities of specific phenotypic outcomes associated with specific variants.
Initiate widespread translation of current CNV knowledge into clinical genetics laboratories in order to better inform families of the genetic findings underlying their child's condition.
Devise experiments to determine the actual mechanism of effect of specific susceptibility variants in order to increase our understanding of the pathways and biology underlying ASDs and how these interact with environmental factors.
Acknowledgments
We thank Dr Nuala Sykes for helpful comments on the content of this article. The work was supported by Autistica, the Medical Research Council (UK) and the Wellcome Trust (grant number 075491/Z/04).
The authors declare that they have no conflict of interest.
For more information
The Autism Genome Project: http://www.autismgenome.org/
Autism Speaks: http://www.autismspeaks.org/
Autistica: http://www.autistica.org.uk/
The National Autistic Society: http://www.autism.org.uk/
The UK10K Project: http://www.uk10k.org/
The Simons Foundation Autism Research Initiative: http://sfari.org/
References
- Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci. 2008;31:137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Anderson GM, Freedman DX, Cohen DJ, Volkmar FR, Hoder EL, McPhedran P, Minderaa RB, Hansen CR, Young JG. Whole blood serotonin in autistic and normal subjects. J Child Psychol Psychiatry. 1987;28:885–900. doi: 10.1111/j.1469-7610.1987.tb00677.x. [DOI] [PubMed] [Google Scholar]
- Anney R, Klei L, Pinto D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Sykes N, Pagnamenta AT, et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010;19:4072–4082. doi: 10.1093/hmg/ddq307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, Rea A, Guy M, Lin S, Cook EH, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008;82:160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley-Koch A, Wolpert CM, Menold MM, Zaeem L, Basu S, Donnelly SL, Ravan SA, Powell CM, Qumsiyeh MB, Aylsworth AS, et al. Genetic studies of autistic disorder and chromosome 7. Genomics. 1999;61:227–236. doi: 10.1006/geno.1999.5968. [DOI] [PubMed] [Google Scholar]
- Ashley-Koch AE, Jaworski J, Ma de Q, Mei H, Ritchie MD, Skaar DA, Robert Delong G, Worley G, Abramson RK, Wright HH, et al. Investigation of potential gene–gene interactions between APOE and RELN contributing to autism risk. Psychiatr Genet. 2007;17:221–226. doi: 10.1097/YPG.0b013e32809c2f75. [DOI] [PubMed] [Google Scholar]
- Auranen M, Vanhala R, Varilo T, Ayers K, Kempas E, Ylisaukko-oja T, Sinsheimer JS, Peltonen L, Järvelä I. A genomewide screen for autism-spectrum disorders: evidence for a major susceptibility locus on chromosome 3q25-27. Am J Hum Genet. 2002;71:777–790. doi: 10.1086/342720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autism Genome Project Consortium. Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Baird G, Simonoff E, Pickles A, Chandler S, Loucas T, Meldrum D, Charman T. Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: the special needs and autism project (SNAP) Lancet. 2006;368:210–215. doi: 10.1016/S0140-6736(06)69041-7. [DOI] [PubMed] [Google Scholar]
- Bakkaloglu B, O'Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, Chawarska K, Klin A, Ercan-Sencicek AG, Stillman AA, et al. Cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet. 2008;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett S, Beck JC, Bernier R, Bisson E, Braun TA, Casavant TL, Childress D, Folstein SE, Garcia M, Gardiner MB, et al. An autosomal genomic screen for autism. Am J Med Genet B Neuropsychiatr Genet. 1999;88:609–615. doi: 10.1002/(sici)1096-8628(19991215)88:6<609::aid-ajmg7>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci. 2005;23:183–187. doi: 10.1016/j.ijdevneu.