

Abstract
Mutations in LAMA2 cause a severe form of congenital muscular dystrophy, called MDC1A. Studies in mouse models have shown that transgenic expression of a designed, miniaturized form of the extracellular matrix molecule agrin (‘mini-agrin’) or apoptosis inhibition by either overexpression of Bcl2 or application of the pharmacological substance omigapil can ameliorate the disease. Here, we tested whether mini-agrin and anti-apoptotic agents act on different pathways and thus exert additive benefits in MDC1A mouse models. By combining mini-agrin with either transgenic Bcl2 expression or oral omigapil application, we show that the ameliorating effect of mini-agrin, which acts by restoring the mechanical stability of muscle fibres and, thereby, reduces muscle fibre breakdown and concomitant fibrosis, is complemented by apoptosis inhibitors, which prevent the loss of muscle fibres. Treatment of mice with both agents results in improved muscle regeneration and increased force. Our results show that the combination of mini-agrin and anti-apoptosis treatment has beneficial effects that are significantly bigger than the individual treatments and suggest that such a strategy might also be applicable to MDC1A patients.
Keywords: cell death, extracellular matrix, laminin-211, merosin, muscle regeneration
INTRODUCTION
Laminin-α2-deficient congenital muscular dystrophy (MDC1A) is the most prevalent form of congenital muscular dystrophies. Patients suffer from severe muscle degeneration often leading to death in early childhood. Moreover, the disease is accompanied by a rather mild neuropathy that is based on the demyelination of the peripheral nerve (Miyagoe-Suzuki et al, 2000; Tome & Fardeau, 1998). On the cellular level, muscles of MDC1A patients are characterized by marked variation in muscle fibre size, extensive fibrosis and proliferation of adipose tissue. MDC1A is caused by mutations in LAMA2, the gene that encodes the laminin-α2 chain, which is an important component of the extracellular matrix of skeletal and heart muscle as well as peripheral nerve. In muscle, the most prevalent form of laminin that contains laminin-α2 is laminin-211 (LM-211), a heterotrimer consisting of the α2, the β1 and the γ1 chain. LM-211 is tightly attached to the basement membrane and interacts with α-dystroglycan and α7β1 integrin expressed on the muscle fibre surface (Colognato & Yurchenco, 2000). Laminin-α2 deficiency, therefore, disrupts the mechanical linkage between the basement membrane and the muscle fibre. In addition, there is evidence that the lack of LM-211 prevents activation of several signalling cascades (Langenbach & Rando, 2002; Laprise et al, 2003). Although skeletal muscle fibres of MDC1A patients and mouse models thereof show increased levels of LM-411, the α4 chain seems not to be sufficient to fully compensate for the loss of LM-211. The reason for this lack of compensation might be the failure of LM-411 to induce laminin self-polymerization and to bind to α-dystroglycan (Colognato & Yurchenco, 1999).
Despite the knowledge of molecular events underlying the disease, there are no treatments available to date (Collins & Bonnemann, 2010). Because the formation of basal lamina and receptor-mediated signalling are important in the maintenance of muscle, impairment of those functions probably determines the severity of MDC1A. Consequently, several mechanisms contribute to the pronounced muscle wasting observed in MDC1A. First, the lack of extracellular matrix–cytoskeletal linkage and/or intracellular signalling results in the inability of muscle fibres to withstand the mechanical forces imposed on them during contraction and leads to their degeneration. Secondly, regeneration of skeletal muscle after injury has been shown to be abortive in mouse models for MDC1A (Bentzinger et al, 2005; Kuang et al, 1999; Meinen et al, 2007). Thirdly, apoptosis of myofibres was shown to contribute to the dystrophic phenotype of MDC1A mouse models (Bentzinger et al, 2005; Kuang et al, 1999; Miyagoe et al, 1997).
Agrin is an extracellular matrix molecule that is well known for its function to induce postsynaptic specializations at the neuromuscular junction (Bezakova & Ruegg, 2003). Alternatively spliced forms of agrin that are expressed in non-neuronal tissue, including muscle, lack this postsynapse-inducing activity but bind with very high affinity to laminins via their N-terminal end (Denzer et al, 1997) and to α-dystroglycan via their C-terminal part (Gesemann et al, 1998). These binding data indicate that agrin could potentially link LM-411 to α-dystroglycan and thus reverse the structural deficit of MDC1A muscle. Indeed, preclinical proof-of-concept studies in dyW/dyW mice, a well-characterized mouse model for MDC1A (Kuang et al, 1998b), have shown that overexpression of a miniaturized form of agrin (called mini-agrin) in skeletal muscle increased the tolerance of muscle to mechanical load and improved regeneration of muscle (Bentzinger et al, 2005; Moll et al, 2001) at any stage of the disease (Meinen et al, 2007). Mini-agrin was also successfully delivered to skeletal muscles by systemic application of adeno-associated viral vectors (Qiao et al, 2005).
Despite strong improvement, transgenic expression of mini-agrin did not remove all of the disease symptoms. The main reason for this is probably the lack of expression of mini-agrin in peripheral nerve and in the central nervous system, but only partial restoration of muscle function may also contribute. For example, mini-agrin is not known to bind to integrins and thus, any function mediated by the binding of LM-211 to integrins cannot be compensated for. Indeed, laminin interactions with integrins were shown to activate several pathways that prevent apoptosis (Burkin & Kaufman, 1999; Kuang et al, 1998a, 1998b; Laprise et al, 2002; Vachon et al, 1996, 1997). Interestingly, muscle-specific overexpression of the apoptosis inhibitor Bcl2 (Dominov et al, 2005) or deletion of the pro-apoptotic factor Bax (Girgenrath et al, 2004) significantly improved the dystrophic phenotype and muscle function, and prolonged lifespan in MDC1A mice. Moreover, the apoptosis inhibitors doxycycline and omigapil have been shown to affect disease course in dyW/dyW mice (Erb et al, 2009; Girgenrath et al, 2009). Because mutations in LAMA2 cause a multitude of phenotypes that are based on LM-211 affecting multiple pathways, we now examined whether a combined treatment exerts additional benefits and could increase treatment efficacy.
In a first ‘proof-of-concept’ study, we generated dyW/dyW mice that overexpressed both mini-agrin and the apoptosis inhibitor protein Bcl2 in skeletal muscle. In a step towards application of a combined treatment, we also tested whether the orally bioavailable apoptosis inhibitor omigapil (N(dibenz(b,f)oxepin-10-ylmethyl)-N-methyl-N-prop-2-ynylamine maleate), when combined with mini-agrin, would add further benefit. We report here that the combined treatment with mini-agrin and apoptosis inhibitors indeed provides additive benefit to single treatment on the disease progression in dyW/dyW mice. In particular, we show that mini-agrin prevents muscle degeneration due to mechanical load and thus reduces the replacement of muscle with fibrotic tissue, whereas, inhibition of apoptosis allows the muscle to maintain a near-normal number of fibres. Together, apoptosis inhibitors and mini-agrin potentiate the regeneration capacity and allow diseased muscle fibres to increase in diameter. Most importantly, the dual treatment resulted in a marked increase in muscle force.
RESULTS
Bcl2 and mini-agrin affect different parameters in muscle of dyW/dyW mice
To see whether inhibition of apoptosis and stabilization of the connection between basement membrane and muscle sarcolemma would have an additive benefit for the muscle disease, we mated heterozygous dyW/wt mice with mice transgenic for Bcl2 (Dominov et al, 2005; Girgenrath et al, 2004) or mini-agrin (Moll et al, 2001). The resulting offsprings with the correct genotype were then further mated to eventually obtain mice that were deficient for laminin-α2 and transgenic for Bcl2 (called dyW/Bcl), mini-agrin (dyW/mag) or both transgenes (dyW/Bcl/mag). To assure that expression of all transgenes was not altered by the genotype, protein levels were determined in the triceps muscle from mice with the different genotypes. Indeed, Bcl2 and mini-agrin were expressed at high levels in the transgenic mice as determined by Western blot analysis (Fig S1A of Supporting information).
To determine the effect of the transgenes on the muscle pathology, we analysed the fast-twitch foreleg muscle triceps brachii (Fig 1) and the slow-twitch hindleg muscle soleus (Fig 2). The latter becomes progressively paralysed in dyW/dyW mice as a consequence of peripheral nerve demyelination (Kuang et al, 1998b). Haematoxylin & Eosin (H & E; Merck) and Masson's trichrome stainings of triceps muscle of 12- (Fig 1A) and 16-week-old animals (Fig 1B) are shown. They revealed extensive fibrosis and a high proportion of small, rounded fibres in dyW/dyW mice (Fig 1A). Expression of mini-agrin (dyW/mag) largely impeded the replacement of muscle with fibrotic tissue while Bcl2 expression (dyW/Bcl) seemed to increase fibrosis in dyW/dyW muscle (Fig 1A), a finding that was confirmed by quantification of the relative fibrotic area (Fig 1C) and by determining the amount of the collagen-specific amino acid hydroxyproline, which is a measure of fibrosis (Fig S2A of Supporting information). However, co-expression of mini-agrin (dyW/Bcl/mag) eliminated the fibrotic impact of Bcl2 (Fig 1A and C and Fig S2A of Supporting information). The high degree of fibrosis in Bcl2 transgenic dyW/dyW mice coincided with a strong increase in the number of macrophages in triceps of dyW/Bcl mice (Fig S2C and D of Supporting information). Like the fibrosis, inflammation was reduced by the co-expression of mini-agrin. The same histological results were also obtained in the soleus muscle of 12-week-old mice (Fig 2A and B and Fig S2B of Supporting information), indicating that the progressed paralysis of the hindlegs did not affect fibrosis.
Figure 1. Histological analysis of triceps brachii muscle from 12- to 16-week-old mice.
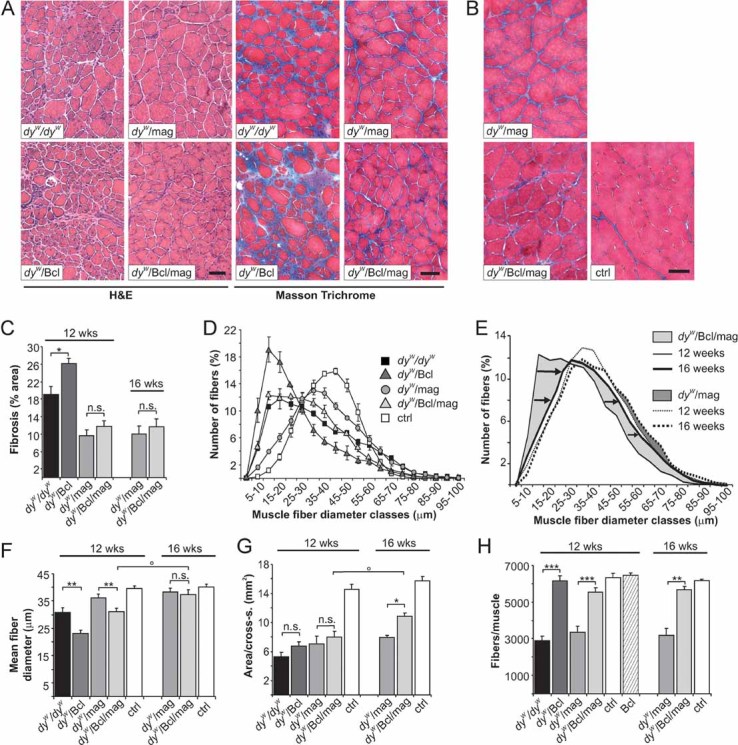
- H & E and Masson's trichrome staining of cross-sections from 12-week old mice.
