

Abstract
The innate immune system recognizes microbial components through pattern-recognition receptors (PRRs), including membrane-bound Toll-like receptors and cytosolic receptors such as RIG-I-like receptors and deoxyribonucleic acid (DNA) sensors. These PRRs trigger distinct signal transduction pathways that culminate in induction of an array of cytokines and other mediators required for host defense. The tripartite motif (TRIM) family is a diverse family of RING finger domain-containing proteins, which are involved in a variety of cellular functions. Importantly, recent studies have shown that they are also involved in the regulation of innate immune responses through the modulation of PRR signalling pathways.
Keywords: innate immunity, signal transduction, TRIM, ubiquitin, virus infection
INTRODUCTION
The innate immune system is the first line of defensive mechanisms that protect hosts against invading microbial pathogens. Host cells express various pattern-recognition receptors (PRRs) that sense a variety of pathogen components called pathogen-associated molecular patterns (PAMPs), including lipids, lipoproteins, proteins and nucleic acids (Kawai & Akira, 2009). PRRs activate intracellular signalling pathways that culminate in the induction of inflammatory cytokines, chemokines and type I interferons (IFNs), as well as the upregulation of co-stimulatory molecules.
PRRs can be grouped in several families. Members of the Toll-like receptor (TLR) family expressed on antigen-presenting cells such as dendritic cells (DCs) and macrophages serve as key PRRs with central roles in the induction of innate immune responses as well as the subsequent development of adaptive immune responses. To date, 10 and 12 functional TLRs have been identified in humans and mice, respectively, with each TLR recognizing distinct pathogen components (Blasius & Beutler, 2010; Kawai & Akira, 2010). The second group of PRRs is composed of cytoplasmic RIG-I-like receptors (RLRs), including RIG-I, Mda5 and LGP2, which are ribonucleic acid (RNA) helicases that contain a DExD/H box RNA helicase domain (Barbalat et al, 2011; Yoneyama & Fujita, 2009). Unlike the membrane-bound TLRs, RLRs reside in the cytoplasm and recognize RNA species produced in the cytoplasm of a variety of cell types, including both immune and non-immune cells. The third group of PRRs is the NLR family with more than 20 members, several of which form inflammasomes to respond to various pathogen components, small particles and diverse cellular stresses. They trigger inflammatory responses, including the secretion of interleukin (IL)-1β via the activation of caspase-1 (Bauernfeind et al, 2011; Davis et al, 2011). In addition, double-stranded (ds) deoxyribonucleic acid (DNA) derived from DNA viruses and bacteria as well as damaged host cells triggers innate immune responses through TLR- and RLR-independent pathways (Barber, 2011). In this respect, DAI and IFI16 were identified as candidate DNA sensors that mediate type I IFN induction. In addition, AIM2 was shown to recognize dsDNA and contribute to the secretion of IL-1β rather than type I IFNs.
The signalling through TLRs, RLRs, DNA sensors and several NLRs culminates in the activation of the nuclear factor κB (NF-κB) and IFN regulatory factor (IRF) families of transcription factors via distinct signalling pathways (Kawai & Akira, 2009). Recent studies have shown that the activations of these pathways are regulated by multiple molecules, including the tripartite motif (TRIM) family, which is one of the largest families of RING domain-containing E3 ligases that mediate post-translational modifications such as protein ubiquitination (Fig 1).
Figure 1. Schematic representation of human TRIM family proteins.

The TRIM family proteins are classified into 11 subgroups (C-I to C-XI) as defined by Short & Cox, 2006, Ozato et al, 2008 and Carthagena et al, 2009. COS, C-terminal subgroup one signature; FN3, fibronectin type 3; PHD, plant homeodomain; FIL, filamin-type Ig; NHL, NCL-1, HT2A and LIN-41; MATH, meprin and TRAF homology; ARF, ADP ribosylation factor-like; TM, transmembrane; UN, unclassified.
TLRs AND SIGNALLING PATHWAYS
TLRs are type I transmembrane proteins composed of leucine rich repeats in the ectodomain, which mediate the recognition of their respective PAMPs, a transmembrane domain and an intracellular domain homologous to that of the IL-1 receptor known as the Toll/IL-1R (TIR) domain, which is required for initiating downstream signalling pathways. There are 10 and 12 functional TLRs in human (TLR1-10) and mice (TLR1-9 and 11-13), respectively, for most of which the ligands were identified through investigation of mice deficient in individual TLRs (Kawai & Akira, 2010). TLRs recognize a variety of PAMPs including lipids, lipoproteins, proteins and nucleic acids derived from a wide range of microbes such as viruses, bacteria, parasites and fungi (Box 1). Depending on their cellular localization, TLRs are largely classified into two subgroups. TLR1, TLR2, TLR4–TLR6 and TLR11 are expressed on cell surfaces and mainly recognize microbial membrane components while TLR3 and TLR7–TLR9 are expressed exclusively in intracellular vesicles such as the endoplasmic reticulum (ER), endosomes, lysosomes and endolysosomes, where they encounter microbial nucleic acids.