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Bekirov IH, Nagy V, Svoronos A, Huntley GW, Benson DL. Cadherin-8 and N-cadherin differentially regulate pre- and postsynaptic development of the hippocampal mossy fiber pathway. Hippocampus. 2008;18:349–363. doi: 10.1002/hipo.20395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, Endris V, Roberts W, Szatmari P, Pinto D, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010;42:489–491. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- Betancur C, Corbex M, Spielewoy C, Philippe A, Laplanche JL, Launay JM, Gillberg C, Mouren-Siméoni MC, Hamon M, Giros B, et al. Serotonin transporter gene polymorphisms and hyperserotonemia in autistic disorder. Mol Psychiatry. 2002;7:67–71. doi: 10.1038/sj.mp.4001923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi F, Bacchelli E, Pesaresi G, Carone S, Bailey AJ, Maestrini E The International Molecular Genetic Study of Autism Consortium (IMGSAC) Absence of coding mutations in the X-linked genes Neuroligin 3 and Neuroligin 4 in individuals with autism from the IMGSAC collection. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:220–221. doi: 10.1002/ajmg.b.30287. [DOI] [PubMed] [Google Scholar]
- Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora E, Beyer KS, Lamb JA, Parr JR, Klauck SM, Benner A, Paolucci M, Abbott A, Ragoussis I, Poustka A, et al. Analysis of reelin as a candidate gene for autism. Mol Psychiatry. 2003;8:885–892. doi: 10.1038/sj.mp.4001310. [DOI] [PubMed] [Google Scholar]
- Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, Alvarez Retuerto AI, Imielinski M, Hadley D, Bradfield JP, et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5:e1000536. doi: 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick KE, DeRosse P, Kane JM, Lencz T, Malhotra AK. Association of genetic variation in the MET proto-oncogene with schizophrenia and general cognitive ability. Am J Psychiatry. 2010;167:436–443. doi: 10.1176/appi.ajp.2009.09050615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Silverman JM, Smith CJ, Kilifarski M, Reichert J, Hollander E, Lawlor BA, Fitzgerald M, Greenberg DA, Davis KL. Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am J Hum Genet. 2001;68:1514–1520. doi: 10.1086/320588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, Elia M, Schneider C, Melmed R, Sacco R, et al. A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci USA. 2006;103:16834–16839. doi: 10.1073/pnas.0605296103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DB, Li C, Sutcliffe JS, Persico AM, Levitt P. Genetic evidence implicating multiple genes in the MET receptor tyrosine kinase pathway in autism spectrum disorder. Autism Res. 2008;1:159–168. doi: 10.1002/aur.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor RM, Kono N, Duvall JA, Alvarez-Retuerto A, Stone JL, Alarcón M, Nelson SF, Geschwind DH. Replication of autism linkage: fine-mapping peak at 17q21. Am J Hum Genet. 2005;76:1050–1056. doi: 10.1086/430278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova MF, van Kooten IAJ, Switala AE, van Engeland H, Heinsen H, Steinbusch HWM, Hof PR, Trippe J, Stone J, Schmitz C. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006;112:287–303. doi: 10.1007/s00401-006-0085-5. [DOI] [PubMed] [Google Scholar]
- Ching MSL, Shen Y, Tan W-H, Jeste SS, Morrow EM, Chen X, Mukaddes NM, Yoo S-Y, Hanson E, Hundley R, et al. Deletions of NRXN1 (Neurexin-1) predispose to a wide spectrum of developmental disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:937–947. doi: 10.1002/ajmg.b.31063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian SL, Brune CW, Sudi J, Kumar RA, Liu S, KaraMohamed S, Badner JA, Matsui S, Conroy J, McQuaid D, et al. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol Psychiatry. 2008;63:1111–1117. doi: 10.1016/j.biopsych.