

Abstract
Epithelial ovarian carcinoma (EOC) is an aggressive neoplasm, which mainly disseminates to organs of the peritoneal cavity, an event mediated by molecular mechanisms that remain elusive. Here, we investigated the expression and functional role of neural cell adhesion molecule (NCAM), a cell surface glycoprotein involved in brain development and plasticity, in EOC. NCAM is absent from normal ovarian epithelium but becomes highly expressed in a subset of human EOC, in which NCAM expression is associated with high tumour grade, suggesting a causal role in cancer aggressiveness. We demonstrate that NCAM stimulates EOC cell migration and invasion in vitro and promotes metastatic dissemination in mice. This pro-malignant function of NCAM is mediated by its interaction with fibroblast growth factor receptor (FGFR). Indeed, not only FGFR signalling is required for NCAM-induced EOC cell motility, but targeting the NCAM/FGFR interplay with a monoclonal antibody abolishes the metastatic dissemination of EOC in mice. Our results point to NCAM-mediated stimulation of FGFR as a novel mechanism underlying EOC malignancy and indicate that this interplay may represent a valuable therapeutic target.
Keywords: fibroblast growth factor receptor, neural cell adhesion molecule, ovarian carcinoma, signalling, tumour progression
INTRODUCTION
Epithelial ovarian carcinoma (EOC) is the fourth most common cause of cancer-related death among women in the western world and is the most lethal tumour type among gynecological malignancies. The difficulty of early diagnosis and the rapid dissemination represent outstanding clinical challenges associated with EOC and contribute to the frequent failure of current treatment protocols. Even upon extensive surgical debulking and chemotherapy, relapse is a common event and, in most cases, recurrent EOC is resistant to any treatment (Lengyel, 2010).
EOC is commonly thought to develop from the ovarian surface epithelium (OSE), a single layer of poorly differentiated epithelial cells that lines the ovary, although the fallopian tube epithelium and the peritoneal mesothelium are also considered as potential sites of EOC origin (Lengyel, 2010). While various genetic lesions involved in the pathogenesis of EOC have been identified, our understanding of the biology of this neoplasm is still incomplete. This is partially due to the peculiar molecular and clinico-pathological properties of EOC, which prevents from transferring to ovarian carcinogenesis many of the concepts established in other epithelial tumours. For example, unlike most solid tumours, the metastatic dissemination of EOC very rarely occurs through the blood circulation. In most cases, EOC metastases arise from small clusters of cancer cells that are shed from the ovary and adhere to the surface of the peritoneal cavity and abdominal organs (Bast et al, 2009).
Cell adhesion molecules mediating either cell–cell interactions or cell–matrix adhesion have emerged as key players throughout the natural history of EOC development, in that they have been implicated both in cancer cell survival upon detachment from the primary tumour and in the subsequent adhesion to and invasion of metastatic sites (Naora & Montell, 2005; Sundfeldt, 2003). Besides their structural function, adhesion molecules also play an important role in signalling, either directly or by modulating the activity of signalling receptors. While the deregulation of these functional properties has been implicated in cancer progression (Cavallaro & Christofori, 2004), its specific contribution to EOC development remains to be defined.
Neural cell adhesion molecule (NCAM) is a cell-surface glycoprotein with an extracellular portion composed of five Ig domains and two fibronectin type-III repeats. NCAM function has been extensively characterized in the nervous system, where it regulates intercellular adhesion, neurite outgrowth and neuronal migration. These activities are mediated both by homophilic interactions and by heterophilic binding of NCAM to a number of different membrane proteins or components of the extracellular matrix (Hinsby et al, 2004). Among the heterophilic partners of NCAM, the fibroblast growth factor receptor (FGFR) has attracted the attention of many investigators due to its functional implications. The four members of the FGFR family (FGFR1–4) consist of receptor tyrosine kinases that are classically activated upon binding of cognate growth factors, the FGFs. FGFR stimulation results in the recruitment and activation of specific effectors that, in turn, trigger a set of signalling pathways (Beenken & Mohammadi, 2009). The functional interaction between NCAM and FGFR was originally reported in neurons, where it was implicated in neurite outgrowth (Williams et al, 1994). Thereafter, we and others have provided extensive evidence of a physical association between the two proteins on different, non-neural cell types (Cavallaro et al, 2001; Francavilla et al, 2007; Kos & Chin, 2002; Sanchez-Heras et al, 2006). These data were further confirmed and extended by surface plasmon and magnetic resonance studies, in which the direct binding of NCAM to FGFR1 and FGFR2 was demonstrated (Christensen et al, 2006; Francavilla et al, 2009; Kiselyov et al, 2003). This approach also revealed that the FGFR-binding motifs are located in the two fibronectin type-III repeats of the NCAM ectodomain (Hansen et al, 2010). We and others have demonstrated that the interaction of NCAM with FGFR results in the stimulation of FGFR signalling in various cell types (Cavallaro et al, 2001; Francavilla et al, 2009; Kiselyov et al, 2003; Williams et al, 1994). Nevertheless, the biological significance of the NCAM/FGFR interplay, especially in non-neural contexts, remains to be established.
Aberrant FGFR activity has been implicated in the onset and/or progression of various cancer types, and several FGFR inhibitors are currently in early phases of clinical development (reviewed in Turner & Grose, 2010). The deregulation of FGFR signalling in cancer can result from different mechanisms, including genomic alterations that lead to ligand-independent signalling as well as uncontrolled receptor stimulation by FGFs (Turner & Grose, 2010). All four members of the FGFR family and various FGFs have been found in EOC tissue (Birrer et al, 2007; Cole et al, 2010; Valve et al, 2000), suggesting that dysregulated FGFR signalling contributes to ovarian carcinogenesis (Castellano et al, 2006; De Cecco et al, 2004; Valve et al, 2000) and, therefore, it may represent a suitable therapeutic target (Ivan & Matei, 2010).
Based on the ability of NCAM to modulate FGFR function and on the proposed role of FGFR activity in ovarian cancer, we hypothesized that the NCAM/FGFR signalling axis is causally involved in EOC development. Screening of tumour biopsies revealed that NCAM is expressed in a significant proportion of EOC samples, but not in normal tissue, where its levels are increased at the invasive front and correlate with tumour grade. Furthermore, we show that the NCAM/FGFR interplay induces EOC invasion and peritoneal dissemination, and that interfering with this interaction represents a promising strategy to inhibit EOC progression. Our findings, therefore, provide novel insights into molecular mechanisms involved in EOC aggressiveness and offer the possibility to explore innovative therapeutic approaches for EOC treatment.
RESULTS
The expression of NCAM in human EOC
The expression of NCAM was investigated in a panel of surgically resected specimens, including 20 normal ovaries, 4 benign lesions (cystadenoma), 252 primary EOC and 206 metastatic lesions (see Materials and Methods Section for details on selection and analysis of patients). Immunohistochemical staining with anti-NCAM antibody showed no signal in the surface epithelium of normal ovaries (Fig 1A) or in pre-neoplastic lesions (not shown). In contrast, 60 (23.8%) primary EOC (Fig 1B) and 71 (34.5%) metastases (Fig 1D) were clearly positive for NCAM, thus indicating that NCAM expression occurs specifically in transformed ovarian epithelial cells. Interestingly, increased levels of NCAM were frequently observed at the invasive front of the tumour lesions (Fig 1B and C). This led to the hypothesis that NCAM promotes EOC invasion, an issue that we have investigated at the cellular level as well as in mouse models (see below). We also asked whether NCAM expression correlated with a series of clinicopathological parameters and found a statistically significant association with higher tumour grade (Table 1). NCAM expression was also more frequent in tumours at advanced International Federation of Gynecology and Obstetrics (FIGO) stages as compared to early stages, although the correlation did not reach statistical significance (Table 1). Taken together, these data point to NCAM as a novel biomarker of EOC associated with clinicopathological features of cancer aggressiveness.
Figure 1. Expression of NCAM in EOC.
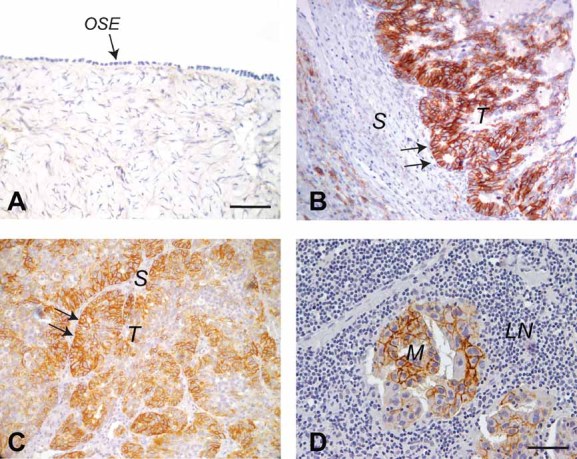
Tissue sections were stained with anti-NCAM antibody.
- A. Normal ovary. The arrow indicates the NCAM-negative OSE. Scale bar, 100 µm.
- B,C. Primary EOC. The arrows indicate the invasive edges of the tumour lesions. T, tumour; S, stroma. Scale bars, 100 µm.
- D. Lymph node metastasis of EOC. M, metastasis; LN, lymph node. Scale bar, 50 µm.
Table 1.
Clinicopathological features and NCAM expression in EOC patients
| Ovarian tissues | p-Value | ||
|---|---|---|---|
| NCAM-positive | NCAM-negative | ||
| Normal OSE | 0 (0%) | 20 (100%) | |
| Cystadenoma | 0 (0%) | 4 (100%) | 0.0003 |
| EOC | 60 (23.8%) | 192 (76.2%) | (OSE vs. EOC) |
| Metastases | 71 (34.5%) | 135 (65.5%) | |
| Histotype | |||
| Serous | 45 (26.6%) | 124 (73.4%) | NS |
| Endometrioid | 8 (18.6%) | 35 (81.4%) | |
| Mixed | 7 (28%) | 18 (72%) | |
| Clear cell | 0 (0%) | 8 (100%) | |
| Mucinous | 0 (0%) | 5 (100%) | |
| Transitional | 0 (0%) | 1 (100%) | |
| Grade | |||
| G1 | 0 (0%) | 12 (100%) | |
| G2 | 19 (27.9%) | 49 (72.1%) | 0.033 |
| G3 | 41 (24.4%) | 127 (75.6%) | (G1 vs. G2/G3) |
| FIGO Stage | |||
| Low (I–II) | 6 (16.2%) | 31 (83.8%) | NS |
| High (III–IV) | 54 (25.1%) | 161 (74.9%) | |
| Nodal status | |||
| Negative | 42 (24.6%) | 129 (75.4%) | NS |
| Positive | 18 (30%) | 63 (77.8%) | |
NS, not significant.