PAMPs RECOGNITION BY TLRs
TLR2 recognizes a wide range of PAMPs derived from various pathogens such as bacteria, fungi, parasites and viruses. Specifically, these include triacyl lipopeptides from bacteria and mycobacteria, diacyl lipopeptides from mycoplasma, peptidoglycan and lipoteichoic acid from Gram-positive bacteria, lipoarabinomannan from mycobacteria, zymosan from fungi and Trypanosoma GPI-mutin and haemagglutinin (HA) protein from measles virus. To discriminate the molecular structure of the ligand, TLR2 generally forms a heterodimer with TLR1 or TLR6. TLR2–TLR6 recognizes the mycobacterial diacylated lipopeptide, lipoteichoic acid and zymosan, whereas, TLR2–TLR1 recognizes the bacterial triacylated lipopeptide. TLR4 recognizes lipopolysaccharide (LPS), a major component of the outer membrane of Gram-negative bacteria. TLR5 recognizes flagellin, a protein component of bacterial flagella. Mouse TLR11 recognizes uropathogenic bacteria products, although a specific ligand has not been identified yet, and a parasite component. Specifically, a soluble fraction of Toxoplasma gondii tachyzoites contains a potent inducer for IL-12 known as soluble Toxoplasma antigen (STAg), which is recognized and of which the active component is a profilin-like molecule that is known to function as an actin-binding protein and implicated in parasite motility or invasion.
Unlike these TLRs that are expressed on the cell surface, TLR3 and TLR7–TLR9 are expressed by intracellular compartments such as the endosome, lysosome or the ER. These intracellular TLRs appear to be sensors of foreign nucleic acids, and trigger anti-viral innate immune responses by producing type I IFN and inflammatory cytokines (Blasius & Beutler, 2010). TLR3 recognizes viral double stranded RNA as well as a synthetic analog of dsRNA polyinosinic–polycytidylic acid (poly IC). TLR7 was originally identified to recognize imidazoquinoline derivatives such as imiquimod and resiquimod (R-848), and guanine analogues such as loxoribine, all of which have anti-viral and anti-tumour properties. Guanosine-rich and uridine-rich ssRNA derived from human immunodeficiency virus (HIV) or influenza virus and synthetic poly-uridine ssRNA are also recognized by TLR7. Human TLR8 recognizes R-848, bacterial RNA and ssRNA although TLR8-deficient mice respond normally to these molecules. TLR9 was originally identified to recognize unmethylated 2′-deoxyribo cytidine–phosphate–guanosine (CpG) DNA motifs frequently present in bacteria and viruses. TLR9 also recognizes haemozoin derived from Plasmodium falciparum that is an insoluble crystal generated as a by-product of the detoxification process after parasite digestion of host haemoglobin.
 |
Dimerization of TLRs initiates the signalling cascade, which then facilitates the recruitment of TIR-domain-containing adapter proteins, including myeloid differentiation primary response gene 88 (MyD88), TIR domain containing adaptor protein (TIRAP), TIR-domain-containing adapter-inducing IFN-β (TRIF) and TRIF-related adaptor molecule (TRAM; Kawai & Akira, 2010; Fig 2). Depending on the type of PAMP, these adapters are selectively recruited to their respective TLR and elicit appropriate responses. Interestingly, MyD88 interacts with all TLRs with the exception of TLR3 and initiates NF-κB and mitogen-activated protein kinase (MAPK) activation to control inflammatory responses. In case of TLR2 and TLR4, the adaptor TIRAP is recruited and functions as a sorting molecule that recruits MyD88. In contrast, TRIF is used by TLR3 and TLR4 and initiates an alternative pathway including IRF3, NF-κB and MAPK to induce type I IFN and inflammatory cytokines. Here, TRAM selectively serves to link TRIF to TLR4, but not TLR3. Therefore, TLR signalling pathways are largely classified as either MyD88-dependent or TRIF-dependent pathway.
Figure 2. Regulation of TLR signalling by TRIM proteins.

Upon LPS stimulation, TLR4 recruits TRAF6 via the adapters TIRAP and MyD88 to activate the TAK1 complex (TAK1, TAB2 and TAB3) for NF-κB activation. Alternatively, TLR4 recruits the adapters TRAM and TRIF to activate NF-κB via TRAF6 as well as IRF3 via TBK1/IKKi. TLR3 recognizes viral dsRNA and activates the TRIF-dependent pathway. TLR7 and TLR9, which recognize viral RNA and DNA, respectively, activate IRF7 via MyD88. TRIM30α negatively regulates NF-κB activation by targeting the TAK1 complex for degradation. TRIM23 activates NEMO and TRIM27 inhibits IKKα/IKKβ and TBK1/IKKi. TRIM21 inhibits IRF5 and IRF8 activation and influences IRF3 and IRF8 activation.