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D, Pichard N, Tordjman S, Baumann C, Burglen L, Excoffier E, Lazar G, Mazet P, Pinguier C, Verloes A, et al. Specific genetic disorders and autism: clinical contribution towards their identification. J Autism Dev Disord. 2005;35:103–116. doi: 10.1007/s10803-004-1038-2. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook EH, Jr, Courchesne R, Lord C, Cox NJ, Yan S, Lincoln A, Haas R, Courchesne E, Leventhal BL. Evidence of linkage between the serotonin transporter and autistic disorder. Mol Psychiatry. 1997;2:247–250. doi: 10.1038/sj.mp.4000266. [DOI] [PubMed] [Google Scholar]
- Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Pierce K. Why the frontal cortex in autism might be talking only to itself: local over-connectivity but long-distance disconnection. Curr Opin Neurobiol. 2005;15:225–230. doi: 10.1016/j.conb.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Coutinho AM, Sousa I, Martins M, Correia C, Morqadinho T, Bento C, Marques C, Ataíde A, Miquel TS, Moore JH, et al. Evidence for epistasis between SLC6A4 and ITGB3 in autism etiology and in the determination of platelet serotonin levels. Hum Genet. 2007;121:243–256. doi: 10.1007/s00439-006-0301-3. [DOI] [PubMed] [Google Scholar]
- Devlin B, Cook EH, Jr, Coon H, Dawson G, Grigorenko EL, McMahon W, Minshew N, Pauls D, Smith M, Spence MA, et al. Autism and the serotonin transporter: the long and short of it. Mol Psychiatry. 2005;10:1110–1116. doi: 10.1038/sj.mp.4001724. [DOI] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S, Sinha S, Ghosh S, Chatterjee A, Ahmed S, Usha R. Genetic analysis of reelin (RELN) SNPs: no association with autism spectrum disorder in the Indian population. Neurosci Lett. 2008;441:56–60. doi: 10.1016/j.neulet.2008.06.022. [DOI] [PubMed] [Google Scholar]
- Feng J, Schroer R, Yan J, Song W, Yang C, Bockholt A, Cook EH, Jr, Skinner C, Schwartz CE, Sommer SS. High frequency of neurexin-1beta signal peptide structural variants in patients with autism. Neurosci Lett. 2006;409:10–13. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, Joseph-George AM, Mackay S, Whitten K, Noble B, et al. Phenotypic spectrum associated with de novo and inherited deletions and duplication at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet. 2010;47:195–203. doi: 10.1136/jmg.2009.069369. [DOI] [PubMed] [Google Scholar]
- Folstein S, Rutter M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry. 1977;18:297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- Fombonne E. Epidemiology of autistic disorder and other pervasive developmental disorders. J Clin Psychiatry. 2005;66:3–8. [PubMed] [Google Scholar]
- Friedman JI, Vrijenhoek T, Markx S, Janssen IM, van der Vliet WA, Faas BHW, Knoers NV, Cahn W, Kahn RS, Edelmann L, et al. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry. 2008;13:261–266. doi: 10.1038/sj.mp.4002049. [DOI] [PubMed] [Google Scholar]
- Gauthier J, Siddiqui TJ, Huashan P, Yokomaku D, Hamdan FF, Champagne N, Lapointe M, Spiegelman D, Noreau A, Lafrenière RG, et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum Genet; 2011 doi: 10.1007/s00439-011-0975-z. DOI: 10.1007/s00439-011-0975-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, Vives L, Walsh T, McCarthy SE, Baker C, et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilmatre A, Dubourg C, Mosca AL, Legallic S, Goldenberg A, Drouin-Garraud V, Layet V, Rosier A, Briault S, Bonnet-Brilhault F, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism and mental retardation. Arch Gen Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M, Foomani G, Dobrzeniecka S, Krebs M-O, Joober R. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet. 2010;87:671–678. doi: 10.1016/j.ajhg.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriksen JG, Vles JS. Neuropsychiatric disorders in males with duchenne muscular dystrophy: frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive–compulsive disorder. J Child Neurol. 2008;23:477–481. doi: 10.1177/0883073807309775. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Grim C, Kraft R, Goldbaum O, Wrede A, Nolte C, Hanisch UK, Richter-Landsberg C, Brück W, Kettenmann H, et al. TRPM3 is expressed in sphingosine-responsive myelinating oligodendrocytes. J Neurochem. 2010;114:654–665. doi: 10.1111/j.1471-4159.2010.06644.x. [DOI] [PubMed] [Google Scholar]