We also stained a subset of primary EOC samples for FGFR1 and detected the receptor in 75 out of 77 samples (97%), in agreement with previous studies that reported widespread expression of FGFR1, FGFR2 and FGFR4 in EOC (Steele et al, 2001; Valve et al, 2000). NCAM expression was found in 28 of the 75 FGFR1-positive samples (Table S1 and Fig S1 of Supporting Information). Furthermore, we found a highly significant correlation between the expression of NCAM and that of the individual FGFR genes in a published cDNA microarray dataset of 255 EOC samples (Crijns et al, 2009; Fig S2 of Supporting Information). Therefore, the presence of NCAM in EOC tissue is accompanied by the expression of FGFRs, thus supporting the functional role of the NCAM–FGFR interaction in EOC as described below.
NCAM-dependent FGFR signalling is required for EOC cell migration and invasion
To investigate whether NCAM is involved in the malignant phenotype of EOC cells, we utilized the MOVCAR cell line, originally isolated from the cancer tissue of MISIIR-TAg transgenic mice, a genetic model of ovarian carcinoma (Connolly et al, 2003). This cell line expresses high levels of NCAM, which is properly localized at the cell surface (Fig S3A of Supporting Information). MOVCAR cells were retrovirally transduced with a short-hairpin RNA that targets the murine NCAM mRNA (MOVCAR-shNCAM cells), resulting in efficient knockdown of NCAM expression (Fig S3A and B of Supporting Information). To rule out any off-target effect of the NCAM shRNA, MOVCAR-shNCAM cells were reconstituted with human NCAM, which is not affected by the shRNA (Fig S3A and B of Supporting Information). The loss of NCAM had no effect on MOVCAR cell proliferation (Fig S3C of Supporting Information). In contrast, the migratory activity of MOVCAR-shNCAM was twofold lower as compared to cells transduced with a control shRNA (Fig 2A). This effect was specifically due to NCAM gene silencing, since the expression of human NCAM restored the migratory potential of MOVCAR-shNCAM cells (Fig 2A).
Figure 2. NCAM and its interplay with FGFR are required for EOC cell migration and invasion.
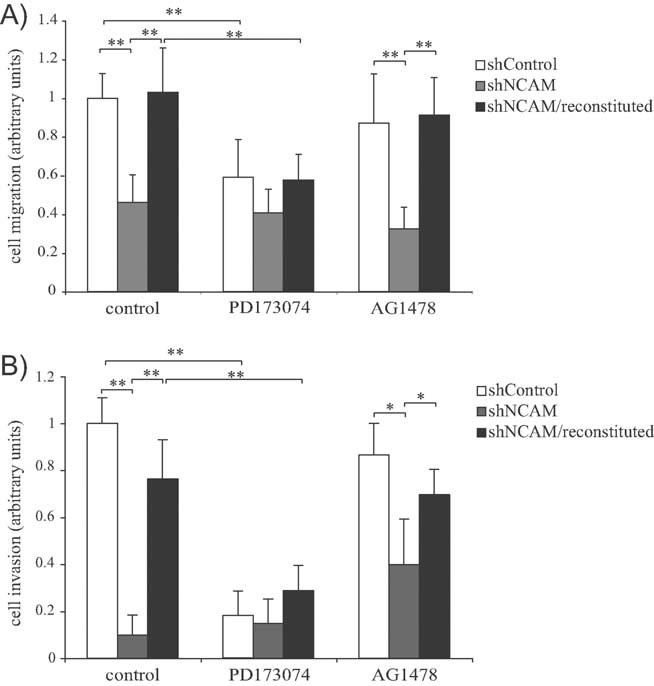
MOVCAR cells were transduced with either a control short-hairpin RNA (shControl) or a short-harpin RNA against mouse NCAM, either expressed alone (shNCAM) or in combination with human NCAM (shNCAM/reconstituted).
- Migration assay in the presence of either vehicle (‘control’), PD173074 or AG1478. Values are expressed in arbitrary units as fold changes over the migration or invasion of MOVCAR-shControl cells treated with vehicle alone. Error bars represent SEM. **p < 0.005.
- Matrigel invasion assays in the presence of either vehicle (‘control’), PD173074 or AG1478. Values are expressed as in panel A. **p < 0.005 and *p < 0.05. The difference between untreated (left grey bar) and AG1478-treated shNCAM cells (right grey bar) is not statistically significant.
Based on the notion that NCAM binds to FGFR and modulates FGFR activity (see Introduction Section), we asked whether this interplay is involved in the NCAM-dependent migration of MOVCAR cells. The latter express functional FGFRs, as demonstrated by their proliferative response to FGF-2 stimulation (Fig S3C of Supporting Information). To determine the contribution of FGFR signalling to NCAM-dependent cell migration, we took advantage of the FGFR inhibitor PD173074 (Skaper et al, 2000). This compound reduced the migration of control MOVCAR cells to a level comparable to untreated MOVCAR-shNCAM cells (Fig 2A). Moreover, the knockdown of NCAM expression and the inhibition of FGFR activity showed no additive effect on MOVCAR cell migration, suggesting that the two molecules are active in the same pathway. The defect in cell migration of MOVCAR-shNCAM cells was rescued upon ectopic expression of human NCAM. However, the ability of human NCAM to restore cell migration was abolished by PD173074 (Fig 2A), thus confirming that FGFR signalling is required for NCAM-induced ovarian cancer cell migration. It should be noted that FGF-2 had no effect on the migration of either control of NCAM-knockdown MOVCAR cells (not shown), as previously shown for different cell types (Francavilla et al, 2009). Finally, MOVCAR cell migration was also measured in the presence of AG1478, a chemical inhibitor of epidermal growth factor receptor (EGFR). In spite of EGFR expression in MOVCAR cells (data not shown), AG1478 had a negligible effect on NCAM-dependent cell migration (Fig 2A), implying that EGFR activity plays no or very little role downstream of NCAM and supporting the specificity of the NCAM/FGFR interplay.
We also employed the Matrigel invasion assay to determine whether NCAM and its functional interaction with FGFR were required for the invasive potential of MOVCAR cells. As shown in Fig 2B, the knockdown of NCAM resulted in the abrogation of MOVCAR cell invasion, which was restored upon reconstitution with human NCAM. As observed for cell migration, PD173074 not only reduced the basal invasiveness of control MOVCAR cells, but also abolished the pro-invasive effect of human NCAM (Fig 2B). Taken together, these results indicate that NCAM is required for both migration and invasion of MOVCAR cells and exerts its function via FGFR activity.
NCAM stimulates EOC cell migration via its interaction with FGFR
Following the observation that NCAM/FGFR interplay is necessary for EOC cell migration and invasion, we asked whether it is also sufficient. To address this question, we selected two human EOC cell lines, SKOV3 and OVCA-433, which express no endogenous NCAM (Figs S4 and S5 of Supporting Information, panels A and B). Both cell lines express various FGFR family members (our unpublished observations; Chandler et al, 1999; Valve et al, 2000), thus providing a suitable experimental system to investigate the impact of ectopically expressed NCAM on FGFR activity. SKOV3 and OVCA-433 cells were stably transfected with full-length NCAM (Figs S4 and S5 of Supporting Information, panels A and B). SKOV3 cells were also retrovirally transduced with a GFP-encoding vector (Fig S4A and B of Supporting Information) for xenotransplantation purposes (see below). In agreement with the data on NCAM silencing in MOVCAR cells, ectopic expression of NCAM in human EOC cells had no effect on cell proliferation (Fig S4C of Supporting Information and data not shown). Rather, NCAM-expressing SKOV3 and OVCA-433 cells exhibited a remarkable increase in their migratory activity as compared to mock-transfected cells (Fig 3A and Fig S5C of Supporting Information). Previous studies have established that the interaction with FGFR involves the two FNIII repeats of NCAM (referred to as FN1 and FN2; Kiselyov et al, 2003). Therefore, to determine the relative contribution of FGFR binding in NCAM-induced EOC cell migration, SKOV3 and OVCA-433 cells were transfected with a mutant version of NCAM lacking FN2 (ΔFN2), in which the interaction with FGFR is disrupted (Francavilla et al, 2007). Mutant NCAM was expressed at a level comparable to full-length NCAM and retained the localization at the cell surface (Figs S4 and S5 of Supporting Information, panels A and B), as well as the ability to induce cell–cell adhesion (not shown), which is mediated by distal Ig domains (Soroka et al, 2003). Furthermore, deletion of the FN2 domain did not alter folding, localization or the adhesive properties of NCAM. The ability of NCAM to stimulate FGFR signalling has been mostly characterized on FGFR1 (Francavilla et al, 2009; Kiselyov et al, 2003), a member of the FGFR family that is prominently expressed in both SKOV3 and OVCA433 cells (Fig S6B of Supporting Information and data not shown). Therefore, we focused on FGFR1 to define the impact of NCAM expression on FGFR activation. First, we confirmed that full-length NCAM, but not ΔFN2, forms a complex with FGFR1 in EOC cells (Fig S6A of Supporting Information). Accordingly, only full-length NCAM was able to induce autophosphorylation of FGFR1 in SKOV3 cells (Fig S6B of Supporting Information), confirming that also in EOC cells NCAM stimulates FGFR activation through its FN domains. In both SKOV3 and OVCA-433 cells, NCAM-ΔFN2 failed to promote cell migration (Fig 3A and Fig S5C of Supporting Information), thus implicating the interaction with FGFR as a pre-requisite for NCAM-induced migration of EOC cells. To further confirm this notion, SKOV3 cells expressing NCAM or a control vector were either treated with the FGFR inhibitor PD173074 or transfected with a dominant-negative version of FGFR1. In both cases, FGFR inhibition resulted in the abrogation of NCAM-dependent migration (Fig 3A) supporting the results obtained with NCAM-ΔFN2. By analogy to MOVCAR cells (see above), FGF-2 showed no pro-migratory activity on SKOV3 cells, a situation that was not changed by the ectopic expression of NCAM (not shown). This suggests that FGF does not cooperate with NCAM in the stimulation of FGFR-mediated EOC cell migration.