Glossary
Chemokines and cytokines
Soluble proteins that regulate the immune response, by serving as, for example, chemoattractants or activators of immune cells.
Dendritic cells (DCs)
Immune cells forming part of the mammalian immune system. Their main function is to process antigen material and present it on the surface to other cells of the immune system. That is, they function as antigen-presenting cells. They act as messengers between the innate and adaptive immunity.
Innate immune system
A branch of the immune system composed of cells, such as DCs, macrophages, neutrophils and NK cells, with germline-encoded receptors specific for markers of host perturbation (e.g. infection, damage or loss of expression of self-molecules). Triggering of these receptors results in a rapid but short-lived inflammatory response. In addition to the cellular component, complement proteins and barriers such as the skin are constituents of the innate branch of immunity.
PAMPs
Pathogen-associated molecular patterns are molecules associated with groups of pathogens that are recognized by cells of the innate immune system. These molecules can be referred to as small molecular motifs conserved within a class of microbes. They are recognized by TLRs and other pattern recognition receptors (PRRs) in both plants and animals.
Pattern recognition receptor (PRR)
Patterns of the innate immune system that are not randomly generated (i.e. germline) that recognize makers of pathogens or cell stress/damage. These include TLRs, RLRs, NLRs and DNA sensors.
Protein ubiquitination
Ubiquitination is a covalent modification that regulates a wide range of biochemical processes (Liu & Chen, 2011). Ubiquitin covalently attaches to one or more lysine residues on target proteins through the sequential activation of a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2) and a ubiquitin ligase (E3). Ubiquitin is composed of 76 amino acids, and contains seven lysine residues (K6, K11, K27, K29, K33, K48 and K63). Each lysine residue can be conjugated to the C-terminal Gly residue of ubiquitin to create polyubiquitin chains. K48-linked polyubiquitination is well recognized as a targeting signal that allows modified proteins to undergo 26S proteasome-dependent degradation. On the other hand, K63-linked polyubiquitination is involved in signal transduction pathways and trafficking of targeted proteins (Bhoj & Chen, 2009). K27-linked polyubiquitination was reported to be a unique modification involved in antiviral immunity (Arimoto et al, 2010). In addition, monoubiquitination and linear ubiquitination have been ascribed as post-translational modifications involved in receptor internalization, trafficking and signal transduction (Haas, 2009).
Type I IFN
Type I IFNs are cytokines released upon pathogen infection. Mammalian type I IFNs include IFNα, IFNβ, IFNκ, IFNδ, IFNε, IFNτ, IFNω and IFNζ. They trigger defensive mechanisms against virus infection through the transcription of hundreds of IFN-stimulated genes (ISGs) in autocrine or paracrine manners to establish antiviral state. Examples of ISGs known to have antiviral properties include GTPases, myxoma resistance (Mx) proteins, zinc finger antiviral protein (ZAP), Friend virus susceptibility factor 1 (Fv1), viperin, cytidine deaminase APOBECs, protein kinase PKR, 2′5′-oligoadenylate synthetase (OAS), adenosine deaminases, ISG15 and microRNAs (Randall & Goodbourn, 2008). In addition, type I IFNs have immunomodulatory functions. Briefly, they enhance natural killer cell activation as well as DC maturation to facilitate virus-specific antibody production and the differentiation of cytotoxic T lymphocytes (Theofilopoulos et al, 2005).
Ubiquitin ligase
Enzyme, which attaches ubiquitin covalently onto a lysine residue of a target protein. Ubiquitin is a small, regulatory protein that is covalently attached onto a protein and may induce a number of alterations in protein stability, function and/or localization.
Interestingly, TLR4 utilizes all four adapters and activates both the MyD88- and TRIF-dependent pathways. It initially recruits TIRAP at the plasma membrane and subsequently facilitates the recruitment of MyD88 to trigger the initial activation of NF-κB and MAPK. Then, TLR4 is internalized and forms a signalling complex with TRAM and TRIF to initiate the TRIF-dependent pathway leading to IRF3 activation as well as the late phase activation of NF-κB and MAPK (Barton & Kagan, 2009). Importantly, activation of both the MyD88- and TRIF-dependent pathways is necessary for induction of inflammatory cytokines via TLR4 signalling. It remains unclear why activation of either pathway alone is insufficient for the induction of inflammatory cytokines through this TLR.