- Masson's trichrome staining from 16-week-old mice. Pathological changes observed in dyW/dyW mice, such as fibrosis, variation of muscle fibre diameters, increase in small fibres and collagen-containing tissue (blue in Masson Trichrome staining), are reduced in dyW/mag and dyW/Bcl/mag mice but are similar or stronger in dyW/Bcl mice.
- Percentage of fibrotic area per triceps cross-section. Mini-agrin reduces (p = 0.007), whereas Bcl2 increases (p = 0.03) fibrosis in dyW/dyW muscle. Fibrosis is comparable in dyW/mag and dyW/Bcl/mag muscles at 12- (p = 0.22) and at 16- (p = 0.14) weeks of age. N ≥ 3 ≤ 5.
- Muscle fibre size distribution of 12-week-old mice. Values represent relative numbers of fibres in a given diameter class (5 µm/class). DyW/Bcl muscles contain many more small fibres than dyW/dyW muscles. The fibre size distribution of dyW/Bcl/mag is shifted to smaller fibres compared to dyW/mag or control (ctrl) muscles; N = 4.
- Change in fibre size distribution from 12- to 16-week-old dyW/Bcl/mag and dyW/mag mice. Percentage of fibres with larger diameters increases from 12 to 16 weeks of age in dyW/Bcl/mag (arrows) but not in dyW/mag mice; N = 4.
- Mean fibre diameter. The size of dyW/Bcl/mag fibres increases from 12 to 16 weeks of age (two-way ANOVA: p = 0.02) and becomes similar to the mean fibre size in dyW/mag muscle (p = 0.22); N = 4.
- Muscle area (mm2) of mid-belly triceps cross-sections. Control muscle has nearly twice the size compared to all the other genotypes at 12 weeks of age. Although not significantly different over all time points (two-way ANOVA: p = 0.058), muscle area of dyW/Bcl/mag mice increases from 12 to 16 weeks of age (two-way ANOVA: p = 0.026) and becomes significantly larger than in dyW/mag mice (p = 0.03) at 16 weeks of age; N = 4.
- Total number of fibres per triceps cross-section. At 12 weeks of age, dyW/dyW (p < 0.001) and dyW/mag (p = 0.001) muscles have a significantly reduced number of fibres, whereas, the fibre number is near normal in dyW/Bcl (p = 0.8) and dyW/Bcl/mag (p = 0.08) mice. In 16-week-old dyW/Bcl/mag mice, the number of fibres is similar to controls (p = 0.86). Two-way ANOVA analysis confirmed the normalization of the fibre number over all ages in dyW/Bcl/mag when compared to dyW/mag muscle (p < 0.0001). Bcl2 expression did not increase the number of fibres on WT background (Bcl); N ≥ 3 ≤ 5. All values represent the mean ± SEM; N indicates the animal number per experimental group. p-Values are Student's t-test (***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05; n.s. p > 0.05) or two-way ANOVA if indicated (°p ≤ 0.05). Size bars = 100 µm.
Figure 2. Histological analysis of soleus muscle from 12-week-old mice.
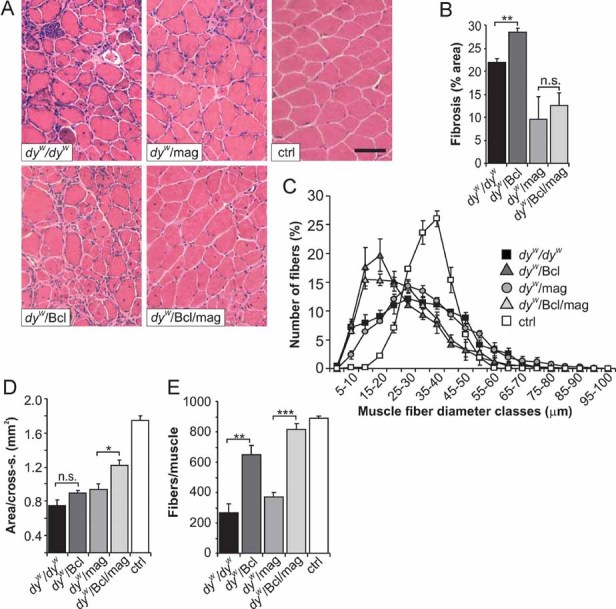
- H & E staining of soleus cross-sections. Whereas, mini-agrin reduces fibrosis (dyW/mag and dyW/Bcl/mag), small fibres are very frequent in dyW/Bcl muscle.
- Percentage of fibrotic area in soleus cross-sections. Fibrosis is more prominent in dyW/Bcl than in dyW/dyW (p = 0.002) muscle. In dyW/mag and dyW/Bcl/mag mice, fibrosis is similar (p = 0.49) and is lower compared to dyW/dyW muscle (dyW/mag: p = 0.004; dyW/Bcl/mag: p = 0.005); N ≥ 3 ≤ 4.
- Muscle fibre size distribution. Values represent relative numbers of fibres in a given diameter class (5 µm/class). None of the transgenes was able to normalize the fibre size distribution; N = 4.
- Muscle area (mm2) of mid-belly soleus cross-sections. Area is increased in dyW/Bcl/mag muscle compared to dyW/mag (p = 0.014) muscle; N ≥ 3 ≤ 4.
- Total number of fibres per soleus cross-section. The number of fibres is increased in dyW/Bcl and dyW/Bcl/mag mice compared to dyW/dyW and dyW/mag mice. The number of fibres in dyW/Bcl/mag mice reaches that found in WT controls, whereas, mini-agrin alone does not strongly influence fibre numbers; N ≥ 3 ≤ 4. N indicates the animal number per experimental group. p > 0.05). Size bars = 100 µm.
As respiratory insufficiency is one of the clinical symptoms of MDC1A patients, we also examined the general histopathology of the diaphragm by H & E staining (Fig S3A of Supporting information) and measured fibrosis using the hydroxyproline assay (Fig S3B of Supporting information). The diaphragm of 12-week-old dyW/dyW mice was thin, fibrotic and showed a clearly reduced fibre number per width when compared to the other genotypes. Fibrosis was also prominent in the diaphragm of dyW/Bcl mice. In dyW/mag and dyW/Bcl/mag mice, the diaphragms were wider and less fibrotic. Moreover, the phenotype did not significantly worsen between the age of 12 and 16 weeks (Fig S3A and B of Supporting information). These results are largely the same as those obtained in triceps brachii and soleus muscles.
Combined treatment with mini-agrin and Bcl2 enables the dyW/dyW muscle to grow
Change in muscle fibre size distribution is a hallmark of MDC1A and dyW/dyW mice. Expression of Bcl2 increased the number of small fibres even further, both in triceps (Fig 1D) and soleus (Fig 2C), whereas, expression of mini-agrin normalized the fibre size distribution. Interestingly, co-expression of mini-agrin with Bcl2 reversed the effect of Bcl2 alone and resulted in the normalization of fibre size distribution in 16-week-old mice, reaching the same profile as seen in dyW/mag mice (Fig 1E and F). This normalization was also seen in the mean size of muscle fibres (Fig 1F). In contrast to triceps muscle, no effect of mini-agrin on muscle fibre size distribution was observed in soleus muscle (Fig 2C). Because mini-agrin and Bcl2 are not expressed in peripheral nerve (Dominov et al, 2005; Moll et al, 2001), the paralysis of the hindlegs was not prevented at this age. Thus, the small fibre size in transgenic mice is probably due to the atrophy induced by the paralysis of the hindlegs.
During our analysis, we realized that the total area of mid-belly cross-sections of triceps (Fig 1G) and soleus (Fig 2D) muscle from 12-week-old dyW/dyW mice was much smaller than in age-matched controls. Overexpression of Bcl2, mini-agrin or the combination of both caused a small but non-significant increase in the cross-sectional area (CSA; Fig 1G and Fig 2D). Interestingly, when mini-agrin was co-expressed with Bcl2, this trend to increase the CSA became significant in triceps muscle from 16-week-old mice (Fig 1G). Because of the peripheral neuropathy that causes a progressive atrophy of the hindleg muscles in dyW/dyW (Miyagoe-Suzuki et al, 2000) as well as in dyW/Bcl, dyW/mag and dyW/Bcl/mag mice, this effect was not examined in soleus muscles from 16-week-old mice. However, the results obtained from triceps indicate that muscle of dyW/Bcl/mag mice has the capability to grow.
Bcl2 enables the maintenance of the original muscle fibre number in dyW/dyW mice
Next, we tested whether the change in muscle area was due to changes in the total number of muscle fibres. While 12-week-old dyW/dyW and dyW/mag mice contained about 40% fewer fibres than controls (Fig 1H and 2E), the number of fibres was largely normalized in dyW/Bcl and dyW/Bcl/mag mice, both in the triceps (Fig 1H) and soleus muscle (Fig 2E). Moreover, the number of fibres remained normal in the triceps of 16-week-old dyW/Bcl/mag mice. Interestingly, Bcl2 expression on a wild-type (WT) background (Bcl) did not increase the fibre number (Fig 1H), suggesting that Bcl2-mediated inhibition of apoptosis may allow the dyW/dyW muscle to maintain the original fibre number, including the small fibres that would have disappeared in the absence of Bcl2.
To verify the role of apoptosis in the loss of fibres in dyW/dyW mice, we used TdT-mediated dUTP nick end labelling (TUNEL) which recognizes nuclear in situ DNA-strand breaks that occur in nuclei undergoing apoptosis. In dyW/dyW and dyW/mag muscles, apoptotic TUNEL-positive muscle fibres could be identified (Fig 3A, arrows) and represented 1.7 and 1% of all fibres, respectively (Fig 3B). In contrast, no TUNEL-positive myonuclei were found upon Bcl2 expression. The TUNEL-positive nuclei detected in dyW/Bcl and dyW/Bcl/mag mice all seemed to belong to non-muscle cells (Fig 3A, arrowheads). Apoptosis has been suggested to increase during abortive muscle regeneration in MDC1A patients (Hayashi et al, 2001) and mouse models thereof (Bentzinger et al, 2005; Dominov et al, 2005; Girgenrath et al, 2004; Kuang et al, 1999). To account for this, we also counted the number of TUNEL-positive, centrally localized myonuclei. Central nucleation has been shown to be indicative of regenerative processes. As shown in Fig 3C, triceps muscle from dyW/dyW and dyW/mag contained more regenerating fibres that were TUNEL-positive than those expressing Bcl2. These results indicate that Bcl2 largely prevents the death of fibres both in normal and regenerating muscle, which may maintain the number of fibres to the levels of control mice.