Figure 3. NCAM induces EOC cell migration by interacting with FGFR.
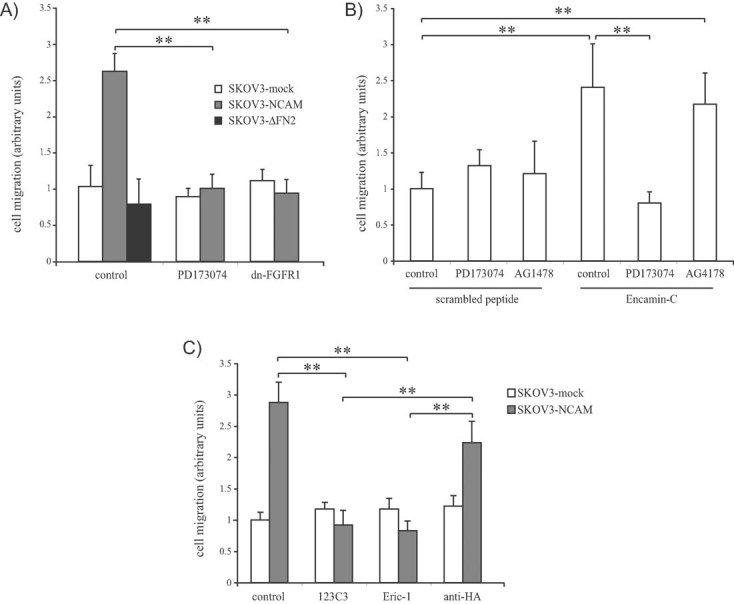
- SKOV3 cells transfected with empty vector (mock), with full-length NCAM or with NCAM-ΔFN2 were subjected to migration assays in the presence or absence of PD173074. A set of cells was co-transfected with dn-FGFR1 as indicated. Values are expressed in arbitrary units as fold changes over the migration of SKOV3-mock cells treated with vehicle.
- Parental SKOV3 cells were treated with Encamin-C or with a control scrambled peptide, either in the absence or in the presence of PD173074 or AG1478, followed by migration assay. Values are expressed in arbitrary units as fold changes over the migration of cells treated with the control peptide.
- C SKOV3 cells transfected with empty vector (mock) or with full-length NCAM were subjected to migration assays in the presence of the mAb 123C3, Eric-1 or anti-HA (as an isotype-matched control antibody). Values are expressed in arbitrary units as fold changes over the migration of untreated SKOV3-mock cells.
To test whether NCAM stimulation of FGFR function per se is sufficient to induce EOC cell migration, we took advantage of the Encamin-C peptide, derived from the FN1 module of NCAM, which has been recently demonstrated to bind to and activate FGFR1 (Hansen et al, 2008). First, we confirmed the FGFR-activating properties of Encamin-C in EOC cells. Encamin-C-treated SKOV3 cells displayed time-dependent autophosphorylation of FGFR1 (Fig S6C of Supporting Information), while a control, scrambled peptide had no effect (not shown). Encamin-C also enhanced the migratory potential of SKOV3 cells, an effect that was suppressed by PD173074 (Fig 3B), confirming that the peptide acts through FGFR signalling. In contrast, the EGFR inhibitor AG1478 showed no significant effect on Encamin-C-induced migration (Fig 3B), supporting the specificity of the peptide interaction with FGFR. These findings demonstrated that the interaction with FGFR is sufficient for NCAM to promote EOC cell migration.
Finally, as an alternative approach to determine the contribution of the NCAM/FGFR interplay in EOC cell migration, we performed migration assays using NCAM-transfected SKOV3 cells treated with monoclonal antibodies (mAb) directed against the FNIII repeats of NCAM. The mAb 123C3, which recognizes an epitope formed by the two FNIII domains of human NCAM (Gerardy-Schahn & Eckhardt, 1994), has been reported to block NCAM-induced neuritogenesis, most likely by interfering with the binding of NCAM to FGFR (Anderson et al, 2005). The mAb Eric-1 is directed against an epitope corresponding or closely related to 123C3 (Gerardy-Schahn et al, 1994). The specificity of 123C3 and Eric-1 for the FNIII domains of NCAM was confirmed by our immunoblotting analysis on recombinant NCAM fragments (Fig S7A of Supporting Information), in line with the notion that these protein modules exhibit a very stable conformation resistant to the denaturing conditions of SDS–polyacrylamide gel electrophoresis (SDS–PAGE; Gerardy-Schahn et al, 1994). Importantly, both 123C3 (Fig S7B and C of Supporting Information) and Eric-1 (not shown) repressed NCAM-stimulated activation of FGFR1, thus strengthening the rationale for using them as functional inhibitors of the NCAM/FGFR interplay. When added to SKOV3 cells, both 123C3 and Eric-1 blocked the migration induced by NCAM expression (Fig 3C), which confirmed that preventing the binding of NCAM to FGFR is a suitable strategy to inhibit EOC cell motility. A control, isotype-matched mAb showed no significant effect. Notably, neither 123C3 nor Eric-1 had any effect on NCAM-dependent cell–cell adhesion (not shown), which involves the most distal Ig domains of the protein (Soroka et al, 2003). Thus, we provide evidence that two different mAbs, which recognize the FGFR-binding modules of NCAM can repress NCAM-induced activation of FGFR and inhibit the migratory potential of NCAM-expressing EOC cells.
NCAM stimulates EOC cell invasion via its interaction with FGFR
Tumour cell invasion is a key step during cancer progression and, therefore, we determined the role of the NCAM/FGFR interaction in the ability of EOC cells to invade Matrigel, a reconstituted basement membrane. The ectopic expression of full-length NCAM resulted in a dramatic increase of the invasive potential of both SKOV3 and OVCA-433 cells (Fig 4A and Fig S5D of Supporting Information). In both cell lines, however, the expression of NCAM-ΔFN2 failed to stimulate Matrigel invasion (Fig 4A and Fig S5D of Supporting Information), implying that the association with FGFR is required for NCAM-dependent EOC cell invasion. The functional involvement of FGFR was further supported by the reduced invasive capability of NCAM-expressing SKOV3 cells treated with PD173074 (Fig 4A). The NCAM/FGFR interplay was not only required but also sufficient for SKOV3 cell invasion, as indicated by the potent pro-invasive effect of the Encamin-C peptide (Fig 4B).
Figure 4. NCAM induces EOC cell invasion by interacting with FGFR.
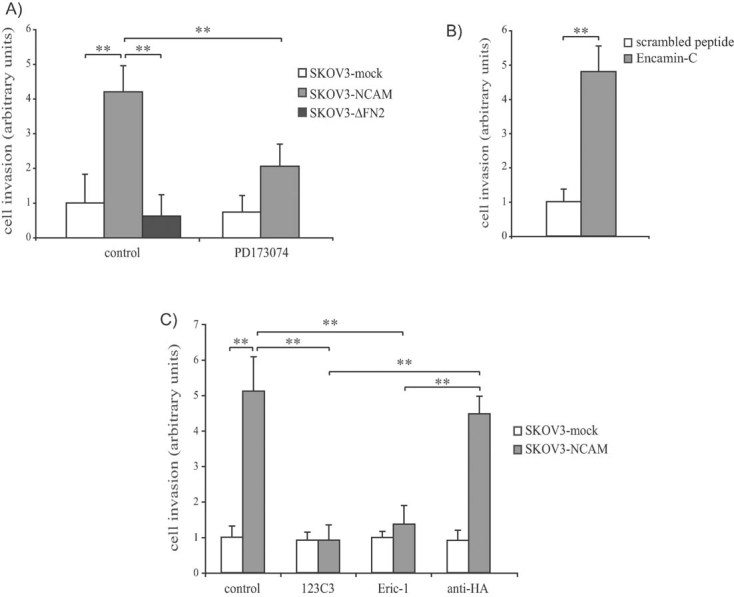
Error bars represent SEM. **p < 0.005.
- SKOV3 cells transfected with empty vector (mock), with full-length NCAM or with NCAM-ΔFN2 were subjected to Matrigel invasion assays in the presence or absence of PD173074. Values are expressed in arbitrary units as fold changes over the invasion of SKOV3-mock cells treated with vehicle.
- Parental SKOV3 cells were treated with Encamin-C or with a control scrambled peptide prior to Matrigel invasion assay. Values are expressed in arbitrary units as fold changes over the invasion of cells treated with the control peptide.
- SKOV3 cells transfected with empty vector (mock) or with full-length NCAM were subjected to Matrigel invasion assays in the presence of the mAb 123C3, Eric-1 or anti-HA (as an isotype-matched control antibody). Values are expressed in arbitrary units as fold changes over the invasion of untreated SKOV3-mock cells.
Finally, we tested whether mAbs that prevent the binding of NCAM to FGFR had any impact on NCAM-dependent EOC cell invasion. By analogy to cell migration (see above), the stimulation of Matrigel invasion by ectopic NCAM expression was abolished by either 123C3 or Eric-1 mAbs (Fig 4C). This supports the hypothesis that interfering with the NCAM/FGFR association has a dramatic impact on the pro-malignant function of NCAM in EOC cells.
The NCAM/FGFR interaction enhances local invasion and peritoneal dissemination of xenotransplanted EOC cells
To determine whether the role of the NCAM/FGFR complex in EOC cell migration and invasion translates into a functional contribution to tumour progression in vivo, we performed EOC xenograft experiments in mice. Local invasion was assessed in an orthotopic model of EOC consisting of SKOV3 cells transfected with an empty vector, with full-length NCAM or with ΔFN2-NCAM, that were injected under the ovarian bursa of immunodeficient mice. Two weeks after injection, all three cell lines formed tumours within the ovary. However, while SKOV3-mock tumours grew with smooth margins showing a poorly invasive phenotype (Fig 5A), NCAM-expressing cells formed tumour masses that extensively infiltrated the ovarian parenchyma (Fig 5B). Furthermore, we also found SKOV3-NCAM cell clusters in extra-ovarian tissue (Fig 5B and Figs S8D and S9A of Supporting Information, arrowheads), implying that NCAM enhances the ability of EOC cells to detach from the primary tumour mass. Both primary and disseminated tumour masses retained NCAM expression (Fig S8 of Supporting Information). The role of NCAM in EOC dissemination was further supported by the detection of multiple intravascular neoplastic emboli in mice injected with SKOV3-NCAM cells (Fig S9B–E of Supporting Information). These tumour lesions were specifically observed within the lymphatics (Fig S9D and E of Supporting Information), but not within blood vessels (not shown), in line with the notion that EOC metastasizes preferentially via the lymphatic circulation (Sundar et al, 2006; Ueda et al, 2005). Notably, SKOV3-ΔFN2 cells showed a very similar behaviour to SKOV3-mock cells, with no major signs of tumour invasion or dissemination (Fig 5C and Fig S8E and F of Supporting Information), thus pointing to the crucial role of the NCAM/FGFR interaction in these processes.