MyD88 then recruits the IL-1 receptor-associated kinases (IRAK) IRAK4, IRAK1, IRAK2 and IRAK-M. Initially, IRAK4 is activated, which plays an essential role in NF-κB and MAPK activation downstream of MyD88, while IRAK1 and IRAK2 are activated sequentially. Activation of both of these kinases is required for NF-κB and MAPK activation. Generally, IRAK activation results in recruitment of tumour necrosis factor receptor (TNFR) associated factor 6 (TRAF6), an E3 ubiquitin ligase that catalyses the formation of polyubiquitin chains linked to lysine (K) 63 on target proteins, which include TRAF6 itself and IRAK1, in conjunction with the E2 ubiquitin-conjugating enzymes Ubc13 and Uev1A (Liu & Chen, 2011). The K63-linked polyubiquitin chains then bind to TGF-beta activated kinase 1 (TAK1)/MAP3K7 binding protein 2 (TAB2) and TAB3, the regulatory components of the TAK1 complex, to activate the TAK1 and to NF-κB essential modulator (NEMO), an essential regulatory component of the IκB kinase (IKK) complex required for NF-κB activation. Thus, the K63 polyubiquitin chains might be responsible for recruiting TAK1 to the IKK complex, which would allow TAK1 to phosphorylate IKKβ, a catalytic subunit of the IKK complex, through its close proximity to the IKK complex. This leads to phosphorylation and subsequent degradation of IκB proteins, which allows NF-κB to translocate into the nucleus to regulate target gene expressions. At the same time, TAK1 induces phosporylation of MKKs, which activates the MAPKs ERK1, ERK2, p38 and JNK, which then activate AP-1.
TRIF recruits TRAF6 and TAK1 to activate NF-κB, most likely through mechanisms similar to those in the MyD88-dependent pathway (Kawai & Akira, 2010), resulting in a complex, which also includes tumour necrosis factor receptor type 1-associated Death domain protein (TRADD), Pellino-1 and RIP1. In addition to NF-κB activation, the TRIF-dependent pathway leads to the activation of IRF3 and IFNβ transcription through the recruitment of a signalling complex involving the non-canonical IKKs TANK-binding kinase 1 (TBK1) and IKKi (also known as IKKε), which catalyse the phosphorylation of IRF3 and induce its nuclear translocation. The activation of TBK1-IKKi also requires TRAF3, which is also incorporated into the TRIF complex.
The TLR7 and TLR9 signalling pathways in plasmacytoid DCs (pDCs) are unique in that they both induce type I IFN in a MyD88-dependent fashion (Kawai & Akira, 2010). In this context, MyD88 binds to IRF7, which is constitutively expressed by pDC, and forms a multiprotein signalling complex with IRAK4, TRAF6, TRAF3, IRAK1, IKKa, Dock2 and OPNi (Gotoh et al, 2010; Kawai & Akira, 2010). IRF7 is then phosphorylated by IRAK1 and/or IKKα, dissociates from the complex and translocates into the nucleus. While IRAK1, IKKα and TRAF3 are specifically involved in the activation of IRF7, MyD88, IRAK4 and TRAF6 are critical for the activation of both IRF7 and NF-κB. In other cell types, such as conventional DCs, TLR9 also induces type I IFN. In this case, IRF1, which is recruited to MyD88, is involved (Kawai & Akira, 2010). Interestingly, IRF5 is also involved in TLR signalling by binding MyD88 and TRAF6 and subsequent translocation to the nucleus after phosphorylation. There, it binds IFN-sensitive response element (ISRE) motifs in the promoter region of genes encoding inflammatory cytokines and induces their expression presumably via collaborative activation with NF-κB.
RLRs AND SIGNALLING PATHWAYS
The RLR family consists of three members: RIG-I, MDA5 and LGP2 (Yoneyama & Fujita, 2010). These sensors recognize the RNA from RNA viruses in the cytoplasm of infected cells and induce expression of type I IFN and inflammatory cytokines. RIG-I and MDA5 contain tandem caspase recruitment domain (CARD)-like regions at their N-termini, which mediate interaction with other CARD-containing proteins, and a central DExD/H helicase domain, which has an ATP-binding motif. Interestingly, LGP2 also contains the helicase domain but lacks the CARD domain.
RIG-I mediates the recognition of various ssRNA viruses, which include influenza A virus, new castle disease virus, vesicular stomatitis virus, Japanese encephalitis virus and hepatitis C virus (HCV; Takeuchi & Akira, 2010; Yoneyama & Fujita, 2010) while MDA5 is required for the recognition of other RNA viruses, including picornaviruses such as encephalomyocarditis virus (EMCV), Mengo virus and Theiler's virus (Takeuchi & Akira, 2010; Yoneyama & Fujita, 2010). Furthermore, dengue virus and West Nile virus are recognized by both RIG-I and MDA5. RIG-I and MDA5 also recognize synthetic ligands. RIG-I recognizes short dsRNA bearing a 5′-triphospate, whereas MDA5 recognizes long dsDNA (Takeuchi & Akira, 2010; Yoneyama & Fujita, 2010) and was shown to recognize polyinosinic:polycytidylic acid (poly IC). However, shortened poly IC is preferentially recognized by RIG-I rather than by MDA5 (Kato et al, 2008), suggesting that RIG-I and MDA5 recognize different lengths of RNA. LGP2 might facilitate the accessibility of viral RNA by RIG-I or MDA5 to induce robust responses (Satoh et al, 2010).