Figure 3. Apoptosis in triceps brachii.
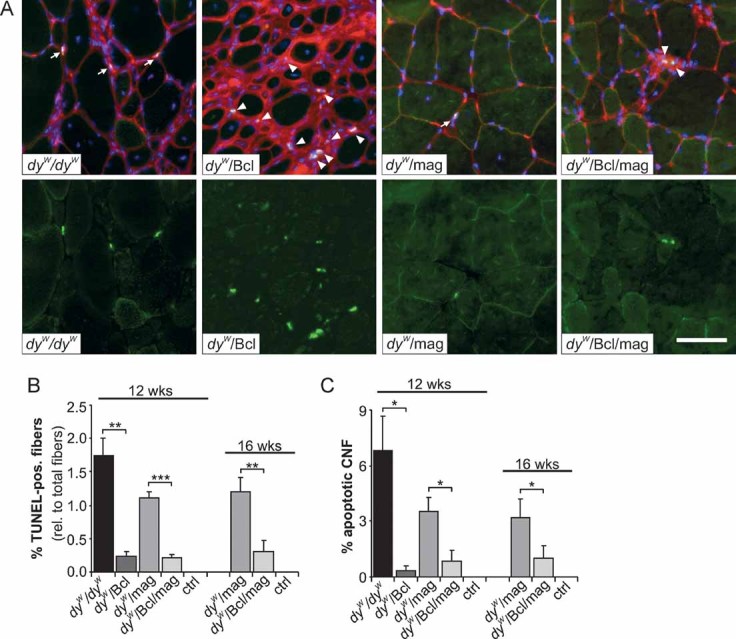
- Apoptotic nuclei recognized by TUNEL staining (green, separately shown in the bottom row) of triceps brachii cross-sections from 12-week-old mice. Muscles were counterstained with WGA (red) to visualize membranes and fibrotic areas and with DAPI (blue) to stain nuclei. Muscle fibres containing apoptotic nuclei are found in dyW/dyW and to a lower extent in dyW/mag muscle (arrows). Apoptotic nuclei recognized in dyW/Bcl and dyW/Bcl/mag muscle reside outside of the muscle fibres (arrowheads).
- Relative number of the TUNEL-positive muscle fibres. Bcl2 expression inhibits apoptosis in muscle fibres of dyW/dyW (p = 0.004). Similarly, Bcl2 prevents apoptosis in dyW/mag mice at 12 weeks (p ≤ 0.001), at 16 weeks (p = 0.002) as well as over both ages (two-way ANOVA: p < 0.0001); N ≥ 3 ≤ 4.
- Percentage of centrally localized nuclei that undergo apoptosis. In dyW/dyW muscles almost 7% and in dyW/mag muscles 3.5% of the regenerating fibres are TUNEL-positive. Apoptosis of regenerating fibres is strongly decreased by expression of Bcl2 in muscles of dyW/dyW (p = 0.02) and dyW/mag mice (12 weeks: p = 0.03; 16 weeks: p = 0.04; two-way ANOVA over both ages: p = 0.0027); N ≥ 3 ≤ 4. All values represent the mean ± SEM; N indicates the animal number per experimental group. p-Values are Student's t-test (***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05; n.s. p > 0.05) or two-way ANOVA as noted above. Size bars = 50 µm.
Bcl2 enhances the regeneration efficiency in dyW/mag muscle
Besides its function in stabilizing muscle fibres during contraction, LM-211 has also been shown to be important for the successful regeneration of muscle (Bentzinger et al, 2005; Kuang et al, 1999). In addition, regeneration is improved in dyW/mag mice (Meinen et al, 2007). Regenerating muscle fibres transiently express the developmental form of myosin heavy chain (dMyHC; Novocastra, NCL-MHCd) and newly regenerated fibres have their nuclei in the centre and not in the periphery. Thus, the number of dMyHC-positive and centrally nucleated fibres (CNF) is a measure of ongoing and successful regeneration. To assess the role of apoptosis inhibition in the regeneration process, we first counted the number of CNF in the different mouse models. As shown earlier, the number of CNF was increased in dyW/dyW mice due to ongoing muscle degeneration when compared to WT controls (Fig 4A). Expression of Bcl2 resulted in a further threefold increase in the number of CNF, suggesting that regeneration of muscle fibres is more effective. Similarly, the number of CNF was increased in dyW/mag mice compared to dyW/dyW but was lower than in dyW/Bcl mice. This finding is consistent with the previous interpretation that mini-agrin enables successful regeneration but primarily prevents muscle degeneration by its linking of the basement membrane to α-dystroglycan (Moll et al, 2001). Interestingly, expression of both Bcl2 and mini-agrin resulted in a further increase of the number of CNF in 12- and 16-week-old mice compared to dyW/mag mice. To test for ongoing regeneration, we also stained for dMyHC. As with CNF, the number of dMyHC-positive fibres in triceps muscle was several fold higher in dyW/Bcl than in dyW/dyW mice and the effect of Bcl2 was also seen when combined with mini-agrin (Fig 4B and C).
Figure 4. Spontaneous and injury-induced muscle regeneration.
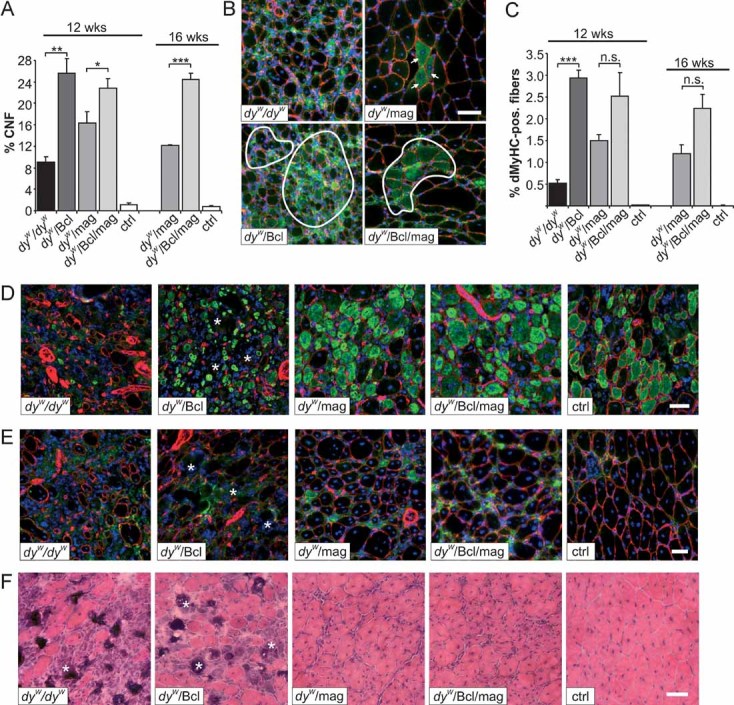
- A. Percentage of CNF. In dyW/dyW mice, only 9% of the muscle fibres are centrally nucleated, indicative of impaired regeneration. In dyW/mag muscle the number of CNF is increased and is further elevated by co-expression of Bcl2 in both 12- and 16-week-old dyW/Bcl/mag mice (two-way ANOVA over all ages: p < 0.0001); N = 4.
- B. Spontaneous muscle regeneration in 12-week-old muscles. Antibodies to dMyHC (green) and laminin-γ1 (red) were used. DAPI (blue) visualizes nuclei. Only few dMyHC-positive fibres are found in dyW/dyW mice. Some more dMyHC-positive fibres are present in dyW/mag (arrows) muscle, whereas, many more are detected upon Bcl2 expression in dyW/Bcl and dyW/Bcl/mag (encircled areas).
- C. Quantification of dMyHC-positive fibres. The number of dMyHC-positive fibres is highest in Bcl2 transgenic mice (dyW/Bcl and dyW/Bcl/mag) followed by dyW/mag and dyW/dyW mice. Although not significant at 12 or 16 weeks of age (t-test), the increase of dMyHC-positive fibres in dyW/Bcl/mag compared to dyW/mag muscles becomes significant when measured over all ages (two-way ANOVA: p = 0.022). No spontaneous regeneration was detected in control muscles (ctrl); N ≥ 4 ≤ 6.
- D.–F. Regenerative response of tibialis anterior muscle of 6-week-old mice after notexin injection.
- D. Muscle cross-sections were stained with antibodies to dMyHC (green) and laminin-γ1 (red), and with the nuclear marker DAPI (blue) 1 week after notexin injection. Only very few dMyHC-positive fibres are detected in dyW/dyW mice, while many muscle fibres are dMyHC-positive in dyW/Bcl, dyW/mag, dyW/Bcl/mag and control (ctrl) mice. Note that muscle fibres in dyW/Bcl mice are small; N ≥ 4 ≤ 5.
- E. Regeneration status of tibialis anterior 2 weeks after notexin injection. Antibodies to dMyHC (green), laminin-γ1 (red) and the nuclear marker DAPI (blue) were used. Absence of dMyHC-positive fibres and large areas devoid of laminin-γ1 staining (asterisks) are indicators of the failed regeneration in dyW/dyW and dyW/Bcl muscles. The presence of few dMyHC-positive fibres and the non-disrupted laminin-γ1 staining indicate successful muscle regeneration in dyW/mag, dyW/Bcl/mag and control mice; N ≥ 3 ≤ 4.
- F. H & E staining of tibialis anterior 2 weeks after notexin injection shows the failure in regeneration leading to fibrosis in dyW/dyW and dyW/Bcl muscles (asterisks) and the successful regeneration in dyW/mag, dyW/Bcl/mag and control mice; N ≥ 3 ≤ 4. All values represent the mean ± SEM; N indicates the animal number per each experimental group. p-Values are Student's t-test (***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05; n.s. p > 0.05) or two-way ANOVA as noted in the text. Size bars = 50 µm.