Figure 5. NCAM promotes local invasion and dissemination in an orthotopic EOC model.
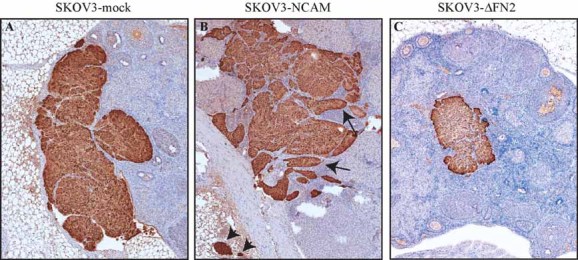
GFP-expressing SKOV3 cells transfected with an empty vector, full-length NCAM or ΔFN2-NCAM were injected under the ovarian bursa of immunodeficient mice. After 14 days, the mice were sacrificed and ovarian tissue sections were stained with anti-GFP antibodies. Arrows indicate areas of tumour infiltration into the ovarian parenchyma. Arrowheads indicate disseminated tumour lesions within extra-ovarian and fibro-adipose tissue. The pictures show representative images from one mouse per group. Four mice per group were used with similar results.
- Ovarian tumours in mice injected with SKOV3-mock cells.
- Ovarian tumours in mice injected with SKOV3-NCAM cells.
- Ovarian tumours in mice injected with SKOV3-ΔFN2 cells (ΔFN2 is a mutant version of NCAM that cannot bind to FGFR).
To further characterize the role of the NCAM/FGFR complex in EOC dissemination to distant organs, SKOV3 cells were injected intraperitoneally into immunodeficient mice, a widespread model for EOC peritoneal metastasis (Arlt et al, 2006; Donahue et al, 2011; Shaw et al, 2004; Slack-Davis et al, 2009). For this purpose, we used SKOV3 cells co-expressing GFP and either an empty vector, full-length NCAM or NCAM-ΔFN2. The expression of NCAM, either full-length or ΔFN2, did not affect tumour volume, as assessed by measuring the neoplastic mass that formed in the omentum, which is the primary site colonized by SKOV3 cells upon intraperitoneal (i.p.) injection (Sawada et al, 2008; data not shown). After 5 weeks, peritoneal dissemination was assessed as the formation of GFP-positive tumour masses attached to the bowel, liver and diaphragm, all typical sites of EOC metastasis in patients. As shown in Fig 6A and B, NCAM expression resulted in increased number of bowel metastases as compared to mock-transfected cells (see Fig S10 of Supporting Information for additional images). NCAM also enhanced the dissemination of SKOV3 cells to the liver and the diaphragm, although the increase in liver metastasis did not reach statistical significance (Fig 6C). Notably, no enhancement of metastatic dissemination to organs of the peritoneal cavity was observed using cells expressing NCAM-ΔFN2 (Fig 6A–C). Rather, these cells gave rise to a number of bowel metastases that was even lower than that of mock-transfected cells (Fig 6B), a phenomenon that was not further investigated. Taken together, these findings implicate that the association of NCAM with FGFR is a prerequisite for EOC spreading in vivo.
Figure 6. NCAM promotes peritoneal dissemination of EOC through its FGFR-binding domain.
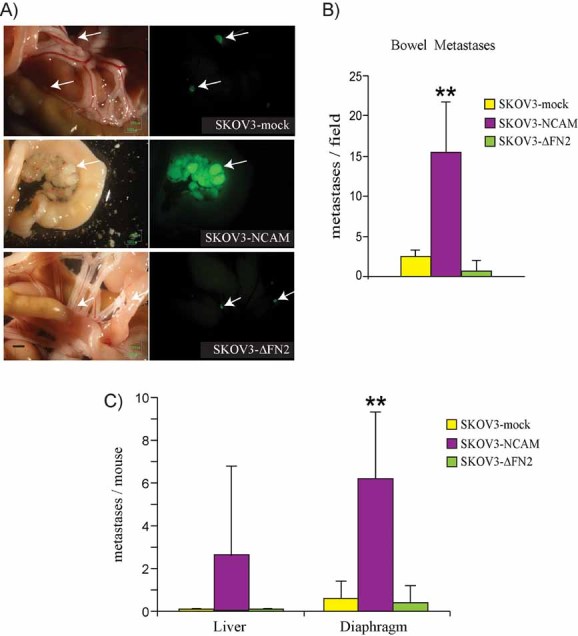
GFP-expressing SKOV3 cells transfected with empty vector (mock), full-length NCAM or NCAM-ΔFN2 were injected into the peritoneum of nude mice. Tumour dissemination was then assessed as described in Materials and Methods Section. **p < 0.005 and *p < 0.05.
- Brightfield (left panels) and fluorescence images (right panels) of the same fields showing bowel metastases. Arrows indicate GFP-positive tumour masses. Scale bar, 1 mm.
- Quantification of bowel metastases, represented as number of GFP-positive tumour masses per microscopic field. Data are expressed as means ± SD (n = 10 for each group).
- Metastatic dissemination of GFP-positive cells to the liver and to the diaphragm was determined by counting the total number of GFP-positive lesions per organ as described in Materials and Methods Section. Values are expressed as the mean percentage (±SD; n = 10 for each group) of GFP-positive cells over the total number of cells analysed.
We therefore asked if this interaction could be specifically targeted to interfere with tumour dissemination. To address this question, mice xenotransplanted with SKOV3-NCAM cells were subjected to i.p. treatment with the mAb 123C3 that, as described above, interferes with NCAM-mediated activation of FGFR. The dissemination of SKOV3-NCAM cells to bowel and diaphragm was dramatically reduced in 123C3-treated mice, and a significant decrease was also observed in liver metastasis. In contrast, a control mAb showed no effect in SKOV3 cell dissemination (Fig 7). The mAb 123C3 does not cross-react with mouse NCAM (not shown), thus its inhibitory function on EOC metastasis is due to the neutralization of NCAM in cancer cells rather than in the microenvironment. These findings indicate that antibody-mediated targeting of the NCAM/FGFR interplay suppresses the metastatic potential of EOC cells, an observation that has relevant therapeutic implications.
Figure 7. Antibody-mediated inhibition of NCAM/FGFR interplay reduces EOC peritoneal dissemination.
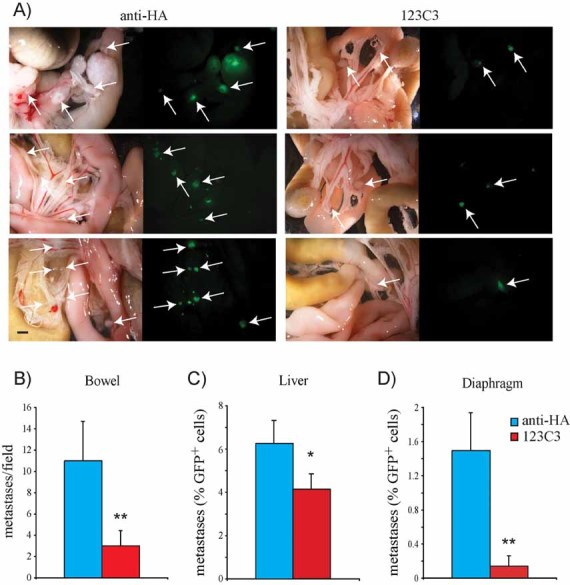
GFP-expressing SKOV3 cells transfected with full-length NCAM were injected into the peritoneum of nude mice. Mice were then treated with 123C3 or anti-HA antibodies, as described in Materials and Methods Section. **p < 0.005 and *p < 0.05.
- A. Brightfield (left panels) and fluorescence images (right panels) of the same fields showing bowel metastases. Arrows indicate GFP-positive tumour masses. Scale bar, 1 mm.
- B. Quantification of bowel metastases, represented as the average number of GFP-positive tumour masses per microscopic field. Data are expressed as means ± SD (n = 6 for each group).
- C,D. Metastatic dissemination of GFP-positive cells to the liver (C) and to the diaphragm (D) was determined by FACS analysis as described in Materials and Methods Section. Values are expressed as the mean percentage (±SD, n = 6 for each group) of GFP-positive cells over the total number of cells analysed.
DISCUSSION
In this study, we show that NCAM is not detectable in normal OSE but is expressed de novo in EOC, an event that correlates with tumour aggressiveness. Our results are in line with the only screening published so far on NCAM expression in EOC, where NCAM was reported to be absent in healthy ovarian epithelium, poorly expressed in low grade/early stage tumours, but highly enriched in advanced EOC (Cho et al, 2006).
The main novelty of our study lies in the functional contribution of this adhesion molecule to EOC progression. NCAM gene silencing and ectopic expression approaches supported the notion that NCAM is both necessary and sufficient to promote a migratory and invasive phenotype in EOC cells, with no major effect on cell proliferation. Interestingly, we have observed this specific function of NCAM in cell motility but not in cell proliferation also in other cell types, including fibroblasts (Francavilla et al, 2007) and different epithelial cell lines (Francavilla et al, 2009), suggesting that it is a rather general phenomenon. Along this line, NCAM has been recently implicated in cell migration in the context of epithelial-to-mesenchymal transition (Lehembre et al, 2008). In the same study, NCAM was found to be upregulated in tumour cells at the invasive front of neoplastic lesions, in agreement with our findings in EOC samples. Taken together, these observations point to NCAM as a novel pro-invasive factor with a causal role in cancer progression.