RLR signalling leads to activation of NF-κB and IRF3 for the induction of type I IFN and inflammatory cytokines (Fig 3). RIG-I and MDA5 recruit the adapter IPS-1 (also known as MAVS, Cardif or VISA; Yoneyama & Fujita, 2010). IPS-1 contains an N-terminal CARD-like domain responsible for the interaction with the RLRs and a transmembrane domain at the C-terminus, which is required for mitochondrial targeting as well as signalling. Downstream of IPS-1, are the TBK1-IKKi and IKK complexes. TRAF3 and TRAF6 are involved in the activation of TBK1-IKKi and IKK complexes via interaction with IPS-1, respectively. The induced, secreted IFNs subsequently induce IFN-stimulated genes (ISGs) via the type I IFN receptor (IFNAR)-signal transducer and activator of transcription (STAT)1/STAT2/IRF9 axis.
Figure 3. Regulation of RIG-I and DNA sensor signalling by TRIM proteins.

Upon recognition of RNA viruses, RIG-I induces the synthesis of inflammatory cytokines and type I IFNs. The secreted IFNs activate the STAT1/STAT2/IRF9 complex to induce ISGs, including TRIM proteins. RIG-I recruits the adapter IPS-1 and activates NF-κB (p50/RelA heterodimer) and IRF3 via IKKα/IKKβ/NEMO and TBK1/IKKi, respectively. TRIM25 induces the ubiquitination of RIG-I to promote downstream signalling. TRIM23 enhances NEMO function by promoting ubiquitination and TRIM27 interferes with IKKα/IKKβ and TBK1/IKKi activation. TRIM21 is involved in the inhibition of IKKβ and IRF7 and positive or negative regulation of IRF3 and IRF8. TRIM56 enhances STING function during DNA sensor signalling pathways for the induction of type I IFNs.
DNA SENSORS
Double-stranded DNA released into the cytoplasm upon infection with DNA viruses or bacteria potently induces type I IFN and inflammatory cytokines via TLR- and RLR-independent pathways (Barbalat et al, 2011). So far, two cytosolic DNA sensors responsible for type I IFN induction have been discovered. DNA-dependent activator of IFN-regulatory factors (DAI) was identified as a candidate DNA sensor that recognizes transfected dsDNA (Takaoka et al, 2007). DAI has the ability to bind to DNA and associate with IRF3. However, type I IFN production by embryonic fibroblast cells and DCs following dsDNA stimulation was not impaired in DAI-deficient mice (Ishii et al, 2008), suggesting a redundancy. IFN-inducible IFI16, which contains a HIN200 domain, was identified as another DNA sensor although its physiological function in terms of type I IFN induction remains unclear (Unterholzner et al, 2010).
Stimulator of IFN genes (STING also known as MITA, ERIS or MPYS) is an essential component of DNA-mediated immune responses that resides in the ER (Barber, 2011). Importantly, cells deficient in STING fail to induce type I IFN and inflammatory cytokines in response to dsDNA and DNA virus infection. In response to dsDNA stimulation, STING relocalizes to punctate structures, to which TBK1 is recruited (Saitoh et al, 2009; Fig 3). This recruitment is likely to be essential for TBK1 activation, which results in IRF3 activation and the transcription of type I IFN genes. However, it remains to be defined how STING activation is controlled.
AIM2 (absent in melanoma 2) was identified as the sensor for dsDNA that mediates IL-1β production rather than type I IFN upon dsDNA stimulation (Barbalat et al, 2011). It has a HIN200 DNA-binding domain and a pyrin domain that interacts with the adapter protein ASC. ASC interacts with and activates Caspase-1, leading to processing pro-IL-1β to mature IL-1β. Cells from AIM2-deficient mice are unable to release IL-1β in response to DNA.
STRUCTURE AND EXPRESSION OF TRIM PROTEINS
The TRIM family has more than 70 members in humans (Fig 1; Meroni & Diez-Roux, 2005; Nisole et al, 2005; Ozato et al, 2008; Sardiello et al, 2008; Short & Cox, 2006). They contain an N-terminal RBCC motif composed of a RING domain, one or two B-boxes, a coiled-coil domain as well as C-terminal regions with different structures. The B-box is a zinc-binding motif but its function is unclear while the coiled-coil domain mediates protein interactions. Although the C-terminal regions differ among individual members, the majority of the TRIM proteins contain a PRY or SPRY domain or both domains (also referred to as the B30.2 domain), which mediates protein–protein interactions. The other C-terminal domains include COS, fibronectin type 3 (FN3), plant homeodomain (PHD), bromodomain, filamin type Ig (FIL), NCL-1, HT2A and LIN-41 (NHL) repeats, meprin and TRAF homology (MATH), ADP ribosylation factor-like (ARF) and transmembrane domains (Ozato et al, 2008; Sardiello et al, 2008).