To follow muscle regeneration directly, we next injected the myotoxin notexin into tibialis anterior muscle and examined muscles over time. One week post-injection, regenerating dMyHC-expressing fibres were rare and small in dyW/dyW muscle while many fibres were dMyHC-positive in all the other genotypes (Fig 4D). Strikingly, the regenerating fibres in dyW/Bcl mice were very small and the muscle included large regions with mononucleated cells and damaged tissue (Fig 4D, asterisks). In contrast, regenerating fibres in dyW/mag and dyW/Bcl/mag mice were rather large and reached a similar size as in controls. Two weeks after notexin injection, the structure of the entire muscle was re-built in dyW/mag, dyW/Bcl/mag and control muscles, whereas, the muscle in dyW/Bcl and dyW/dyW mice contained large fibrotic regions (asterisks) and the fibres remained unconnected (Fig 4E and F). Thus, although the muscle of dyW/Bcl mice shows early signs of a successful regeneration, late steps in this process are not completed. In contrast, these late steps are successfully completed if mice are also transgenic for mini-agrin.
Effect of co-expression of Bcl2 and mini-agrin on behaviour and survival in dyW/dyW mice
We next evaluated the effect of the different treatments on overall health. As a single transgene, Bcl2 had a moderate but significant effect on survival, whereas, mini-agrin tripled mean survival compared to dyW/dyW mice (Fig 5A). Although mice that expressed both mini-agrin and Bcl2 survived a little bit longer than dyW/mag mice, the difference was not significant. Bcl2 had also a small effect on body weight (Fig 5B), overall health (Fig 5C), locomotory activity (Fig 5D) and grip strength (Fig 5E). Compared to dyW/dyW mice, the effect of Bcl2 was significant over all time points (see figure legend). Transgenic expression of Bcl2 was also beneficial in dyW/mag mice in all those paradigms and the effect was significant over all time points for the body weight (Fig 5B) and the grip strength (Fig 5E) but not for the locomotion (Fig 5D). Interestingly though, locomotion reached significance at the last time point of 16 weeks (compare dyW/Bcl/mag with dyW/mag). Thus, these data confirm previous work using Bcl2 (Dominov et al, 2005; Girgenrath et al, 2004) but also show that the effect of mini-agrin is much larger than that of Bcl2 and that the additional benefit gained by co-expressing Bcl2 with mini-agrin is rather small and becomes significant at the older age.
Figure 5. Survival and behaviour of mice.
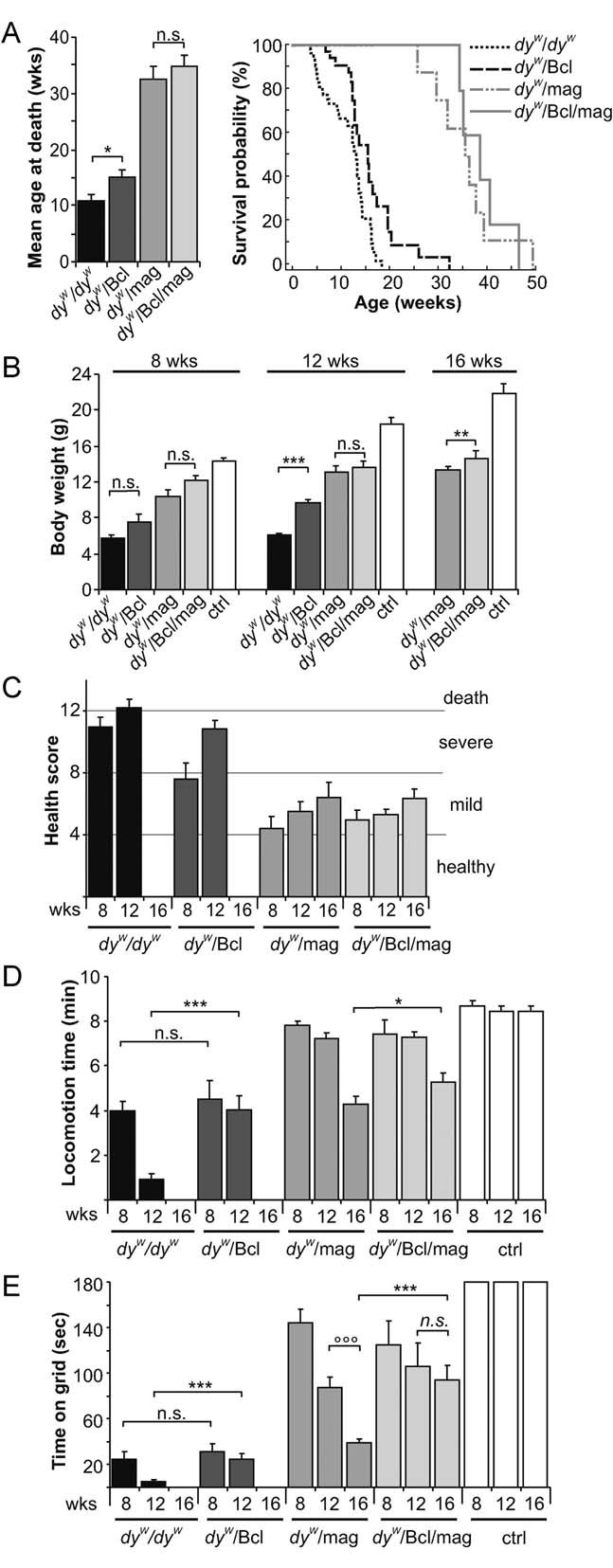
- Average survival (left panel) and Kaplan–Meier cumulative survival plot (right panel). Average survival of dyW/dyW mice increases by 30% upon Bcl2 expression (p = 0.02). Expression of mini-agrin triples the mean survival time (p ≤ 0.001). In the few (N = 5) double transgenic dyW/Bcl/mag mice tested, the survival was not prolonged compared to dyW/mag mice; N = 35, 22, 8 and 5 for dyW/dyW, dyW/Bcl, dyW/mag and dyW/Bcl/mag, respectively.
- Body weight at the ages of 8, 12 and 16 weeks. Both transgenes increase the body weight of dyW/dyW mice. Although the effect of Bcl2 is rather small in comparison to the effect exerted by mini-agrin, Bcl2 significantly elevates body weight in dyW/dyW mice over all time points (two-way ANOVA: p < 0.0001). In addition, Bcl2 expression increases body weight over all ages in dyW/mag mice (two-way ANOVA: p = 0.002); N ≥ 4 ≤ 11.
- Overall health status of mice was judged by the assessment of activity, appetite, posture, fur and eyes of 8, 12 and 16-week-old mice. For details on the score sheet used see Materials and Methods Section; N ≥ 4 ≤ 11.
- Open-field locomotion during 10 min. Bcl2 expression delays the loss of locomotory activity in dyW/dyW mice over all time points (two-way ANOVA: p ≤ 0.001) but does not reach the same efficacy as mini-agrin. Although not significant over all time points (two-way ANOVA: p = 0.28), locomotion becomes improved in 16-week-old dyW/Bcl/mag when compared to dyW/mag mice (p = 0.048); N ≥ 4 ≤ 11.
- Grip strength was measured by placing the mice on a vertical grid. The test was stopped after 180 s. DyW/Bcl mice are stronger than dyW/dyW mice measured over all time points (two-way ANOVA: p = 0.005). In 16-week-old dyW/mag mice, co-expression of Bcl2 prevents the loss of grip strength (p ≤ 0.001). Over all time points, Bcl2 increased the muscle strength of dyW/mag mice (two-way ANOVA: p = 0.014); N ≥ 4 ≤ 11. All values represent the mean ± SEM; N indicates the animal number per experimental group; p-values are Student's t-test (***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05; n.s. p > 0.05) or two-way ANOVA if indicated (°°°p ≤ 0.001; °°p ≤ 0.01; °p ≤ 0.05; n.s. p > 0.05).
Muscle function is improved in dyW/Bcl/mag mice
To test directly whether the difference on grip strength reflected an improvement in the force of muscles, we next measured the contractile properties in isolated extensor digitorum longus (EDL; Table 1, Fig 6A, C and E) and soleus (Table 1; Fig 6B, D and F) muscles. To avoid falsification of the results by the MDC1A-typical progressing hindleg paralysis (Miyagoe-Suzuki et al, 2000), we analysed the muscles in young, 4-week-old animals, when paralysis was not yet prominent and muscle histology of dyW/mag mice was still largely indistinguishable from control mice (Fig S4 of Supporting information). In both muscles, the wet weight, optimal contraction length (L0), CSA and the twitch-to-tetanus ratio (Pt/P0) increased from dyW/dyW, dyW/Bcl, dyW/mag and dyW/Bcl/mag to control mice (Table 1). There was some improvement in most of the parameters from dyW/dyW to dyW/Bcl mice, although this trend was never statistically significant (Table 1 and Fig 6). Interestingly, when Bcl2 and mini-agrin were co-expressed (dyW/Bcl/mag) there was a significant increase in the absolute twitch force (Pt) compared to dyW/mag mice in both EDL and soleus muscle (Table 1 and Fig 6A and B). Similarly, the normalized, maximum isometric tetanic force (Po) was higher in dyW/Bcl/mag mice than in dyW/mag mice (Fig 6C and D). However, upon normalization of Po to muscle size [i.e. maximum specific tetanic force (sPo)], the significance of the difference between dyW/mag and dyW/Bcl/mag mice got lost (Fig 6E and F). The fact that the difference between dyW/Bcl/mag and dyW/mag mice is higher for the absolute force than the specific force is in agreement with our finding (Fig 1H and E) that the number of fibres is much increased in dyW/Bcl/mag mice. Thus, although the force generated per muscle fibre is not much higher in dyW/Bcl/mag mice than in dyW/mag mice, the total force is significantly increased. These data show that expression of Bcl2 in dyW/mag mice increases muscle force by up to 50% and thus indicates that dyW/dyW mice do benefit from a combined treatment with mini-agrin and inhibition of apoptosis.
Table 1.