- Holt R, Barnby G, Maestrini E, Bacchelli E, Brocklebank D, Sousa I, Mulder EJ, Kantojärvi K, Järvelä I, Klauck SM, et al. Linkage and candidate gene studies of autism spectrum disorders in European populations. Eur J Hum Genet. 2010;18:1013–1019. doi: 10.1038/ejhg.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JR. Autism: The first firm finding = underconnectivity. Epilepsy Behav. 2007;11:20–24. doi: 10.1016/j.yebeh.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Hussman JP, Chung R-H, Griswold AJ, Jaworski JM, Salyakina D, Ma D, Konidari I, Whitehead PL, Vance JM, Martin ER, et al. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol Autism. 2011;2:1. doi: 10.1186/2040-2392-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont ML, Sanlaville D, Redon R, Raoul O, Cormier-Daire V, Lyonnet S, Amiel J, Le Merrer M, Heron D, de Bois MC, et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet. 2006;43:843–849. doi: 10.1136/jmg.2006.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg CI, Soderstrom H, Giros B, Leboyer M, Gillberg C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just MA, Cherkassky VL, Keller TA, Minshew NJ. Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain. 2004;127:1811–1821. doi: 10.1093/brain/awh199. [DOI] [PubMed] [Google Scholar]
- Just MA, Cherkassky VL, Keller TA, Kana RK, Minshew NJ. Functional and anatomical cortical underconnectivity in autism: evidence from an fMRI study of an executive function task and corpus callosum morphometry. Cereb Cortex. 2007;17:951–961. doi: 10.1093/cercor/bhl006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Kishikawa S, Higgins AW, Seong IS, Donovan DJ, Shen Y, Lally E, Weiss LA, Najm J, Kutsche K, et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolevzon A, Cai G, Soorya L, Takahashi N, Grodberg D, Kajiwara Y, Willner JP, Tryfon A, Buxbaum JD. Analysis of a purported SHANK3 mutation in a boy with autism: clinical impact of rare variant research in neurodevelopmental disabilities. Brain Res. 2011;1380:98–105. doi: 10.1016/j.brainres.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Koshino H, Kana RK, Keller TA, Cherkassky VL, Minshew NJ, Just MA. fMRI investigation of working memory for faces in autism: visual coding and underconnectivity with frontal areas. Cereb Cortex. 2008;18:289–300. doi: 10.1093/cercor/bhm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CSL, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a sever speech and language disorder. Nature. 2001;413:519–523. doi: 10.1038/35097076. [DOI] [PubMed] [Google Scholar]
- Lamb JA, Barnby G, Bonora E, Sykes N, Bacchelli E, Blasi F, Maestrini E, Broxholme J, Tzenova J, Weeks D, et al. Analysis of IMGSAC autism susceptibility loci: evidence for sex limited and parent of origin specific effects. J Med Genet. 2005;42:132–137. doi: 10.1136/jmg.2004.025668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritsen MB, Als TD, Dahl HA, Flint TJ, Wang AG, Vang M, Kruse TA, Ewald H, Mors O. A genome-wide search for alleles and haplotypes associated with autism and related pervasive developmental disorders on the Faroe Islands. Mol Psychiatry. 2006;11:37–46. doi: 10.1038/sj.mp.4001754. [DOI] [PubMed] [Google Scholar]