Our study demonstrates that the functional contribution of NCAM to EOC progression relies on its interaction with and activation of FGFR. The role of FGFR in mediating NCAM function has been originally proposed in neurons, where this interplay was implicated in neurite outgrowth (Williams et al, 1994). Subsequently, we have reported the physical association of NCAM with FGFR-4 on pancreatic beta tumour cells (Cavallaro et al, 2001), and the direct interaction between NCAM and either FGFR-1 (Kiselyov et al, 2003) or FGFR-2 (Christensen et al, 2006), indicating that NCAM is able to bind and activate multiple members of the FGFR family. Besides neurite outgrowth (Williams et al, 1994) and cell–matrix adhesion (Cavallaro et al, 2001), in vitro studies have recently implicated the NCAM/FGFR interplay in Schwann cell growth and process elongation (Mehanna et al, 2009). Our report provides the first evidence that NCAM-mediated stimulation of FGFR activity plays a causal role in cancer cell migration and invasion, two events strictly related to tumour malignancy.
Many lines of evidence indirectly support the biological relevance of NCAM-dependent activation of FGFR in vivo. First, we have established a connection between the loss of NCAM/FGFR-mediated regulation of cell–matrix adhesion and disruption of tissue architecture in an NCAM-knockout model of pancreatic beta-cell carcinoma (Cavallaro et al, 2001). Second, NCAM ablation has only mild effect on the embryonic development of mice; however, the transgenic expression of the soluble NCAM's ectodomain in NCAM−/− mice results in dominant embryonic lethality (Rabinowitz et al, 1996). While this implicates the tight regulation of NCAM heterophilic interactions as a prerequisite for normal embryonic development, it is tempting to speculate that excessive stimulation of FGFR by soluble NCAM contributes to the phenotype of mutant mice. Third, NCAM−/− mice display a series of abnormalities, including a reduced basal phosphorylation of FGFR1 and of several downstream targets in the hippocampus as well as various behavioural and cognitive disorders accompanied by reduced survival and differentiation of hippocampal neurons. This phenotype is reverted by the treatment with an NCAM-derived, FGFR-binding peptide (Aonurm-Helm et al, 2010, 2008; Bisaz et al, 2009), thus highlighting the important contribution of the NCAM/FGFR interplay to brain function. While all these observations support the physiopathological relevance of the NCAM interaction with FGFR, our study represents the first direct demonstration that such an interaction is specifically required for important processes in cultured cells as well as in an animal model.
The involvement of FGFR activity in ovarian tumourigenesis is supported by several studies. Different members of the FGFR family are aberrantly expressed in EOC lesions (Steele et al, 2001; Valve et al, 2000), and dysregulated FGFR signalling has been associated to EOC progression (Castellano et al, 2006; De Cecco et al, 2004) and chemoresistance (Cole et al, 2010). Besides the constitutive activation caused by mutations or over-expression, the aberrant FGFR activity in EOC can result from the uncontrolled stimulation by its classical ligands, the FGFs (De Cecco et al, 2004; Steele et al, 2001; Valve et al, 2000). Our results imply that this view should be revisited, and de novo expression of NCAM should be considered as an additional mechanism of FGFR activation in EOC. Although we have focused on FGFR1 to analyse NCAM-induced phosphorylation of FGFR, EOC tumours and cell lines express also other members of the FGFR family (Chandler et al, 1999; Steele et al, 2001; Valve et al, 2000), and NCAM has been shown to interact also with FGFR2 (Christensen et al, 2006) and FGFR4 (Cavallaro et al, 2001). It is possible, therefore, that the pro-malignant function of NCAM in EOC involves multiple FGFRs. It is important to stress that NCAM does not appear to stimulate FGFR signalling by enhancing the activity of endogenous FGFs. In fact, not only NCAM is unable to interact with FGF-2, and, therefore, to act as a co-receptor for the growth factor, but it actually inhibits the binding of FGF-2 to FGFR (Francavilla et al, 2007). Rather, previous observations on the direct interaction of NCAM with FGFRs (Christensen et al, 2006; Kiselyov et al, 2003; Kos & Chin, 2002) and the results presented here point to NCAM-autonomous activation of FGFR. In line with this notion, NCAM and FGF-2 induce a different FGFR-mediated response in EOC cells, with NCAM promoting cell migration but not proliferation and FGF-2 showing the opposite effect (this report and data not shown). Such a dichotomy between NCAM and FGF appears to be a rather general phenomenon, as it occurs also in different epithelial and fibroblastic cell types (Francavilla et al, 2009). We have recently characterized the molecular mechanisms that account for the divergent response to NCAM versus FGF-2, by showing that the two ligands promote the activation of different downstream effectors. They also exert a differential regulation on the stability and trafficking of endocytic FGFR, resulting in different signalling kinetics and cellular response (Francavilla et al, 2009). This suggests that similar mechanisms underlie the divergent effect of NCAM and FGF on EOC cells. In this context, we have previously reported that the NCAM/FGFR interplay regulates β1 integrin-dependent cell adhesion to the extracellular matrix (Cavallaro et al, 2001). Cell–matrix adhesion plays a key role in the crosstalk between EOC cells and their microenvironment, and β1 integrins, in particular, have been implicated in various processes related to peritoneal dissemination (Ahmed et al, 2005; Casey et al, 2001; Kenny et al, 2008; Sawada et al, 2008; Shield et al, 2007; Strobel & Cannistra, 1999). Analogously, we have identified Src as a key mediator of NCAM/FGFR-stimulated cell migration (Francavilla et al, 2009), and Src signalling is known to contribute to EOC malignancy (Han et al, 2006; Konecny et al, 2009). Together with the correlation between NCAM expression and peritoneal metastasis in EOC patients (Cho et al, 2006), and with our observation on the NCAM/FGFR-dependent invasion of EOC cells into abdominal organs, these findings point to β1 integrin and Src as potential effectors downstream of NCAM/FGFR during peritoneal dissemination.
The observation that an antibody suppressing NCAM-induced activation of FGFR prevents peritoneal metastasis of EOC, on one hand, lends further support to the physiopathological relevance of this signalling axis in ovarian cancer malignancy. On the other hand, it has dramatic therapeutic implications, providing a strong rationale to explore the targeting of the NCAM/FGFR interplay in the context of novel strategies against EOC dissemination. As for other tumour types, targeted therapy has progressively emerged in ovarian cancer as a suitable approach to complement the conventional chemotherapy protocols and, where possible, overcome some of their main limitations (e.g. chemoresistance and toxicity; Blagden & Gabra, 2009; Yap et al, 2009). Furthermore, experimental evidence in breast cancer supports the notion that targeting FGFR activity represses tumour growth and dissemination (Dey et al, 2010). Thus, based on our data with the 123C3 mAb, it is conceivable that the specific inhibition of the NCAM/FGFR interaction contributes to counteract EOC dissemination and recurrence. Notably, antibody-mediated targeting of another immunoglobulin-like adhesion molecule, L1CAM, proved efficient in repressing the i.p. growth and dissemination of SKOV3 cells (Arlt et al, 2006).
In summary, this study shows the aberrant expression of NCAM in human EOC tissue and its association with cancer aggressiveness, and that the interplay of NCAM with FGFR enhances the migratory and invasive potential of EOC cells in vitro and their metastatic ability in vivo. Finally, we report that interfering with NCAM-mediated activation of FGFR results in a dramatic reduction of EOC malignancy, providing the rationale for further assessing this approach as a novel therapeutic strategy.
MATERIALS AND METHODS
Antibodies
The following antibodies were used in this study: mouse mAb anti-human NCAM, clone 123C3, either purchased from Santa Cruz Biotechnology (Santa Cruz, CA) or purified from the conditioned medium of hybridoma cells (kindly provided by J. Hilkens, Netherlands Cancer Institute, Amsterdam); mouse mAb anti-human NCAM, clone Eric-1 (Santa Cruz Biotechnology); mouse mAb anti-mouse NCAM, clone NCAM13 (Becton Dickinson, Franklin Lakes, NJ); rat mAb anti-mouse NCAM, clone H28 (kindly provided by M. Schachner, Hamburg, Germany); mouse mAb anti-HA tag (Santa Cruz Biotechnology, Inc); anti-phospho-FGFR (Tyr653/654; Cell Signaling Technology, Danvers, MA). Goat anti-human Fc fragment and mouse anti-tubulin (Sigma–Aldrich, St. Louis, MO); and anti-GFP (Roche Diagnostics, Indianapolis, IN). Myc-tagged single-chain variable fragment (scFv) against FGFR1 (Francavilla et al, 2007) was isolated by screening the ETH-2 Gold library (Silacci et al, 2005) against recombinant FGFR1-Fc (R&D Systems, Minneapolis, MN) and mAb anti-myc, clone 9E10 (Santa Cruz Biotechnology), was used for the detection of the bound scFv antibody on cell lysates as described (Francavilla et al, 2007).
Chemicals
Recombinant FGF2 was purchased from Peprotech (London, UK); FGFR inhibitor PD173074 (used at 100 nM) was kindly provided by Pfizer (Groton, CT); and EGFR inhibitor AG1478 (Sigma–Aldrich, used at 20 nM). NCAM-derived peptide Encamin-C (KAEWKSLGEEAWHSK) and its scrambled version (KEEHSLAEKWGASWK), kindly provided by ENKAM Pharmaceuticals (Copenhagen, Denmark), were previously described (Hansen et al, 2008) and used at 20 µg/ml in serum free medium.
Selection of the patients and tissue microarray (TMA) analysis
Two-hundred and fifty two ovarian cancer patients undergoing surgery at the European Institute of Oncology (Milano, Italy) from 1995 to 2004 were enrolled in the study. All patients underwent first surgery at the European Institute of Oncology and received no neoadjuvant treatment. All tissues were collected via standardized operative procedures approved by the Institutional Ethical Board, and informed consent was obtained for all tissue samples linked with clinical data.
Representative areas were first selected on haematoxylin–eosin-stained tumour sections by a trained pathologist; two representative core biopsies for every tumour block were included in the TMAs. TMAs were assembled on a custom-built tissue arrayer (Beecher Instruments, Sun Prairie, WI, USA) as previously described (Zecchini et al, 2008).
Routine immunohistochemical staining was performed with anti-human NCAM mAb (clone 123C3) on paraffin-embedded TMA sections and on whole sections from 20 normal ovaries and 4 benign cystoadenomas. Whole sections of randomly chosen tumours included in the TMA were also stained to confirm immunoreactivity was preserved on the TMAs cores. A subset of 77 tumours was also stained with anti-FGFR polyclonal antibody (Santa Cruz Biotechnology).