In vitro function of EDL and soleus muscles
| dyW/dyW | dyW/Bcl | dyW/mag | dyW/Bcl/mag | ctrl | |
|---|---|---|---|---|---|
| Body weight (g) | 8.4 ± 1.4 | 9.1 ± 1.2 | 14.3 ± 0.1 | 15.9 ± 1.1 | 21.3 ± 1.6 |
| EDL | |||||
| Nmuscles | 6 | 5 | 5 | 7 | 6 |
| Wet muscle mass (mg) | 3.5 ± 0.5 | 3.8 ± 0.3 | 6.3 ± 0.8 | 6.7 ± 0.2 | 9.0 ± 0.4 |
| L0 (mm) | 7.9 ± 0.3 | 8.4 ± 0.7 | 10.4 ± 0.2 | 10.4 ± 0.3 | 10.9 ± 0.4 |
| CSA (mm2) | 0.42 ± 0.04 | 0.42 ± 0.01 | 0.54 ± 0.04 | 0.62 ± 0.04 | 0.78 ± 0.04 |
| Pt (mN) | 2.9 ± 1.5 | 4.3 ± 1.4 | 11.9 ± 1.7 | 24.6 ± 4.6 | 51.5 ± 5.3 |
| sPt (mN/mm2) | 5.9 ± 2.2 | 10.0 ± 3.2 | 20.5 ± 1.8 | 38.7 ± 6.7 | 67.5 ± 8.3 |
| TPt (ms) | 12.0 ± 0.7 | 11.0 ± 0.4 | 10.8 ± 0.6 | 12.3 ± 0.9 | 12.5 ± 0.7 |
| RT1/2 (ms) | 37.4 ± 4.2 | 34.2 ± 3.5 | 30.9 ± 2.8 | 28.9 ± 0.9 | 30.8 ± 2.9 |
| P0 (mN) | 20.9 ± 8.0 | 30.9 ± 5.3 | 84.2 ± 5.3 | 120.2 ± 11.0 | 227.5 ± 21.1 |
| sP0 (mN/mm2) | 42.3 ± 8.4 | 72.5 ± 11.8 | 146.2 ± 15.0 | 193.2 ± 24.4 | 297.2 ± 32.9 |
| Pt/P0 | 0.118 ± 0.02 | 0.129 ± 0.03 | 0.146 ± 0.02 | 0.195 ± 0.02 | 0.228 ± 0.02 |
| soleus | |||||
| Nmuscles | 5 | 5 | 6 | 6 | 5 |
| Wet muscle mass (mg) | 3.46 ± 0.7 | 3.6 ± 0.3 | 3.8 ± 0.2 | 4.8 ± 0.2 | 7.3 ± 0.3 |
| L0 (mm) | 8.0 ± 0.2 | 7.9 ± 0.3 | 8.25 ± 0.2 | 8.75 ± 0.3 | 9.1 ± 0.2 |
| CSA (mm2) | 0.41 ± 0.08 | 0.42 ± 0.03 | 0.431 ± 0.03 | 0.52 ± 0.02 | 0.76 ± 0.03 |
| Pt (mN) | 1.8 ± 0.6 | 3.3 ± 0.6 | 5.4 ± 1.2 | 9.7 ± 1.3 | 21.6 ± 3.5 |
| sPt (mN/mm2) | 3.9 ± 0.8 | 7.6 ± 1.2 | 12.0 ± 2.2 | 20.7 ± 3.3 | 24.6 ± 2.6 |
| TPt (ms) | 24.0 ± 0.9 | 26.4 ± 2.5 | 29.0 ± 2.0 | 24.0 ± 1.0 | 23.8 ± 1.7 |
| RT1/2 (ms) | 56.4 ± 4.0 | 59.2 ± 4.8 | 62.8 ± 3.5 | 57.4 ± 2.4 | 56.8 ± 5.3 |
| P0 (mN) | 22.2 ± 7.9 | 30.4 ± 4.1 | 45.5 ± 8.5 | 70.2 ± 5.5 | 105.2 ± 14.6 |
| sP0 (mN/mm2) | 47 ± 13 | 71 ± 8 | 102 ± 14 | 124 ± 11 | 138 ± 17 |
| Pt/P0 | 0.092 ± 0.01 | 0.112 ± 0.02 | 0.115 ± 0.00 | 0.155 ± 0.03 | 0.164 ± 0.04 |
The table shows the in vitro contractile properties at 30°C of EDL and soleus muscle. Data (mean ± SEM) include: number of tested muscles (N(muscles)), animal body weight (g), wet muscle weight (mg), optimal muscle length (L0, mm), CSA (mm2), peak twitch force (Pt; mN), specific twitch force represents Pt normalized to CSA (sPt; mN/mm2), time to peak twitch tension (TPt; ms), half-relaxation time (RT1/2; ms), maximum isometric tetanic force (P0; mN), maximum specific tetanic force (sP0; mN/mm2) and twitch tetanus ratio (Pt/P0).
Figure 6. Contraction properties of EDL and soleus muscle.
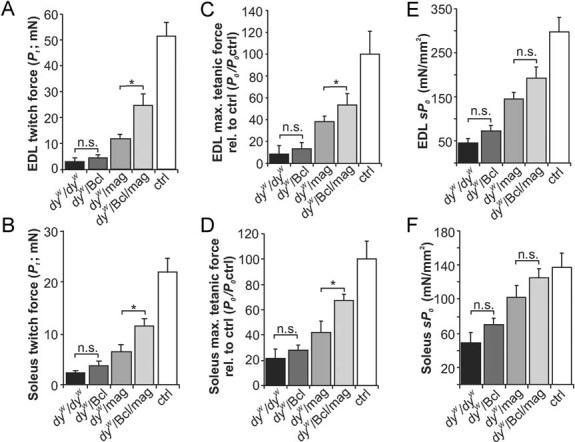
EDL (A, C, E) and soleus (B, D, F) muscles from mice with the indicated genotypes.
- A,B. Absolute twitch force (Pt), measured by a single electrical stimulation at 15 V, is similar in dyW/dyW and dyW/Bcl mice (EDL: p = 0.52; soleus: p = 0.15) but increases from dyW/Bcl to dyW/mag (EDL: p = 0.009; soleus: p = 0.05), from dyW/mag to dyW/Bcl/mag (EDL: p = 0.018; soleus: p = 0.03) and from dyW/Bcl/mag to control (EDL: p = 0.006; soleus: p = 0.001).
- C,D. Maximum isometric tetanic force (P0) of EDL (C) or soleus muscle (D) expressed as percentage of controls (P0/P0 ctrl). Co-expression of Bcl2 (dyW/Bcl/mag) increases the maximal tetanic force of dyW/mag in EDL (p = 0.03) and in soleus (p = 0.02).
- E,F. The maximum specific tetanic force (sP0) increases gradually from dyW/dyW to control mice in both EDL (E) and soleus (F). However, no significant change of sP0 is seen between dyW/Bcl/mag and dyW/mag mice in EDL (E; p = 0.07) or in soleus (F; p = 0.24) muscle. All values represent the mean ± SEM; N numbers are shown in Table 1. p-values are Student's t-test (***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05; n.s. p > 0.05).
Pharmacological inhibition of apoptosis enhances amelioration by mini-agrin
We have recently shown that oral application of the pharmacological apoptosis inhibitor omigapil to dyW/dyW mice ameliorates MDC1A pathology (Erb et al, 2009). Based on the finding that transgenic expression of Bcl2 increased the therapeutic effect of mini-agrin, we next tested whether the combination of mini-agrin and systemic application of omigapil would be of benefit. To this end, dyW/mag mice were treated with a daily dose of 0.1 mg/kg omigapil starting at the age of 15 days. The first week of treatment, omigapil was injected into the peritoneum followed by oral gavage after weaning. Mice were analysed at the age of 12 weeks. Visual inspection of H & E-stained cross-sections from triceps muscle did not reveal a striking difference between omigapil- (dyW/mag-omigapil) and vehicle- (dyW/mag-vehicle) treated dyW/mag mice (Fig 7A). Quantitative assessment of changes showed, however, lowered creatine kinase (CK) levels in the blood (Fig 7B), normalized muscle fibre size distribution (Fig 7C), a trend to increasing the mean fibre size (Fig 7D) and to enlarging muscle area in dyW/mag mice (Fig 7E). In addition, omigapil also reduced the muscle fibre loss in dyW/mag mice (Fig 7F). Importantly, many of the functional parameters were significantly improved by omigapil. Those included body weight gain (Fig 7G), locomotive activity in the open-field test (Fig 7H) and grip strength (Fig 7I). However, we could not detect a difference in the overall survival (Fig 7J). Together, treatment of dyW/mag mice with omigapil further improved several of the disease parameters but did not affect survival.
Figure 7. Effects of omigapil treatment in 12-week-old dyW/mag mice.
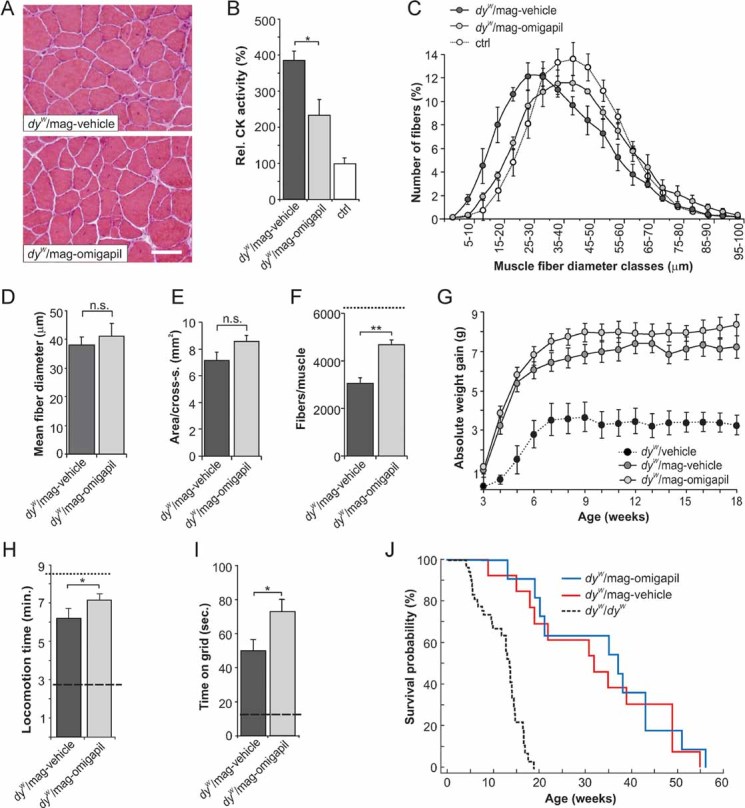
- H & E staining of cross-sections from triceps brachii muscle of dyW/mag mice treated daily with 0.1 mg/kg omigapil (dyW/mag-omigapil) or vehicle (dyW/mag-vehicle).
- Blood CK levels expressed as percentage of control. Omigapil treatment in dyW/mag mice significantly lowers the CK levels (p = 0.023); N ≥ 6 ≤ 8.
- Muscle fibre size distribution of triceps brachii. Values represent relative numbers of fibres in a given diameter class (5 µm/class). Muscles from vehicle-treated dyW/mag mice contain more small-caliber fibres than muscles from age-matched dyW/mag-omigapil mice; N = 5.
- Mean fibre diameter. Omigapil treatment slightly but not significantly (p = 0.07) elevates the mean fibre size in dyW/mag muscles; N = 5.
- Muscle area (mm2) of mid-belly triceps cross-sections. Omigapil treatment tends to enlarge muscle area in dyW/mag mice (p = 0.06); N = 5.
- Total number of fibres per triceps cross-section. Omigapil treatment reduces muscle fibre loss in dyW/mag (p = 0.006) but does not completely normalize fibre numbers, as control muscle contain >6000 fibres (dashed line); N = 5.