- Lenhard W, Breitenbach E, Ebert H, Schindelhauer-Deutscher HJ, Henn W. Psychological benefit of diagnostic certainty for mothers of children with disabilities: lessons from down syndrome. Am J Med Genet. 2005;133A:170–175. doi: 10.1002/ajmg.a.30571. [DOI] [PubMed] [Google Scholar]
- Levinson DF, Duan J, Oh S, Wang K, Sanders AR, Shi J, Zhang N, Mowry BJ, Olincy A, Amin F, et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am J Psychiatry. 2011;168:302–316. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P, Eagleson KL, Powell EM. Regulation of neocortical interneuron development and the implications for neurodevelopmental disorders. Trends Neurosci. 2004;27:400–406. doi: 10.1016/j.tins.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Lord C, Wagner A, Rogers S, Szatmari P, Aman M, Charman T, Dawson G, Durand MV, Grossman L, Guthrie D, et al. Challenges in evaluating psychosocial interventions for autistic spectrum disorders. J Autism Dev Disord. 2005;35:695–708. doi: 10.1007/s10803-005-0017-6. [DOI] [PubMed] [Google Scholar]
- Ma D, Salyakina D, Jaworski JM, Konidari I, Whitehead PL, Anderson AN, Hoffman JD, Slifer SH, Hedges DJ, Cukier HN, et al. A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann Hum Genet. 2009;73:263–273. doi: 10.1111/j.1469-1809.2009.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestrini E, Lai C, Marlow A, Matthews N, Wallace S, Bailey A, Cook EH, Weeks DE, Monaco AP. Serotonin transporter (5-HTT) and gamma-aminobutyric acid receptor subunit beta3 (GABRB3) gene polymorphisms are not associated with autism in the IMGSA families. The International Molecular Genetic Study of Autism Consortium. Am J Med Genet. 1999;88:492–496. doi: 10.1002/(sici)1096-8628(19991015)88:5<492::aid-ajmg11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Marini C, Mei D, Parmeggiani L, Norci V, Calado E, Ferrari A, Moreira A, Pisano T, Specchio N, Vigevano F, et al. Protocadherin 19 mutations in girls with infantile-onset epilepsy. Neurology. 2010;75:646–653. doi: 10.1212/WNL.0b013e3181ed9e67. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley JL, Li C, Jiang L, Olson LM, Crickett G, Gainer K, Folstein SE, Haines JL, Sutcliffe JS. Genome-wide and ordered-subset linkage analyses provide support for autism loci on 17q and 19p with evidence of phenotypic and interlocus genetic correlates. BMC Med Genet. 2005;6:1. doi: 10.1186/1471-2350-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejias R, Adamczyk A, Anggono V, Niranjan T, Thomas GM, Sharma K, Skinner C, Schwartz CE, Stevenson RE, Fallin MD, et al. Gain-of-function glutamate receptor interacting protein 1 variants alter GluA2 recycling and surface distribution in patients with autism. Proc Natl Acad Sci USA. 2011;108:4920–4925. doi: 10.1073/pnas.1102233108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DT, Adam MP, Swaroop A, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM, Yoo S-Y, Flavell SW, Kim T-K, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatrics. 2004;113:e472–e486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- Noor A, Whibley A, Marshall CR, Gianakopoulos PJ, Piton A, Carson AR, Orlic-Milacic M, Lionel AC, Sato D, Pinto D, et al. Disruption at the PTCHD1 locus on Xp22.11 in autism spectrum disorder and intellectual disability. Sci Transl Med. 2010;2:49ra68. doi: 10.1126/scitranslmed.3001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnamenta AT, Khan H, Walker S, Gerrelli D, Wing K, Bonaglia MC, Giorda R, Berney T, Mani E, Molteni M, et al. Rare familial 16q21 microdeletions under a linkage peak implicate cadherin 8 (CDH8) in susceptibility to autism and learning disability. J Med Genet. 2011a;48:48–54. doi: 10.1136/jmg.2010.079426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnamenta AT, Holt R, Yusuf M, Pinto D, Wing K, Betancur C, Scherer SW, Volpi EV, Monaco AP. A family with autism and rare copy number variants disrupting the Duchenne/Becker muscular dystrophy gene DMD and TRPM3. J Neurodevelop Disord. 2011b;3:124–131. doi: 10.1007/s11689-011-9076-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G. Shank3 mutant mice display autisitic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persico AM, D'Agruma L, Maiorano N, Totaro A, Militerni R, Bravaccio C, Wassink TH, Schneider C, Melmed R, Trillo S, et al. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol Psychiatry. 2001;6:150–159. doi: 10.1038/sj.mp.4000850. [DOI] [PubMed] [Google Scholar]
- Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Råstam M, Sponheim E, Coleman M, Zappella M, Aschauer H, Van Maldergem L, et al. Genome-wide scan for autism susceptibility genes. Hum Mol Genet. 1999;8:805–812. doi: 10.1093/hmg/8.5.805. [DOI] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piven J, Tsai GC, Nehme E, Coyle JT, Chase GA, Folstein SE. Platelet serotonin, a possible marker for familial autism. J Autism Dev Disord. 1991;21:51–59. doi: 10.1007/BF02206997. [DOI] [PubMed] [Google Scholar]
- Powell EM, Mars WM, Levitt P. Hepatocyte growth factor/scatter factor is a motogen for interneurons migrating from the ventral to dorsal telencephalon. Neuron. 2001;30:79–89. doi: 10.1016/s0896-6273(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, Kalaydjieva L, McCague P, Dimiceli S, Pitts T, et al. A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet. 1999;65:493–507. doi: 10.1086/302497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo SL, Tsatsanis K. What is known about autism: genes, brain, and behaviour. Am J Pharmacogenomics. 2005;5:71–92. doi: 10.2165/00129785-200505020-00001. [DOI] [PubMed] [Google Scholar]
- Schaefer GB, Mendelsohn NJ, The Professional Practice and Guidelines Committee Clinical genetics evaluation in identifying the etiology of autism spectrum disorders. Genet Med. 2008;10:301–305. doi: 10.1097/GIM.0b013e31816b5cc9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Van Zeeland AA, Abrahams BS, Alvarez-Retuerto AI, Sonnenblick LI, Rudie JD, Ghahremani D, Mumford JA, Poldrack RA, Dapretto M, Geschwind DH, et al. Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci Transl Med. 2010;2:56ra80. doi: 10.1126/scitranslmed.3001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serajee FJ, Zhong H, Mahbubul Huq AH. Association of Reelin gene polymorphisms with autism. Genomics. 2006;87:75–83. doi: 10.1016/j.ygeno.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Shao Y, Wolpert CM, Raiford KL, Menold MM, Donnelly SL, Ravan SA, Bass MP, McClain C, von Wendt L, Vance JM, et al. Genomic screen and follow-up analysis for autistic disorder. Am J Med Genet B Neuropsychiatr Genet. 2002a;114:99–105. doi: 10.1002/ajmg.10153. [DOI] [PubMed] [Google Scholar]
- Shao Y, Raiford KL, Wolpert CM, Cope HA, Ravan SA, Ashley-Koch AA, Abramson RK, Wright HH, DeLong RG, Gilbert JR, et al. Phenotypic homogeneity provides increased support for linkage on chromosome 2 in autistic disorder. Am J Hum Genet. 2002b;70:1058–1061. doi: 10.1086/339765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, Miller KJ, Frazier JA, Silverstein I, Picker J, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:727–735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar DA, Shao Y, Haines JL, Stenger JE, Jaworski J, Martin ER, DeLong GR, Moore JH, McCauley JL, Sutcliffe JS, et al. Analysis of the RELN gene as a genetic risk factor for autism. Mol Psychiatry. 2005;10:563–571. doi: 10.1038/sj.mp.4001614. [DOI] [PubMed] [Google Scholar]