Membrane staining for NCAM in tumour cells was scored as positive and the percentage of positive tumour cells was assessed by two-independent pathologists. The average percentage of NCAM-positive cells of two cores was computed for analysis.
Statistical analysis
Contingency tables and Chi-squared were used to evaluate differences in the frequency of NCAM-positive (more than 10% of immunoreactive cells) tumours with respect to grade, stage, histotype and nodal invasion. t-Test was used to evaluate the differences in the numbers of migrating and invading tumour cells among the represented experimental conditions and to compare the number of metastases formed by tumour cell lines expressing wild-type, mutant or no NCAM.
All statistical analyses were carried out with SAS statistical software (SAS Institute, Inc., Cary, NC) by a trained statistician. A p-value of 0.05 or less was considered to be statistically significant.
Immunohistochemistry
Immunohistochemistry was performed using 5-µm serial sections from formalin-fixed and paraffin-embedded tissue samples. Tissues were deparaffinized in Histolemon (Carlo Erba, Milano, Italy) and hydrated through graded alcohol series. Epitope unmasking was performed in 0.25 mM EDTA (pH 8, at 98°C, for 50 min). Endogenous peroxidases were quenched in 3% H2O2 and slides were pre-incubated for 1 h in blocking solution (PBS, 2% (bovine serum albumin) BSA, 2% goat serum and 0.01% Tween-20), followed by the incubation with 10 µg/ml mouse anti-NCAM (mAb 123C3) overnight at 4°C. Peroxidase-based Dako EnVision+ kit was used as a detection system. Slides were counterstained with haematoxylin for histological evaluation.
Cell culture and transfection
The mouse ovarian carcinoma cell line MOVCAR (Connolly et al, 2003), kindly provided by D. Connolly (Philadelphia, PA) was maintained in DMEM, 5% foetal calf serum (FCS), l-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, 10 µg/ml human insulin, 6 µg/ml human transferrin and 5 mM sodium selenite. The human ovarian carcinoma cell lines SKOV3 (ATCC, Manassas, VA) and OVCA433 (kindly provided by R. Drapkin, Boston, MA) were maintained in RPMI 1640 or DMEM medium (Invitrogen, Carlsbad, CA), respectively, supplemented with 10% FCS, 2 mM l-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. Cell lines were transfected with pcDNA3.1 expression vectors encoding either full-length mouse NCAM or a mutant version of NCAM lacking the second FNIII repeat (ΔFN2, a kind gift of M. Lewerenz and G. Christofori, Basel, Switzerland; Francavilla et al, 2007). The empty pcDNA3.1 vector (‘mock’) was used as control. In one set of experiments, SKOV3 cells were transfected with the cDNA encoding dominant-negative FGFR1 (Werner et al, 1993) cloned into pMex-neo vector. SKOV3 cells were transfected using Lipofectamine Plus (Invitrogen) and following the manufacturer's instructions. Stable transfectants were obtained by selection with 1 mg/ml G418 (Invitrogen) for at least 2 weeks. OVCA433 cells were transfected with Lipofectamine 2000 (Invitrogen) and selected with 400 µg/ml G418 (Invitrogen). To obtain the stable green fluorescent protein (GFP) expression, SKOV3 cells were also transduced with retroviral particles carrying the Pinco-GFP construct (Grignani et al, 1998), followed by selection with 1 µg/ml puromycin. Both transfected and transduced cells were maintained as heterogeneous populations to avoid cloning artefacts.
RNA interference
NCAM expression in MOVCAR cells was silenced by retroviral delivery of pSUPER-retro-neo + GFP (Oligoengine, Seattle, WA) encoding a short-hairpin interfering RNA targeting specifically mouse NCAM (target sequence 5′-AAGTACAAGGCTGAGTGGAAGCTTCCACTCAGCCTTGTAC-3′).
Reconstitution of MOVCAR cells was obtained by stable transfection with human NCAM (140 kD isoform) cloned into pcDNA3.1/Zeo(+) vector (Invitrogen). Cells were transfected with Lipofectamine 2000 (Invitrogen) and selected with 400 µg/ml Zeocin (Invitrogen) for 3 weeks.
The paper explained
PROBLEM
Epithelial ovarian carcinoma (EOC) represents an outstanding challenge for clinical oncologists, due to the difficulty of early diagnosis and to the high rate of relapse that follows the dissemination of cancer cells to the peritoneal cavity. Recurrent tumours often develop chemoresistance, accounting for the poor 5-year survival rate of EOC patients. Therefore, a better understanding of the biological mechanisms and molecular players involved in EOC malignancy is essential to design innovative therapeutic strategies and improve the prognosis for patients.
RESULTS
We have shown that the NCAM is not expressed in normal ovarian epithelium, while high levels were detected in a subset of cancer samples from EOC patients, where NCAM expression was associated with the tumour grade. To understand whether this expression pattern underlies a functional contribution of NCAM to EOC malignancy, we combined loss and gain-of-function approaches in EOC-derived cells. Our results revealed that NCAM confers a migratory and invasive phenotype to cancer cells. This function is mediated by NCAM's ability to interact with and activate fibroblast growth factor receptor (FGFR), a receptor tyrosine kinase that has previously been implicated in EOC progression. Furthermore, by employing mouse models of EOC development, we observed that the NCAM/FGFR interplay promotes both local invasion and metastatic dissemination of EOC to organs of the peritoneal cavity. Notably, a mAb that disrupts the interaction of NCAM with FGFR repressed the pro-metastatic activity of NCAM. Our findings implicated NCAM-mediated stimulation of FGFR function as a novel mechanisms underlying EOC aggressiveness.
IMPACT
Our study outlines the causal role of NCAM in EOC invasion and metastasis, a role mediated by the peculiar ability of this adhesion molecule to bind and activate FGFR. While we have recently described the outcome of the NCAM/FGFR interaction at the molecular and sub-cellular level, this is the first evidence of the contribution of such a signalling axis to cancer aggressiveness.
On one hand, our data highlight novel molecular mechanisms involved in EOC malignancy, allowing for a deeper understanding of the biology of this elusive disease. On the other hand, we provide proof-of-principle evidence that the NCAM/FGFR interplay represents a therapeutic target that can be efficiently inactivated, resulting in repression of EOC metastasis. This supports the rationale for combining NCAM/FGFR-targeted therapy with different treatments as an innovative approach to design more effective anti-EOC strategies.
Immunofluorescence
Cells were cultured on glass coverslips and then fixed in 3% paraformaldehyde, 2% sucrose in PBS and blocked in 5% albumin in TBST (20 mM Tris–HCl pH 7.4, 150 mM NaCl and 0.1% Tween-20). After blocking, cells were stained with primary antibodies (anti-human NCAM, clone 123C3, at 1 µg/ml, anti-mouse NCAM, clone NCAM13, at 1:1000) in blocking solution for 1 h at room temperature, followed by incubation with secondary antibodies conjugated with either FITC or Cy3 (Jackson Immuno Research Laboratories), diluted 1:400 in blocking solution. Cell nuclei were counterstained with 4′-6-diamidino-2-phenylindole (DAPI). Images were acquired on an Olympus BX71 microscope equipped with analySIS software (Olympus Soft Imaging Solutions, Münster, Germany). Images were then processed using NIH ImageJ and Adobe Photoshop CS2 9.0.2 (Adobe Systems, Inc., San Jose, CA) softwares.
Western blotting
Cells were lysed on ice in lysis buffer (10 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 0.1% Tween-20 and 1% Triton X-100) containing protease and phosphates inhibitors. Proteins (20–100 µg each lane) were separated on a 8% SDS–PAGE and transferred onto nitrocellulose membrane and blocked in 4% non-fat milk in TBST. Proteins were detected by specific antibodies, followed by peroxidase-conjugated secondary antibodies and revealed by an enhanced chemiluminescence kit (ECL kit, Amersham Biosciences, Little Chalfont, UK). The following concentrations/dilutions of primary antibodies were used: mouse anti human NCAM (clones 123C3 and ERIC-1), 1.5 µg/ml; mouse anti-mouse NCAM (clone NCAM13), 1:2000; anti-Fc and anti-tubulin (Sigma–Aldrich), 1:10.000; anti-FGFR1, 1:1000; and anti-pFGFR1 1:1000.
Migration and invasion assays
Cell migration assays were performed on Transwell Migration Chambers with 5-µm pores (Costar, Cambridge, MA). Briefly, 1 × 105 serum-starved EOC cells were added to the upper chamber in serum-free medium and 10% FBS medium was added to the lower chamber as the chemoattractant. After 16 h, cells on the top side of the filter were removed with a cotton swab, and cells migrated to the bottom side were fixed in PBS, 3% paraformaldehyde, 2% sucrose and stained with DAPI. After three washes in PBS, filters were gently removed and mounted on microscope slides. Images of DAPI-stained nuclei were acquired on an Olympus Biosystems BX71 microscope. Migrated cells were counted in 10 random fields for each filter. Each experiment was performed in triplicate and repeated at least three times.
Cell invasion assays were performed on Transwell filters (8-µm pores) containing 100 µl of reconstituted basement membrane (Matrigel™; BD Biosciences). Cells were allowed to invade for 24 h. Imaging and quantification were carried out as described for cell migration; experiments were performed in triplicate and repeated at least three times.
In vivo tumourigenesis
All experiments with mice were performed in accordance with the guidelines established in the Principles of Laboratory Animal Care (directive 86/609/EEC) and approved by the Italian Ministry of Health. Orthotopic transplantation of SKOV3 cells was performed as described elsewhere (Cordero et al, 2010). Briefly, 1 × 106 SKOV3-mock, SKOV3-NCAM or SKOV3-ΔFN2 cells were resuspended in 5 µl of PBS and injected under the ovarian bursa of pathogen-free, female athymic nu/nu mice (7–9-week old; 20 g average body weight) from Charles River Laboratories (Wilmington, MA). Mice were sacrificed after 2 weeks for the assessment of local invasion and dissemination, which was performed by immunohistochemical staining of ovarian sections.