The COS domain is responsible for protein binding to microtubules, and the FN3 domain mediates binding to DNA and heparin. The PHD domain is involved in chromatin-mediated gene regulation, and the bromodomain, which is always located downstream of PHD in TRIM proteins, recognizes acetylated lysine residues. These two domains together mediate transcriptional repression. NHL repeats resemble WD40 to mediate protein–protein interactions, and the FIL domain is involved in actin crosslinking. The MATH domain is involved in protein–protein interactions, and ARF regulates intracellular trafficking. Based on the different structures of their C-terminal regions, the TRIM proteins can be classified into 11 subgroups (Carthagena et al, 2009; Ozato et al, 2008; Short & Cox, 2006).
The subcellular localizations vary among the TRIM family members. Overexpression studies indicated that the majority of TRIM proteins are localized in the cytoplasm or nucleus in filamentous, diffuse or speckled patterns although they did not colocalize with previously defined markers of compartments such as endosomes, mitochondria, Golgi and spliceosome (Lerner et al, 2007; Reymond et al, 2001; Short & Cox, 2006). However, TRIMs known to be localized to specific cellular compartments are TRIM5 and TRIM19/PML. TRIM5 is localized in the cytoplasmic structures designated as cytoplasmic bodies and TRIM19 is localized in a specific nuclear domain called nuclear bodies (NBs; also known as PML body, ND10 or POD; Bernardi & Pandolfi, 2007; Campbell et al, 2007). TRIM1/MID2, TRIM9 and TRIM18/MID1 were found to be associated with microtubules and TRIM13 was localized to the ER as well as the nucleus (Lerner et al, 2007; Short & Cox, 2006).
TRIMs AND DISEASES
Mutations of TRIM family members have been reported to be associated with a variety of diseases, including Mendelian inherited disorders, cancer, inflammation and autoimmunity (Table 1). Mutation of TRIM18 is found in Opitz syndrome, which is an X-linked recessive disorder characterized by hypertelorism, genital-urinary defects, lip-palate-laryngotracheal clefts, imperforate anus, developmental delay and congenital heart defects (Quaderi et al, 1997). The mutant TRIM18 proteins with the C-terminal region that are produced in this disease show decreased affinity for microtubules. Defects in TRIM37/MUL1 cause mulibrey nanism, which is an autosomal recessive disorder accompanied by severe growth failure of prenatal onset and constrictive pericardium with consequent hepatomegaly (Avela et al, 2000). A homozygous mutation (D487N) in one of the C-terminal NHL domains of TRIM32 is the cause of limb-girdle muscular dystrophy type 2H, a degenerative myopathy characterized by pelvic girdle, shoulder girdle and quadriceps muscle weakness (Frosk et al, 2002; Saccone et al, 2008). A homozygous mutation (P130S) in the B-box domain of this protein causes Bardet–Biedl syndrome type 11 that is characterized by severe pigmentary retinopathy, early onset obesity, polydactyly, hypogenitalism, renal malformation and mental retardation (Chiang et al, 2006). TRIM63/MURF-1 is implicated in muscle atrophy and sarcopenia (Bodine et al, 2001) while TRIM19 was identified as a protein fused to retinoic acid receptor α (RARα) that blocks the differentiation of haematopoietic progenitor cells and causes acute promyelocytic leukaemia (Bernardi & Pandolfi, 2007; de The et al, 1991). TRIM24/TIF1α and TRIM27/RFP have oncogenic activities when fused to B-RAF kinase and RET tyrosine kinase, respectively (Hasegawa et al, 1996; Le Douarin et al, 1995). Interestingly, the TRIM13 gene is frequently lost in various malignancies, including B-cell chronic lymphocytic leukaemia, suggesting that TRIM13 acts as a tumour suppressor gene (Bullrich et al, 2001). TRIM20/pyrin mutations are found in patients with familial Mediterranean fever (FMF) characterized by recurrent episodic fever and serosal inflammation (French FMF Consortium, 1997). TRIM21/SS-A/Ro52 and TRIM68/SS-56 are known to be autoantigens present in the sera of autoimmune patients, including Sjögren syndrome and systemic lupus erythematosus patients (Ben-Chetrit et al, 1988; Billaut-Mulot et al, 2001).
Table 1.