- Weight gain normalized to the body weight at 15 days of age (onset of treatment). For each week, body weight data were averaged from daily measurements. Body weight at 15 days of age (starting body weight) was 5.55 ± 0.20 g for dyW/mag-omigapil mice and 6.88 ± 0.21 g for dyW/mag-vehicle mice. Body weight curve for vehicle-treated dyW/dyW mice (Erb et al, 2009) is shown for comparison (dotted line); N ≥ 9 ≤ 11.
- Locomotion within a 10 min observation period. Locomotive activity of dyW/mag-omigapil mice is significantly increased compared to vehicle-treated dyW/mag mice (p = 0.046). Locomotion of dyW/dyW (dashed line) and WT control mice (dotted line) are given for comparison; N ≥ 9 ≤ 11.
- Grip strength is expressed as time mice were able to hold on a vertical grid. Omigapil treatment significantly increases the grip strength in dyW/mag mice (p = 0.02). Grip strength of dyW/dyW mice is indicated (dashed line). WT control mice stay >180 s on the grid; N ≥ 9 ≤ 11.
- Kaplan–Meier cumulative survival plot. Daily treatment (0.1 mg/kg) with omigapil does not improve the survival probability of dyW/mag mice. All values are mean ± SEM. N indicates the animal number per experimental group. p-Values are Student's t-test (***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05; n.s. p > 0.05). Scale bar = 50 µm.
DISCUSSION
In this work, we tested whether the combination of anti-apoptosis treatment and transgenic expression of mini-agrin has additive effects on the disease progression in a mouse model for MDC1A. Indeed, we find such an additive effect for several of the parameters measured. This was particularly obvious in the grip strength (Fig 5E), the force measurements on isolated muscles (Fig 6), and the number and size of muscle fibres (Fig 1).
Combination of mini-agrin and Bcl2 improves force production in MDC1A muscles
Our data indicate that expression of Bcl2 is the key factor that preserves the number of muscle fibres while expression of mini-agrin is important for the preservation of force in individual muscle fibres. Support for this hypothesis stems from the singly transgenic mice. In dyW/Bcl mice, the muscles contain the same number of fibres as in control mice (Fig 1E). However, the muscle fibres in dyW/Bcl mice are small, even significantly smaller than those in dyW/dyW mice (Fig 1D and F). This decrease can explain the finding that the area of mid-belly cross-sections remains as small as in dyW/dyW muscles (Fig 1G) despite the increase in fibre number and fibrosis (Fig 1C; Fig S2A of Supporting information).
Thus, prevention of apoptosis by Bcl2 seems to allow survival but not the growth of muscle fibres. Mice singly transgenic for mini-agrin, in contrast, show a fibre size distribution that is close to that in control muscles (Fig 1D and E), but individual muscles of the dyW/mag mice contain fewer fibres (Fig 1H). Consequently, the area of mid-belly cross-sections is still smaller than in control muscles (Fig 1G). Thus, mini-agrin seems to allow growth and stabilization of muscle fibres. Intriguingly, the combination of Bcl2 and mini-agrin increases the CSA in triceps muscle up to 70% of that of control muscle (Fig 1G). Importantly, co-expression of Bcl2 and mini-agrin tended to further potentiate the gain of muscle force measured in vitro (Fig 6). The fact that this test was performed in as young as 4-week-old mice, an age at which the typical hindleg paralysis of dyW/dyW mice is not yet prominent and at which muscle histology of dyW/mag mice is comparable to control mice (Fig S4 of Supporting information; Moll et al, 2001), indicates that co-expression of the two transgenes indeed improves muscle strength and not only delays the loss of muscle force in dyW/dyW mice. Together, these results suggest that inhibition of apoptosis by Bcl2 in combination with the mechanical stabilization of muscle by mini-agrin can enhance contractile properties of muscle.
Mini-agrin and Bcl2 potentiate their effect on muscle regeneration in dyW/dyW mice
Besides the function of LM-211 to stabilize muscle fibre integrity, its binding to α7β1 integrin has been suggested to activate signals important for survival (Vachon et al, 1996, 1997). Muscle regeneration is strongly impaired in dyW/dyW and dy3K/dy3K mice (Bentzinger et al, 2005; Kuang et al, 1999; Miyagoe et al, 1997) and Bax levels have been shown to be increased during regeneration (Olive & Ferrer, 2000). Thus, non-productive muscle regeneration in laminin-α2-deficient mice might be due to increased apoptosis and inhibition of apoptosis may thus be particularly important during regenerative processes. Indeed, Bcl2 levels have been shown to be considerably increased in the early period of muscle fibre regeneration (Bulyakova & Azarova, 2006; Shefer et al, 2002).
When we tested this idea, we found that many muscle fibres of dyW/Bcl mice contained centralized myonuclei and re-expressed the regeneration marker dMyHC (Fig 4B). One week after notexin-induced muscle damage, many muscle fibres of dyW/Bcl mice were dMyHC-positive (Fig 4D) but regeneration was not successful after 2 weeks (Fig 4E and F) because most of the initially formed muscle fibres remained unconnected and were replaced by fibrotic tissue. Co-expression of mini-agrin prevented this fibrosis, allowed regenerating fibres to increase in diameter and to re-build a structured muscle tissue (Fig 4E and F). Thus, the combined treatment of apoptosis inhibition and mini-agrin expression increased the regeneration efficiency in dyW/dyW muscles. This effect is based on the Bcl2-mediated prevention of apoptosis at early stages of regeneration and the mini-agrin-mediated reconnection of the muscle fibres to the extracellular matrix, which is essential to complete regeneration. In addition and since notexin leaves the satellite cells unharmed, the added benefit by combining Bcl2 and mini-agrin in the muscle regeneration process might also originate, at least in part, from a protective effect on the satellite cells. Thus, the cumulative action of Bcl2 and mini-agrin during spontaneous muscle regeneration is probably the molecular basis for the overall increase in fibre number and size in the dyW/Bcl/mag mice.
Systemic inhibition of apoptosis prevents the inflammatory phenotype of muscle-restricted Bcl2 expression
Mini-agrin is known to markedly reduce the replacement of muscle with fibrotic tissue. Unlike others (Dominov et al, 2005), we observed prominent fibrosis (Fig 1A–C; Fig 2A and B) and inflammation (Fig 2A and B of Supporting information) in muscles of dyW/Bcl mice. Indeed, Bcl2 expression under control of the muscle-specific MyoD promoter inhibits apoptosis in dyW/dyW muscle fibres (Fig 3A and B), but accumulates other (inflammatory) cell types that frequently are apoptotic (Fig 3A, arrowheads). Together with the finding that Bcl2 impairs the removal of damaged tissue from notexin-injured dyW/dyW muscle (Fig 4D and E), the increased fibrosis in muscles of dyW/Bcl mice may trigger an inflammatory response that activates macrophages. Although the mechanical muscle stabilization by mini-agrin largely counteracts this adverse effect of Bcl2 (Fig 1A–C; Fig 2A and B; Fig S2C and D of Supporting information), only a generalized inhibition of apoptosis can prevent the inflammatory response completely. This is seen in the experiments using oral application of omigapil (Erb et al, 2009) or in the whole-body deletion of the pro-apoptotic gene Bax (Girgenrath et al, 2004). Thus, the increased fibrosis in the Bcl2 transgenic mice might be due to the restricted inhibition of apoptosis to skeletal muscle.
In this context, we also noticed that omigapil already at 12 weeks of age enhanced the ameliorative effect of mini-agrin and improved fibre size distribution (Fig 7F), mean fibre size (Fig 7D), muscle area (Fig 7E), grip strength (Fig 7I) and locomotion (Fig 7H). In contrast, in dyW/Bcl/mag mice these parameters were ameliorated only at the age of 16 weeks (Fig 1E–G; Fig 5D and E). It is well possible that this age-specific difference relies on the fact that inhibition of apoptosis by systemic application of omigapil acts on many cell types, whereas, inhibition of apoptosis by Bcl2 expression is restricted to muscles. Therefore, we think that the increased fibrosis and inflammatory phenotype upon muscle-restricted Bcl2 expression in dyW/dyW mice is a secondary effect that resides in another cell type than muscle fibres and thus might not be of concern for the treatment of MDC1A.
Translational aspects for the treatment of MDC1A by mini-agrin or apoptosis inhibitors
Another important insight of our studies is the observation that the efficacy of mini-agrin is by far greater than that of anti-apoptosis treatment. This is particularly striking in the survival, overall health, the locomotive activity and muscle force measurements (grip strength and force measurements on isolated muscle). For example, although Bcl2 expression or omigapil treatment extend the lifespan of dyW/dyW mice (Fig 5A; Dominov et al, 2005; Erb et al, 2009), no prolongation of lifespan was seen in the dyW/Bcl/mag mice (Fig 5A). This lack of efficacy is probably due to the extended lifespan of dyW/mag mice, which makes it difficult to detect a significant further increase by anti-apoptosis treatment. However, when the mice were forced to hold themselves on a vertical grid, dyW/Bcl/mag mice could hold-on the grid more than twice as long as dyW/mag mice (Fig 5E). Correspondingly, the orally administered anti-apoptotic drug omigapil also improved muscle strength in dyW/mag mice (Fig 7I). The improvement of muscle function is of utmost importance for MDC1A patients.
We hypothesize that the difference in efficacy between mini-agrin and apoptosis inhibition is due to the different levels where the two treatments interfere with the disease process. While mini-agrin will take over at least some of the structural functions of LM-211, apoptosis is a late step in the cascade of events triggered by the absence of the laminin-α2 chain and thus its interference with the disease process is less specific. On the other hand, interference with such late steps has the advantage that potential drugs can be useful for a broad range of muscle dystrophies as those late events are common to several diseases. One possible mechanism underlying the additive effect of mini-agrin and the anti-apoptotic treatment could be the well-described effect of LM-211 to bind to α7β1 integrin. This interaction has been proposed to be important for the prevention of muscle fibre death (Vachon et al, 1996, 1997). As mini-agrin does not bind to this integrin, it can also not prevent apoptosis induced by the loss of LM-211 in dyW/dyW mice.
In conclusion, a combined treatment with apoptosis inhibitors and mini-agrin increases the regeneration efficiency in dyW/dyW muscles and, thereby, maintains muscle integrity. This in turn, enables the muscle to increase in size (Fig 1G and 2D), which results in the improvement of muscle strength and function (Fig 5D and E). Thus, a treatment that makes use of the additive benefit of apoptosis inhibition and mini-agrin might constitute a valuable therapy for MDC1A patients. However, realization of a combined treatment involves the development of a method that delivers mini-agrin to the skeletal muscles of human MDC1A patients, which is not feasible at the moment. Thus, pharmacological treatment options can probably be developed much faster. This is particularly true for omigapil, which shows the same efficacy as transgenic expression of Bcl2 (this work). Its clinical development is well advanced and it was proven to be safe in large clinical trials with Parkinson's disease and amyotrophic lateral sclerosis patients (Miller et al, 2007; Olanow et al, 2006).