The assays for peritoneal metastasis after intra-peritoneal injection of SKOV3 cells and the antibody treatment were performed as described (Arlt et al, 2006) with slight modifications. Briefly, pathogen-free, female athymic nu/nu mice (7–9-week old; 20 g average body weight) from Charles River Laboratories were inoculated intraperitoneally with 5 × 106 SKOV3-mock, SKOV3-NCAM or SKOV3-ΔFN2 cells resuspended in 300 µl of PBS, leading to tumour formation within 5 weeks. Six to ten mice per cell line were used. Where specified, tumour-bearing mice were injected intraperitoneally twice a week for 4 weeks with 10 mg/kg of either anti-NCAM (clone 123C3) or anti-HA mAb, starting 5 days after tumour cell inoculation. After 5 weeks, mice were sacrificed and analysed for tumour formation, metastatic spreading to the bowel, the liver and the diaphragm. Organs were kept in cold PBS and GFP-positive bowel metastases were analysed by fluorescence-dissection microscopy using a Stereotopic Microscope SMZ 1500 (Nikon) equipped with a 0.75X objective. Each field was photographed with a Digital Sight Camera (DS-5Mc), and the number of metastases per field was determined. For livers and diaphragms, instead, the total number of visible metastases was determined.
Acknowledgments
We are grateful to C. Francavilla for critically reading our manuscript, to P. Cattaneo for the characterization of soluble NCAM-derived proteins and to D. Connolly, R. Drapkin, M. Lewerenz, G. Christofori, M. Schachner and Pfizer for providing cell lines and reagents. We are also grateful to S. Boveri, M. Quarto, S. Confalonieri and P. Nuciforo for their contribution to the analysis of TMAs. A. Decio is a fellow of ‘Amiche del Mario Negri’, Milan, Italy. This work was supported by research grants from Associazione Italiana Ricerca sul Cancro, Association for International Cancer Research, Fondazione Cariplo, Fondazione Telethon and Italian Ministry of Health.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
SZ, LB, RG and UC conceived and designed the experiments; SZ, LB and AD performed the experiments; MB performed the immunohistochemistry analysis; MB and GM performed the histopathological analysis; LM performed some immunofluorescence experiments; FS, GA and NC set up the clinical follow-up database; CC and GV provided the samples for the tissue microarrays; EB and VB generated and provided the NCAM-derived peptide Encamin-C; SZ, LB and UC wrote the paper.
For more information
Author Institution homepages:
http://www.ieo.it/Italiano/Ricerca/ExperimentalOncology/Pages/MolecularMed.aspx
http://www.marionegri.it/mn/en/dipLab.html?lab=18
Ovarian Cancer homepage – NCI:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Ahmed N, Riley C, Rice G, Quinn M. Role of integrin receptors for fibronectin, collagen and laminin in the regulation of ovarian carcinoma functions in response to a matrix microenvironment. Clin Exp Metastasis. 2005;22:391–402. doi: 10.1007/s10585-005-1262-y. [DOI] [PubMed] [Google Scholar]
- Anderson AA, Kendal CE, Garcia-Maya M, Kenny AV, Morris-Triggs SA, Wu T, Reynolds R, Hohenester E, Saffell JL. A peptide from the first fibronectin domain of NCAM acts as an inverse agonist and stimulates FGF receptor activation, neurite outgrowth and survival. J Neurochem. 2005;95:570–583. doi: 10.1111/j.1471-4159.2005.03417.x. [DOI] [PubMed] [Google Scholar]
- Aonurm-Helm A, Jurgenson M, Zharkovsky T, Sonn K, Berezin V, Bock E, Zharkovsky A. Depression-like behaviour in neural cell adhesion molecule (NCAM)-deficient mice and its reversal by an NCAM-derived peptide, FGL. Eur J Neurosci. 2008;28:1618–1628. doi: 10.1111/j.1460-9568.2008.06471.x. [DOI] [PubMed] [Google Scholar]
- Aonurm-Helm A, Berezin V, Bock E, Zharkovsky A. NCAM-mimetic, FGL peptide, restores disrupted fibroblast growth factor receptor (FGFR) phosphorylation and FGFR mediated signaling in neural cell adhesion molecule (NCAM)-deficient mice. Brain Res. 2010;1309:1–8. doi: 10.1016/j.brainres.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Arlt MJ, Novak-Hofer I, Gast D, Gschwend V, Moldenhauer G, Grunberg J, Honer M, Schubiger PA, Altevogt P, Kruger A. Efficient inhibition of intra-peritoneal tumor growth and dissemination of human ovarian carcinoma cells in nude mice by anti-L1-cell adhesion molecule monoclonal antibody treatment. Cancer Res. 2006;66:936–943. doi: 10.1158/0008-5472.CAN-05-1818. [DOI] [PubMed] [Google Scholar]
- Bast RC, Jr, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrer MJ, Johnson ME, Hao K, Wong KK, Park DC, Bell A, Welch WR, Berkowitz RS, Mok SC. Whole genome oligonucleotide-based array comparative genomic hybridization analysis identified fibroblast growth factor 1 as a prognostic marker for advanced-stage serous ovarian adenocarcinomas. J Clin Oncol. 2007;25:2281–2287. doi: 10.1200/JCO.2006.09.0795. [DOI] [PubMed] [Google Scholar]
- Bisaz R, Schachner M, Sandi C. Causal evidence for the involvement of the neural cell adhesion molecule, NCAM, in chronic stress-induced cognitive impairments. Hippocampus. 2009;21:56–71. doi: 10.1002/hipo.20723. [DOI] [PubMed] [Google Scholar]
- Blagden S, Gabra H. Promising molecular targets in ovarian cancer. Curr Opin Oncol. 2009;21:412-419. doi: 10.1097/CCO.0b013e32832eab1f. [DOI] [PubMed] [Google Scholar]
- Casey RC, Burleson KM, Skubitz KM, Pambuccian SE, Oegema TR, Jr, Ruff LE, Skubitz AP. Beta 1-integrins regulate the formation and adhesion of ovarian carcinoma multicellular spheroids. Am J Pathol. 2001;159:2071–2080. doi: 10.1016/s0002-9440(10)63058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano G, Reid JF, Alberti P, Carcangiu ML, Tomassetti A, Canevari S. New potential ligand-receptor signaling loops in ovarian cancer identified in multiple gene expression studies. Cancer Res. 2006;66:10709–10719. doi: 10.1158/0008-5472.CAN-06-1327. [DOI] [PubMed] [Google Scholar]
- Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- Cavallaro U, Niedermeyer J, Fuxa M, Christofori G. NCAM modulates tumour-cell adhesion to matrix by inducing FGF-receptor signalling. Nat Cell Biol. 2001;3:650–657. doi: 10.1038/35083041. [DOI] [PubMed] [Google Scholar]
- Chandler LA, Sosnowski BA, Greenlees L, Aukerman SL, Baird A, Pierce GF. Prevalent expression of fibroblast growth factor (FGF) receptors and FGF2 in human tumor cell lines. Int J Cancer. 1999;81:451–458. doi: 10.1002/(sici)1097-0215(19990505)81:3<451::aid-ijc20>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Cho EY, Choi Y, Chae SW, Sohn JH, Ahn GH. Immunohistochemical study of the expression of adhesion molecules in ovarian serous neoplasms. Pathol Int. 2006;56:62–70. doi: 10.1111/j.1440-1827.2006.01925.x. [DOI] [PubMed] [Google Scholar]
- Christensen C, Lauridsen JB, Berezin V, Bock E, Kiselyov VV. The neural cell adhesion molecule binds to fibroblast growth factor receptor 2. FEBS Lett. 2006;580:3386–3390. doi: 10.1016/j.febslet.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Cole C, Lau S, Backen A, Clamp A, Rushton G, Dive C, Hodgkinson C, McVey R, Kitchener H, Jayson GC. Inhibition of FGFR2 and FGFR1 increases cisplatin sensitivity in ovarian cancer. Cancer Biol Ther. 2010;10:495–504. doi: 10.4161/cbt.10.5.12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X, Harris SS, Vanderhyden BC, Hamilton TC. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 2003;63:1389–1397. [PubMed] [Google Scholar]
- Cordero AB, Kwon Y, Hua X, Godwin AK. In vivo imaging and therapeutic treatments in an orthotopic mouse model of ovarian cancer. J Vis Exp. 2010 doi: 10.3791/2125. pii: 2125. DOI: 10.3791/2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crijns AP, Fehrmann RS, de Jong S, Gerbens F, Meersma GJ, Klip HG, Hollema H, Hofstra RM, te Meerman GJ, de Vries EG, et al. Survival-related profile, pathways, and transcription factors in ovarian cancer. PLoS Med. 2009;6:e24. doi: 10.1371/journal.pmed.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco L, Marchionni L, Gariboldi M, Reid JF, Lagonigro MS, Caramuta S, Ferrario C, Bussani E, Mezzanzanica D, Turatti F, et al. Gene expression profiling of advanced ovarian cancer: characterization of a molecular signature involving fibroblast growth factor 2. Oncogene. 2004;23:8171–8183. doi: 10.1038/sj.onc.1207979. [DOI] [PubMed] [Google Scholar]
- Dey JH, Bianchi F, Voshol J, Bonenfant D, Oakeley EJ, Hynes NE. Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res. 2010;70:4151–4162. doi: 10.1158/0008-5472.CAN-09-4479. [DOI] [PubMed] [Google Scholar]
- Donahue RN, McLaughlin PJ, Zagon IS. The opioid growth factor (OGF) and low dose naltrexone (LDN) suppress human ovarian cancer progression in mice. Gynecol Oncol. 2011 doi: 10.1016/j.ygyno.2011.04.009. in press. [DOI] [PubMed] [Google Scholar]
- Francavilla C, Loeffler S, Piccini D, Kren A, Christofori G, Cavallaro U. Neural cell adhesion molecule regulates the cellular response to fibroblast growth factor. J Cell Sci. 2007;120:4388–4394. doi: 10.1242/jcs.010744. [DOI] [PubMed] [Google Scholar]