TRIM proteins associated with diseases
| TRIM | Involvement in disease | References |
|---|---|---|
| TRIM13 (RFP2, LEU5) | Located within the minimal deletion region for B-cell chronic lymphocytic leukaemia | Bullrich et al (2001) |
| TRIM18 (MID1) | Mutated in Opitz syndrome type I | Quaderi et al (1997) |
| TRIM19 (PML) | Fusion with RARα results in acute promyelocytic leukaemia | de The et al (1991) |
| TRIM20 (Pyrin, FMF, MFEV) | Mutated in familial Mediterranean fever | French FMF Consortium (1997) |
| TRIM21 (SSA, Ro52) | Encodes autoantigen in patients with SLE and Sjogren's syndrome | Ben-Chetrit et al (1988) |
| TRIM24 (TIF1α) | Fusion with B-raf | Le Douarin et al (1995) |
| TRIM27 (RFP) | Fusion with RET in papillary thyroid carcinoma | Hasegawa et al (1996) |
| TRIM32 | Mutated in limb-girdle muscular dystrophy type 2H and Bardet–Biedl syndrome type 11 | Frosk et al (2002), Saccone et al (2008), Chiang et al (2006) |
| TRIM37 (MUL) | Mutated in mulibrey nanism | Avela et al (2000) |
| TRIM63 (MuRF1) | Upregulted in muscle atrophy | Bodine et al (2001) |
| TRIM68 (SS-56) | Encodes autoantigen in patients with SLE and Sjogren's syndrome | Billaut-Mulot et al (2001) |
PROTEIN MODIFICATION BY TRIM PROTEINS
The presence of a RING domain suggests that TRIMs function as E3 ubiquitin ligases, and indeed, a number of TRIMs have been demonstrated to have E3 ubiquitin ligase activities (Ozato et al, 2008). Notably, two TRIMs are shown to mediate the conjugation of ubiquitin-related molecules such as small ubiquitin-like modifier (SUMO) and IFN-inducible ISG15. TRIM19 is an essential component of NBs, which are implicated in transcriptional repression and the induction of apoptosis (Bernardi & Pandolfi, 2007). Importantly, SUMO is concentrated in NBs, and TRIM19 associates with the E2 SUMO-conjugating enzyme Ubc9 to mediate protein SUMOylation. TRIM25/EFP was shown to promote protein ISGylation in addition to K63-linked polyubiquitination (Nakasato et al, 2006; Zou & Zhang, 2006). ISG15 is implicated in type I IFN-mediated innate immune responses through modulation of IFNAR signalling pathways (Jeon et al, 2010).
ANTIVIRAL ACTIVITIES OF TRIMs
A number of TRIM family members are induced by type I IFN stimulation (Carthagena et al, 2009; Rajsbaum et al, 2008) and have been found to have antiviral activities (Table 2). TRIM5 is well known to restrict the entry of diverse retroviruses, including HIV-1 and N-tropic murine leukaemia viruses (N-MLV; Nisole et al, 2005; Ozato et al, 2008; Stremlau et al, 2004). TRIM5 restricts HIV-1 strongly in rhesus monkeys but weakly in humans. It binds to the retroviral capsid through the C-terminal SPRY domain and causes premature capsid disassembly. TRIM1, which is closely related to TRIM5, also has an ability to restrict MLV (Yap et al, 2004). TRIM22/Staf50 restricts the replication of HIV through inhibition of long terminal repeat (LTR)-driven transcription, EMCV through degradation of 3C protease and hepatitis B virus through inhibition of RNA synthesis (Eldin et al, 2009; Gao et al, 2009). Intriguingly, TRIM21 was shown to function as a cytosolic IgG receptor that mediates adenovirus neutralization (Keeble et al, 2008; Mallery et al, 2010). It binds to virus-coated IgG inside cells and targets the IgG complex for degradation, suggesting a new mechanism for virus neutralization. TRIM56 was recently identified to associate with the N-terminal protease of bovine diarrhoea virus and inhibit the replication of this virus (Wang et al, 2011). TRIM19 is implicated in the inhibition of a variety of viruses, including vesicular stomatitis virus, influenza A virus, human cytomegalovirus (HCMV), herpes simplex virus-1 and HIV-1, although the mechanisms underlying these restrictions remain unclear (Geoffroy & Chelbi-Alix, 2011; Nisole et al, 2005). TRIM28 displays antiviral activities independently of their IFN-inducibility. It functions as a co-repressor to silence MLV in embryonic stem cells (Wolf & Goff, 2007; Table 2).
Table 2.