MATERIALS AND METHODS
Mice
DyW/dyW mice containing a LacZ insertion in the LAMA2 gene served as the mouse model for MDC1A. Genotyping of dyW/dyW mice was performed as previously described (Kuang et al, 1998a). The chick mini-agrin transgene (mag) was expressed under the control of the muscle-specific CK promoter (Moll et al, 2001). The mice were genotyped as described (Meinen et al, 2007). Transgenic mice expressing full-length human Bcl2 under control of an approximately 7 kb-long fragment of the mouse MyoD promoter (Dominov et al, 2005; Girgenrath et al, 2004; Tapscott et al, 1992) were obtained from Dr Janice Dominov. The MyoD promoter was described to be active in myoblasts, activated satellite cells and in muscle fibres (Charge et al, 2008; Dominov et al, 2005; Girgenrath et al, 2004; Tapscott et al, 1992). Since their generation, all of the transgenic mouse lines were maintained in C57BL/6J mice and whenever possible littermate controls were used. To ensure optimal access of the dystrophic mice to water and food, cages were supplied with long-necked water bottles and wet food. All procedures were performed in accordance with the Swiss regulations for animal experimentation and under the required licenses.
Treatment of dyW/mag mice with omigapil
Homozygous dyW/mag were treated with 0.1 mg/kg omigapil dissolved in 0.5% ethanol as vehicle. For all experiments, treatment started at the age of 15 days. For the first week of drug treatment, omigapil was administered once daily by intraperitoneal injection. After weaning (3 weeks of age), omigapil was applied once daily by oral gavage. Age-matched animals treated with vehicle only were used as controls.
Masson trichrome, haematoxylin & eosin and immunostainings
Triceps brachii and soleus muscles were immersed in 7% gum Tragacanth (Sigma, St. Louis, MO, USA) and rapidly frozen in liquid nitrogen-cooled isopentane (−150°C). Cross-sections of 12 µm thickness were cut and collected on SuperFrost® Plus slides. General histology was performed using H & E staining. To visualize collagenous tissue, Masson's trichrome staining (Luna, 1968) was performed using Trichrome Stain (Masson) Kit (Sigma–Aldrich, HT-15). The antibodies used for immunofluorescence were purchased from the following commercial sources: Monoclonal mouse anti-rat dMyHC, monoclonal rat anti-mouse laminin-γ1 chain (Chemicon, MAB1914) and monoclonal rat anti-mouse F4/80 (Abcam, ab6640). Membrane-bound and extracellular epitopes were visualized with Alexa-488- or Alexa-594 conjugated wheat germ agglutinin (WGA; Molecular Probes). Appropriate secondary antibodies were: Cy3-conjugated (Jackson ImmunoResearch Laboratories), Alexa Fluor® 488-conjugated secondary antibodies (Molecular Probes) and TRITC-labelled streptavidin. Apoptotic myonuclei were detected by ‘TdT-mediated dUTPbiotin nick end labelling’ (TUNEL) using the ‘In Situ Cell Death Detection Kit, Fluorescein’ according to the manufacturer's protocol (Roche Diagnostics Ltd). DAPI (4′,6′-Diamidino-2-phenylindole hydrochloride) was used to stain nuclei. Pictures of stained cross-sections were collected using a Leica DM5000B fluorescence microscope, a digital camera (F-View; Soft Imaging System), and analySIS® software (Soft Imaging System).
Histological quantifications
Mid-belly cross-sections of triceps brachii and soleus muscles were analysed. Muscle area, fibre number and fibre size distribution was evaluated using analySIS® software (Soft Imaging System). The muscle fibre size distribution was quantified on entire WGA-stained cross-sections using the minimum distance of parallel tangents at opposing particle borders (minimal ‘Feret's diameter’) as described (Briguet et al, 2004). Normalization of the number of fibres in each fibre diameter class of 5 µm was based on the total fibre number per muscle. Fibrotic area was evaluated on entire WGA-stained cross-sections, was normalized to muscle area and was expressed relative to controls. Fibres with centrally located nuclei (CNF) and regenerating dMyHC-positive fibres were counted in the entire WGA/DAPI-stained and dMyHC/laminin-γ1/DAPI stained cross-sections, respectively. Only TUNEL- and DAPI-positive nuclei located within muscle fibres were counted as apoptotic myonuclei. F4/80 staining allowed counting of macrophages in the entire cross-sections. In all histological quantification experiments, at least four mice of each testing group were analysed.
Notexin-induced muscle damage
Tibialis anterior of 6-week-old mice was injured by injection of 15 µl notexin (Sigma, 50 µg/ml) as described (Bentzinger et al, 2005). Mice were sacrificed 6 and 14 days post-injection and muscles were isolated and processed as described above.
Western blot
Triceps brachii muscles were homogenized in RIPA protein extraction buffer (Abcam). Equal amounts of protein were separated on 20% (Bcl2, 25 kD) or 12% (mini-agrin, double band ∼120 kD) SDS–PAGE and immunoblotted. Nitrocellulose membrane was incubated with antibody to human Bcl2 (CST#2872) and polyclonal rabbit anti-chick N25C95 mini-agrin (Ab 201), that was produced in-house (Gesemann et al, 1995). For detection, appropriate horse radish peroxidase-conjugated antibodies were used and immunoreactivity was visualized by the ECL detection method (Pierce).
Body condition and behavioural tests, creatine kinase assay
Overall disease score was assessed using a health score sheet that judges activity, appetite, posture, fur and eyes of 8-, 12- and 16-week-old mice. Scores: from 0 to 4 = good health: bright and active attitude; normal posture, movements, fur and eyes; 5–8 = fair health: Several measures are mildly affected; 9–11 = poor health: mouse is obviously sick (inactivity, small size, hunchback, severe uncoordinated movements, dull fur and eyes); 12–14 = death expected in near future: almost unable to move, small, thin, dehydrated, unkempt fur and matted eyes. Body weight was measured at the age of 8, 12 and 16 weeks. Locomotive behaviour was evaluated by placing the mice into a new cage and measuring motor activity (walking, digging and righting up) during 10 min (Moll et al, 2001). Grip strength was determined by measuring the time the animals can hold on a vertical grid. Cut-off time was 180 s. All values were normalized to values obtained from control animals. Blood levels of CK were determined with 2 µl of serum using the CK-NAC Liqui-UV kit (Rolf Greiner Biochemica). In omigapil-treated mice, body weight and death events were recorded daily. The body weight was recorded for each animal from day 15 (onset of the experiment) onwards. For each animal, the average weight gain per week was calculated.
In vitro muscle force assessment
Left and right EDL and soleus muscles of at least three 4-week-old mice per genotype were carefully dissected and mounted into a muscle testing setup (Heidelberg Scientific Instruments) to test force in vitro. By stimulation with a single electrical pulse (4 kHz, 15 Volt, 0.5 ms) using an AD Instruments converter, muscles were adjusted to the optimum length (L0; mm) which is achieved when isometric twitch force is maximal (Pt; mN). Tetanus and force-frequency relationship were evaluated in response to 400 ms pulses at 10–120 Hz in EDL and in response to 1100 ms pulses at 10–150 Hz in soleus. Maximum absolute isometric tetanic force (P0) was determined from the plateau of the frequency–force relationship, which was typically achieved with 150 Hz for EDL, and for soleus muscles with 100 Hz. Since absolute P0 is dependent upon muscle size, P0 values were normalized for CSA [CSA = muscle mass/L0 × 1.06; (Brooks & Faulkner, 1988) and were expressed as specific force (sP0; kN/m2).
The paper explained
PROBLEM
Congenital muscular dystrophies are early onset, often severe diseases of the skeletal muscle that are of heterogeneous genetic origin. MDC1A is the most prevalent form of those congenital muscular dystrophies and is caused by mutations in the gene encoding laminin-α2, which is one of the subunits of LM-211, a major component of the muscle basement membrane. MDC1A patients often cannot stand or walk, suffer from respiratory distress and eventually die in early childhood. There is no treatment option for those patients but recent years have seen quite some progress in the understanding of the disease mechanisms and preclinical studies have suggested a few new treatment options.
RESULTS
This study is based on previous, preclinical experiments that established two independent ways to slow down disease progression in a mouse model for MDC1A, the dyW/dyW mice. The first option uses a miniaturized form of the extracellular matrix molecule agrin (mini-agrin), which is not homologous to LM-211, but shares binding properties with the protein that allow it to restore the mechanical linkage between muscle fibre and basement membrane. The second option uses inhibition of apoptosis, as muscle fibres in MDC1A undergo massive cell death because of their fragility. Although each treatment alone does not alleviate all of the dystrophic symptoms in mice, they both offer distinctive and interesting entry points for a possible treatment of MDC1A patients. We therefore examined in a mouse model for MDC1A whether the combination of both treatments would exert additive benefits and thus increase treatment efficacy.
We find that such a combined treatment with mini-agrin and apoptosis inhibition by either transgenic Bcl2 expression or application of the orally available apoptosis inhibitor omigapil indeed provides additive benefit on the disease progression in dyW/dyW mice. The side-by-side comparison also showed that mini-agrin treatment is superior to apoptosis inhibition, which is consistent with the notion that mini-agrin exerts its function at the early steps of the disease. The combination proved superior to single treatment with mini-agrin in preserving the number of muscle fibres and allowing improved survival of muscle fibres upon damage thereby improving muscle regeneration. Importantly, this improvement resulted in a marked increase in muscle force.
IMPACT
Our work thus represents a proof-of-concept study showing that combination therapies might be beneficial for the treatment of MDC1A patients. Unfortunately, gene therapeutic approaches in patients (which could be used to apply mini-agrin) are still facing challenges. Thus, our work also suggests that the combination of pharmacological inhibition of apoptosis combined with low efficacy application of mini-agrin (e.g. injection of recombinant protein or low efficacy gene therapy) might still be a valuable option to improve the disease in MDC1A patients. The finding that muscle force is clearly improved by the combination treatment in the dyW/dyW mice further suggest that such a therapy might be particularly beneficial to improve muscle function and thus the mobility of MDC1A patients.
Statistical analysis
Quantitative data are expressed as mean ± SEM. To compare the different genotypes, p-values were calculated using the unpaired two-sample Student t-test assuming equal variances. Statistical analysis of values that were measured at two different time points used the two-way ANOVA test. Kaplan–Meier survival curves were generated and compared using the Peto–Peto Wilcoxon test.