- Sousa I, Clark TG, Toma C, Kobayashi K, Choma M, Holt R, Sykes NH, Lamb JA, Bailey AJ, Battaglia A, et al. MET and autism susceptibility: family and case–control studies. Eur J Hum Genet. 2009;17:749–758. doi: 10.1038/ejhg.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa I, Clark TG, Holt R, Pagnamenta AT, Mulder EJ, Minderaa RB, Bailey AJ, Battaglia A, Klauck SM, Poustka F, et al. Polymorphisms in the leucine-rich repeat genes are associated with autism spectrum disorder susceptibility in populations of European ancestry. Mol Autism. 2010;1:7. doi: 10.1186/2040-2392-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa I, Holt R, Pagnamenta A, Monaco M. Unravelling the Genetics of Autism Spectrum Disorders. In: Roth I, Rezaie P, editors. Researching the Autism Spectrum. Cambridge: Cambridge University Press; 2011. pp. 53–111. [Google Scholar]
- Szlarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2010;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanseem I, Nakamura K, Miyachi T, Toyota T, Yamada S, Tsujii M, Tsuchiya KJ, Anitha A, Iwayama Y, Yamada K, et al. Further evidence for the role of MET in autism susceptibility. Neurosci Res. 2010;68:137–141. doi: 10.1016/j.neures.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Vincent JB, Kolozsvari D, Roberts WS, Bolton PF, Gurling HMD, Scherer SW. Mutation screening of X-chromosomal neuroligin genes: no mutations in 196 autism probands. Am J Med Genet B Neuropsychiatr Genet. 2004;129B:82–84. doi: 10.1002/ajmg.b.30069. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PMA, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, Ma SY, Chauhan A, Chuahan V, Bobrowicz TW, et al. The neuropathology of autism: decfects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010;119:755–770. doi: 10.1007/s00401-010-0655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Arking DE and the Gene Discovery Project of John Hopkins and the Autism Consortium. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wermter A-Km, Kamp-Becker I, Strauch K, Schulte-Körne G, Remschmidt H. No evidence for involvement of genetic variants in the X-linked neuroligin genes NLGN3 and NLGN4X in probands with autism spectrum disorder on high functioning level. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:535–537. doi: 10.1002/ajmg.b.30618. [DOI] [PubMed] [Google Scholar]
- Xu C, Macciardi F, Li PP, Yoon I-S, Cooke RG, Hughes B, Parikh SV, McIntyre RS, Kennedy JL, Warsh JJ. Association of the putative susceptibility gene, transient receptor potential protein melastatin type 2, with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:36–43. doi: 10.1002/ajmg.b.30239. [DOI] [PubMed] [Google Scholar]
- Yaspan BL, Bush WS, Torstenson ES, Ma D, Pericak-Vance MA, Ritchie MD, Sutcliffe JS, Haines JL. Genetic analysis of biological pathway data through genomic randomization. Hum Genet. 2011;129:563–571. doi: 10.1007/s00439-011-0956-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonan AL, Alarcón M, Cheng R, Magnusson PKE, Spence SJ, Palmer AA, Grunn A, Juo S-H, Terwilliger H, Liu JD, et al. A genomewide screen of 345 families for autism-susceptibility loci. Am J Hum Genet. 2003;73:886–897. doi: 10.1086/378778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young HK, Barton BA, Waisbren S, Portales Dale L, Ryan MM, Webster RI, North KN. Cognitive and psychological profile of males with Becker muscular dystrophy. J Child Neurol. 2008;23:155–162. doi: 10.1177/0883073807307975. [DOI] [PubMed] [Google Scholar]
- Williams NM, Zaharieva I, Martin A, Langley K, Mantripragada K, Fossdal R, Stefansson H, Stefansson K, Magnusson P, Gudmundsson OO, et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet. 2010;376:1401–1408. doi: 10.1016/S0140-6736(10)61109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Milunsky JM, Newton S, Ko J, Zhao G, Maher TA, Tager-Flusberg H, Bolliger MF, Carter AS, Boucard AA, et al. A neuroligin-4 missense mutation associated with autism impairs neuroligin-4 folding and endoplasmic reticulum export. J Neurosci. 2009;29:10843–10854. doi: 10.1523/JNEUROSCI.1248-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]