- Francavilla C, Cattaneo P, Berezin V, Bock E, Ami D, de Marco A, Christofori G, Cavallaro U. The binding of NCAM to FGFR1 induces a specific cellular response mediated by receptor trafficking. J Cell Biol. 2009;187:1101–1116. doi: 10.1083/jcb.200903030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerardy-Schahn R, Eckhardt M. Hot spots of antigenicity in the neural cell adhesion molecule NCAM. Int J Cancer. 1994;8:38–42. doi: 10.1002/ijc.2910570708. [DOI] [PubMed] [Google Scholar]
- Gerardy-Schahn R, Eckhardt M, Ledermann J, Kemshead JT. Topography of NCAM antigenic epitopes recognized by SCLC-cluster-1 antibodies. A consensus view. Int J Cancer. 1994;8:27–29. doi: 10.1002/ijc.2910570705. [DOI] [PubMed] [Google Scholar]
- Grignani F, Kinsella T, Mencarelli A, Valtieri M, Riganelli D, Lanfrancone L, Peschle C, Nolan GP, Pelicci PG. High-efficiency gene transfer and selection of human hematopoietic progenitor cells with a hybrid EBV/retroviral vector expressing the green fluorescence protein. Cancer Res. 1998;58:14–19. [PubMed] [Google Scholar]
- Han LY, Landen CN, Trevino JG, Halder J, Lin YG, Kamat AA, Kim TJ, Merritt WM, Coleman RL, Gershenson DM, et al. Antiangiogenic and antitumor effects of SRC inhibition in ovarian carcinoma. Cancer Res. 2006;66:8633–8639. doi: 10.1158/0008-5472.CAN-06-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SM, Kohler LB, Li S, Kiselyov V, Christensen C, Owczarek S, Bock E, Berezin V. NCAM-derived peptides function as agonists for the fibroblast growth factor receptor. J Neurochem. 2008;106:2030–2041. doi: 10.1111/j.1471-4159.2008.05544.x. [DOI] [PubMed] [Google Scholar]
- Hansen SM, Li S, Bock E, Berezin V. Synthetic NCAM-derived ligands of the fibroblast growth factor receptor. Adv Exp Med Biol. 2010;663:355–372. doi: 10.1007/978-1-4419-1170-4_22. [DOI] [PubMed] [Google Scholar]
- Hinsby AM, Berezin V, Bock E. Molecular mechanisms of NCAM function. Front Biosci. 2004;9:2227–2244. doi: 10.2741/1393. [DOI] [PubMed] [Google Scholar]
- Ivan M, Matei D. Blockade of FGF signaling: therapeutic promise for ovarian cancer. Cancer Biol Ther. 2010;10:505–508. doi: 10.4161/cbt.10.5.13023. [DOI] [PubMed] [Google Scholar]
- Kenny HA, Kaur S, Coussens LM, Lengyel E. The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J Clin Invest. 2008;118:1367–1379. doi: 10.1172/JCI33775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov VV, Skladchikova G, Hinsby AM, Jensen PH, Kulahin N, Soroka V, Pedersen N, Tsetlin V, Poulsen FM, Berezin V, et al. Structural basis for a direct interaction between FGFR1 and NCAM and evidence for a regulatory role of ATP. Structure (Camb) 2003;11:691–701. doi: 10.1016/s0969-2126(03)00096-0. [DOI] [PubMed] [Google Scholar]
- Konecny GE, Glas R, Dering J, Manivong K, Qi J, Finn RS, Yang GR, Hong KL, Ginther C, Winterhoff B, et al. Activity of the multikinase inhibitor dasatinib against ovarian cancer cells. Br J Cancer. 2009;101:1699–1708. doi: 10.1038/sj.bjc.6605381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos FJ, Chin CS. Costimulation of T cell receptor-triggered IL-2 production by Jurkat T cells via fibroblast growth factor receptor 1 upon its engagement by CD56. Immunol Cell Biol. 2002;80:364–369. doi: 10.1046/j.1440-1711.2002.01098.x. [DOI] [PubMed] [Google Scholar]
- Lehembre F, Yilmaz M, Wicki A, Schomber T, Strittmatter K, Ziegler D, Kren A, Went P, Derksen PW, Berns A, et al. NCAM-induced focal adhesion assembly: a functional switch upon loss of E-cadherin. EMBO J. 2008;27:2603–2615. doi: 10.1038/emboj.2008.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel E. Ovarian cancer development and metastasis. Am J Pathol. 2010;177:1053–1064. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehanna A, Mishra B, Kurschat N, Schulze C, Bian S, Loers G, Irintchev A, Schachner M. Polysialic acid glycomimetics promote myelination and functional recovery after peripheral nerve injury in mice. Brain. 2009;132:1449–1462. doi: 10.1093/brain/awp128. [DOI] [PubMed] [Google Scholar]
- Naora H, Montell DJ. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nat Rev Cancer. 2005;5:355–366. doi: 10.1038/nrc1611. [DOI] [PubMed] [Google Scholar]
- Rabinowitz JE, Rutishauser U, Magnuson T. Targeted mutation of NCAM to produce a secreted molecule results in a dominant embryonic lethality. Proc Natl Acad Sci USA. 1996;93:6421–6424. doi: 10.1073/pnas.93.13.6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Heras E, Howell FV, Williams G, Doherty P. The fibroblast growth factor receptor acid box is essential for interactions with N-cadherin and all of the major isoforms of neural cell adhesion molecule. J Biol Chem. 2006;281:35208–35216. doi: 10.1074/jbc.M608655200. [DOI] [PubMed] [Google Scholar]
- Sawada K, Mitra AK, Radjabi AR, Bhaskar V, Kistner EO, Tretiakova M, Jagadeeswaran S, Montag A, Becker A, Kenny HA, et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008;68:2329–2339. doi: 10.1158/0008-5472.CAN-07-5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther. 2004;10:1032–1042. doi: 10.1016/j.ymthe.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Shield K, Riley C, Quinn MA, Rice GE, Ackland ML, Ahmed N. Alpha2beta1 integrin affects metastatic potential of ovarian carcinoma spheroids by supporting disaggregation and proteolysis. J Carcinog. 2007;6:11. doi: 10.1186/1477-3163-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silacci M, Brack S, Schirru G, Marlind J, Ettorre A, Merlo A, Viti F, Neri D. Design, construction, and characterization of a large synthetic human antibody phage display library. Proteomics. 2005;5:2340–2350. doi: 10.1002/pmic.200401273. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Kee WJ, Facci L, Macdonald G, Doherty P, Walsh FS. The FGFR1 inhibitor PD173074 selectively and potently antagonizes FGF-2 neurotrophic and neurotropic effects. J Neurochem. 2000;75:1520–1527. doi: 10.1046/j.1471-4159.2000.0751520.x. [DOI] [PubMed] [Google Scholar]
- Slack-Davis JK, Atkins KA, Harrer C, Hershey ED, Conaway M. Vascular cell adhesion molecule-1 is a regulator of ovarian cancer peritoneal metastasis. Cancer Res. 2009;69:1469–1476. doi: 10.1158/0008-5472.CAN-08-2678. [DOI] [PubMed] [Google Scholar]
- Soroka V, Kolkova K, Kastrup JS, Diederichs K, Breed J, Kiselyov VV, Poulsen FM, Larsen IK, Welte W, Berezin V, et al. Structure and interactions of NCAM Ig1-2-3 suggest a novel zipper mechanism for homophilic adhesion. Structure. 2003;11:1291–1301. doi: 10.1016/j.str.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Steele IA, Edmondson RJ, Bulmer JN, Bolger BS, Leung HY, Davies BR. Induction of FGF receptor 2-IIIb expression and response to its ligands in epithelial ovarian cancer. Oncogene. 2001;20:5878–5887. doi: 10.1038/sj.onc.1204755. [DOI] [PubMed] [Google Scholar]
- Strobel T, Cannistra SA. Beta1-integrins partly mediate binding of ovarian cancer cells to peritoneal mesothelium in vitro. Gynecol Oncol. 1999;73:362–367. doi: 10.1006/gyno.1999.5388. [DOI] [PubMed] [Google Scholar]
- Sundar SS, Zhang H, Brown P, Manek S, Han C, Kaur K, Charnock MF, Jackson D, Ganesan TS. Role of lymphangiogenesis in epithelial ovarian cancer. Br J Cancer. 2006;94:1650–1657. doi: 10.1038/sj.bjc.6603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundfeldt K. Cell–cell adhesion in the normal ovary and ovarian tumors of epithelial origin; an exception to the rule. Mol Cell Endocrinol. 2003;202:89–96. doi: 10.1016/s0303-7207(03)00068-6. [DOI] [PubMed] [Google Scholar]
- Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- Ueda M, Hung Y-C, Terai Y, Kanda K, Kanemura M, Futakuchi H, Yamaguchi H, Akise D, Yasuda M, Ueki M. Vascular endothelial growth factor-C expression and invasive phenotype in ovarian carcinomas. Clin Cancer Res. 2005;11:3225–3232. doi: 10.1158/1078-0432.CCR-04-1148. [DOI] [PubMed] [Google Scholar]
- Valve E, Martikainen P, Seppanen J, Oksjoki S, Hinkka S, Anttila L, Grenman S, Klemi P, Harkonen P. Expression of fibroblast growth factor (FGF)-8 isoforms and FGF receptors in human ovarian tumors. Int J Cancer. 2000;88:718–725. doi: 10.1002/1097-0215(20001201)88:5<718::aid-ijc6>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Werner S, Weinberg W, Liao X, Peters KG, Blessing M, Yuspa SH, Weiner RL, Williams LT. Targeted expression of a dominant-negative FGF receptor mutant in the epidermis of transgenic mice reveals a role of FGF in keratinocyte organization and differentiation. EMBO J. 1993;12:2635–2643. doi: 10.1002/j.1460-2075.1993.tb05924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EJ, Furness J, Walsh FS, Doherty P. Activation of the FGF receptor underlies neurite outgrowth stimulated by L1, N-CAM, and N-cadherin. Neuron. 1994;13:583–594. doi: 10.1016/0896-6273(94)90027-2. [DOI] [PubMed] [Google Scholar]
- Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer. 2009;9:167–181. doi: 10.1038/nrc2583. [DOI] [PubMed] [Google Scholar]
- Zecchini S, Bianchi M, Colombo N, Fasani R, Goisis G, Casadio C, Viale G, Liu J, Herlyn M, Godwin AK, et al. The differential role of L1 in ovarian carcinoma and normal ovarian surface epithelium. Cancer Res. 2008;68:1110–1118. doi: 10.1158/0008-5472.CAN-07-2897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.