Acknowledgments
We thank Dr Janice Dominov for generously providing the mice expressing human Bcl2 in skeletal muscles. The in vitro force measurements on isolated muscles were performed in the laboratories of Dr Francesco Zorzato. We thank Teppo Huttunen for many helpful discussions. This work was supported by the ‘Muscular Dystrophy Association (MDA)’, 3300 East Sunrise Drive, Tucson, AZ 85718, the Swiss Foundation for Research on Muscle Disease and the Cantons of Basel-Stadt and Baselland.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
SM designed, performed and evaluated most of the experiments; SL helped in the notexin experiments and RT helped to measure muscle force in vitro; ME and TM helped with the application of omigapil and helped with statistics; MAR designed and evaluated the experiments, and wrote the manuscript together with SM. All the authors read and commented on the manuscript.
For more information
Patient organization Cure CMD:
Muscular Dystrophy Association:
Swiss Foundation for Research on Muscle Diseases:
TREAT-NMD Neuromuscular Network:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Bentzinger CF, Barzaghi P, Lin S, Ruegg MA. Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-alpha2-deficient mice. FASEB J. 2005;19:934–942. doi: 10.1096/fj.04-3376com. [DOI] [PubMed] [Google Scholar]
- Bezakova G, Ruegg MA. New insights into the roles of agrin. Nat Rev Mol Cell Biol. 2003;4:295–308. doi: 10.1038/nrm1074. [DOI] [PubMed] [Google Scholar]
- Briguet A, Courdier-Fruh I, Foster M, Meier T, Magyar JP. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul Disord. 2004;14:675–682. doi: 10.1016/j.nmd.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulyakova NV, Azarova VS. Regeneration of skeletal muscles and state of thymus in gamma-irradiated rats under laser therapy of the area of muscle trauma. Minim Invasive Ther Allied Technol. 2006;15:277–285. doi: 10.1080/13645700600803950. [DOI] [PubMed] [Google Scholar]
- Burkin DJ, Kaufman SJ. The alpha7beta1 integrin in muscle development and disease. Cell Tissue Res. 1999;296:183–190. doi: 10.1007/s004410051279. [DOI] [PubMed] [Google Scholar]
- Charge SB, Brack AS, Bayol SA, Hughes SM. MyoD- and nerve-dependent maintenance of MyoD expression in mature muscle fibres acts through the DRR/PRR element. BMC Dev Biol. 2008;8:5. doi: 10.1186/1471-213X-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins J, Bonnemann CG. Congenital muscular dystrophies: toward molecular therapeutic interventions. Curr Neurol Neurosci Rep. 2010;10:83–91. doi: 10.1007/s11910-010-0092-8. [DOI] [PubMed] [Google Scholar]
- Colognato H, Yurchenco PD. The laminin alpha2 expressed by dystrophic dy(2J) mice is defective in its ability to form polymers. Curr Biol. 1999;9:1327–1330. doi: 10.1016/s0960-9822(00)80056-1. [DOI] [PubMed] [Google Scholar]
- Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dyn. 2000;218:213–234. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Denzer AJ, Brandenberger R, Gesemann M, Chiquet M, Ruegg MA. Agrin binds to the nerve-muscle basal lamina via laminin. J Cell Biol. 1997;137:671–683. doi: 10.1083/jcb.137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominov JA, Kravetz AJ, Ardelt M, Kostek CA, Beermann ML, Miller JB. Muscle-specific BCL2 expression ameliorates muscle disease in laminin {alpha}2-deficient, but not in dystrophin-deficient, mice. Hum Mol Genet. 2005;14:1029–1040. doi: 10.1093/hmg/ddi095. [DOI] [PubMed] [Google Scholar]
- Erb M, Meinen S, Barzaghi P, Sumanovski LT, Courdier-Fruh I, Ruegg MA, Meier T. Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-alpha2 deficiency. J Pharmacol Exp Ther. 2009;331:787–795. doi: 10.1124/jpet.109.160754. [DOI] [PubMed] [Google Scholar]
- Gesemann M, Denzer AJ, Ruegg MA. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J Cell Biol. 1995;128:625–636. doi: 10.1083/jcb.128.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesemann M, Brancaccio A, Schumacher B, Ruegg MA. Agrin is a high-affinity binding protein of dystroglycan in non-muscle tissue. J Biol Chem. 1998;273:600–605. doi: 10.1074/jbc.273.1.600. [DOI] [PubMed] [Google Scholar]
- Girgenrath M, Dominov JA, Kostek CA, Miller JB. Inhibition of apoptosis improves outcome in a model of congenital muscular dystrophy. J Clin Invest. 2004;114:1635–1639. doi: 10.1172/JCI22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgenrath M, Beermann ML, Vishnudas VK, Homma S, Miller JB. Pathology is alleviated by doxycycline in a laminin-alpha2-null model of congenital muscular dystrophy. Ann Neurol. 2009;65:47–56. doi: 10.1002/ana.21523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi YK, Tezak Z, Momoi T, Nonaka I, Garcia CA, Hoffman EP, Arahata K. Massive muscle cell degeneration in the early stage of merosin-deficient congenital muscular dystrophy. Neuromuscul Disord. 2001;11:350–359. doi: 10.1016/s0960-8966(00)00203-0. [DOI] [PubMed] [Google Scholar]
- Kuang W, Xu H, Vachon PH, Engvall E. Disruption of the lama2 gene in embryonic stem cells: laminin alpha 2 is necessary for sustenance of mature muscle cells. Exp Cell Res. 1998a;241:117–125. doi: 10.1006/excr.1998.4025. [DOI] [PubMed] [Google Scholar]
- Kuang W, Xu H, Vachon PH, Liu L, Loechel F, Wewer UM, Engvall E. Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J Clin Invest. 1998b;102:844–852. doi: 10.1172/JCI3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang W, Xu H, Vilquin JT, Engvall E. Activation of the lama2 gene in muscle regeneration: abortive regeneration in laminin alpha2-deficiency. Lab Invest. 1999;79:1601–1613. [PubMed] [Google Scholar]
- Langenbach KJ, Rando TA. Inhibition of dystroglycan binding to laminin disrupts the PI3K/AKT pathway and survival signaling in muscle cells. Muscle Nerve. 2002;26:644–653. doi: 10.1002/mus.10258. [DOI] [PubMed] [Google Scholar]
- Laprise P, Poirier EM, Vezina A, Rivard N, Vachon PH. Merosin–integrin promotion of skeletal myofiber cell survival: differentiation state-distinct involvement of p60Fyn tyrosine kinase and p38alpha stress-activated MAP kinase. J Cell Physiol. 2002;191:69–81. doi: 10.1002/jcp.10075. [DOI] [PubMed] [Google Scholar]
- Laprise P, Vallee K, Demers MJ, Bouchard V, Poirier EM, Vezina A, Reed JC, Rivard N, Vachon PH. Merosin (laminin-2/4)-driven survival signaling: complex modulations of Bcl-2 homologs. J Cell Biochem. 2003;89:1115–1125. doi: 10.1002/jcb.10581. [DOI] [PubMed] [Google Scholar]
- Luna L. Manual of Histologic Staining Methods of the Armed Forces Institute of Pathology. New York: McGraw-Hill; 1968. pp. 94–95. [Google Scholar]
- Meinen S, Barzaghi P, Lin S, Lochmuller H, Ruegg MA. Linker molecules between laminins and dystroglycan ameliorate laminin-alpha2-deficient muscular dystrophy at all disease stages. J Cell Biol. 2007;176:979–993. doi: 10.1083/jcb.200611152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R, Bradley W, Cudkowicz M, Hubble J, Meininger V, Mitsumoto H, Moore D, Pohlmann H, Sauer D, Silani V, et al. Phase II/III randomized trial of TCH346 in patients with ALS. Neurology. 2007;69:776–784. doi: 10.1212/01.wnl.0000269676.07319.09. [DOI] [PubMed] [Google Scholar]
- Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, Takeda S. Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 1997;415:33–39. doi: 10.1016/s0014-5793(97)01007-7. [DOI] [PubMed] [Google Scholar]
- Miyagoe-Suzuki Y, Nakagawa M, Takeda S. Merosin and congenital muscular dystrophy. Microsc Res Tech. 2000;48:181–191. doi: 10.1002/(SICI)1097-0029(20000201/15)48:3/4<181::AID-JEMT6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Moll J, Barzaghi P, Lin S, Bezakova G, Lochmuller H, Engvall E, Muller U, Ruegg MA. An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature. 2001;413:302–307. doi: 10.1038/35095054. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Schapira AH, LeWitt PA, Kieburtz K, Sauer D, Olivieri G, Pohlmann H, Hubble J. TCH346 as a neuroprotective drug in Parkinson's disease: a double-blind, randomised, controlled trial. Lancet Neurol. 2006;5:1013–1020. doi: 10.1016/S1474-4422(06)70602-0. [DOI] [PubMed] [Google Scholar]
- Olive M, Ferrer I. Bcl-2 and bax immunohistochemistry in denervation–reinnervation and necrosis-regeneration of rat skeletal muscles. Muscle Nerve. 2000;23:1862–1867. doi: 10.1002/1097-4598(200012)23:12<1862::aid-mus10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Qiao C, Li J, Zhu T, Draviam R, Watkins S, Ye X, Chen C, Xiao X. Amelioration of laminin-alpha2-deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci USA. 2005;102:11999–12004. doi: 10.1073/pnas.0502137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shefer G, Partridge TA, Heslop L, Gross JG, Oron U, Halevy O. Low-energy laser irradiation promotes the survival and cell cycle entry of skeletal muscle satellite cells. J Cell Sci. 2002;115:1461–1469. doi: 10.1242/jcs.115.7.1461. [DOI] [PubMed] [Google Scholar]
- Tapscott SJ, Lassar AB, Weintraub H. A novel myoblast enhancer element mediates MyoD transcription. Mol Cell Biol. 1992;12:4994–5003. doi: 10.1128/mcb.12.11.4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tome FM, Fardeau M. Hereditary inclusion body myopathies. Curr Opin Neurol. 1998;11:453–459. doi: 10.1097/00019052-199810000-00007. [DOI] [PubMed] [Google Scholar]
- Vachon PH, Loechel F, Xu H, Wewer UM, Engvall E. Merosin and laminin in myogenesis; specific requirement for merosin in myotube stability and survival. J Cell Biol. 1996;134:1483–1497. doi: 10.1083/jcb.134.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon PH, Xu H, Liu L, Loechel F, Hayashi Y, Arahata K, Reed JC, Wewer UM, Engvall E. Integrins (alpha7beta1) in muscle function and survival. Disrupted expression in merosin-deficient congenital muscular dystrophy. J Clin Invest. 1997;100:1870–1881. doi: 10.1172/JCI119716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.