Virus restriction by TRIM proteins
| TRIM | Virus | Note | References |
|---|---|---|---|
| TRIM1 | MLV | Inhibits MLV entry, gene expression and release | Yap et al (2004), Uchil et al (2008) |
| TRIM5 | HIV, MLV, SIV, FIV, EIAV | Inhibits HIV and MLV entries. Recognizes capsid | Stremlau et al (2004), Nisole et al (2005), Ozato et al (2008), Uchil et al (2008) |
| TRIM8 | HIV, MLV | Inhibits HIV and MLV entries. Inhibits MLV release | Uchil et al (2008) |
| TRIM10 | HIV | Inhibits HIV entry | Uchil et al (2008) |
| TRIM11 | HIV, MLV | Inhibits HIV entry. Inhibits HIV and MLV gene expression and releases | Uchil et al (2008) |
| TRIM13 | MLV | Inhibits MLV gene expression and release | Uchil et al (2008) |
| TRIM14 | MLV | Inhibits MLV gene expression and release | Uchil et al (2008) |
| TRIM15 | HIV, MLV | Inhibits HIV and MLV release | Uchil et al (2008) |
| TRIM19 | HIV, MLV, VSV, IAV, HCMV, HSV-1, Ebola virus, Lassa fever virus, LCMV, human foamy virus and adenovirus | Inhibits MLV release | Geoffroy & Chelbi-Alix (2011), Nisole et al (2005), Uchil et al (2008) |
| TRIM21 | MLV, adenovirus | Inhibits MLV gene expression and release. Promotes degradation of antibody-bound virus particles | Keeble et al (2008), Mallery et al (2010), Uchil et al (2008) |
| TRIM22 | HIV, EMCV, HBV | Inhibits HIV LTR-driven transcription. Reduces HIV Gag p24 level. Inhibits HBV core promoter activity. Ubiquitinates EMCV 3C protease | Gao et al (2009), Eldin et al (2009) |
| TRIM25 | HIV, MLV | Inhibits MLV entry. Inhibition of HIV and MLV releases | Uchil et al (2008) |
| TRIM26 | HIV, MLV | Inhibits HIV and MLV entries and releases | Uchil et al (2008) |
| TRIM27 | HIV, MLV | Inhibits HIV and MLV releases. Inhibits MLV gene expression | Uchil et al (2008) |
| TRIM28 | MLV, HTLV1 | Inhibits MLV release. Silences transcription of MLV and HLTV-1 in ES cells | Wolf & Goff (2007), Uchil et al (2008) |
| TRIM31 | HIV, MLV | Inhibits HIV and MLV entries. Inhibits MLV gene expression and release | Uchil et al (2008) |
| TRIM32 | HIV, MLV | Inhibits HIV and MLV releases. Inhibits MLV gene expression. | Uchil et al (2008) |
| TRIM35 | MLV | Inhibits MLV release | Uchil et al (2008) |
| TRIM56 | HIV, MLV, BVDV | Inhibits HIV and MLV entries. Inhibits HIV release. Inhibits BVDV replication by binding to the N-terminal protease | Wang et al (2011), Uchil et al (2008) |
| TRIM62 | MLV | Inhibits MLV entry, gene expression and release | Uchil et al (2008) |
A comprehensive screen for antiretroviral activity involving 55 TRIMs (36 human and 19 mouse) revealed approximately 20 TRIMs with abilities to interfere with the entry or release of retroviruses in transiently transfected cells (Uchil et al, 2008). This screening identified that human TRIM5, 11, 26 and 31 as well as mouse TRIM8, 10 and 56 inhibited HIV entry, whereas, human TRIM1, 5, 25, 26 and 62 as well as mouse TRIM8, 25, 31 and 56 inhibited N-MLV entry. Moreover, human TRIM15, 19, 21, 25, 26 and 32 and mouse TRIM8, 11, 25, 27 and 56 suppress HIV release from cells. Human TRIM11 suppresses both HIV gene expression and release. Human TRIM8, 15, 19, 25, 26, 28 and 35 and mouse TRIM19 and 25 inhibit MLV release, whereas, human TRIMs1, 11, 13, 14, 21, 27, 31, 32 and 62 and mouse TRIM8, 11 and 27 inhibit both MLV gene expression and release. These findings suggest that multiple TRIM proteins restrict various stages of retrovirus replication.
In addition to the aforementioned TRIMs that directly target virus-associated molecules to restrict virus replication or budding, TRIMs have emerged that target cellular proteins involved in PRR signalling to influence innate immune responses (Table 3). Below, we describe the details of the TRIM members that modify PRR signalling.
Table 3.
Regulation of innate immune signalling pathways by TRIM proteins
| TRIM | Expression after IFN or LPS stimulation | Note | References |
|---|---|---|---|
| TRIM5 | Upregulation | Recognizes retrovirus capsid and activates TAK1 via generation of K63-linked polyubiquitin chains | Pertel et al (2011) |
| TRIM13 (RFP2, LEU5) | No effect | Causes NF-κB activation when overexpressed | Matsuda et al (2003) |
| TRIM20 | Upregulation | Supports preteasomal degradation of IκBs and enhances NF-κB nuclear trasnlocation | Chae et al (2008) |
| Promotes monoubiquitination and degradation of IKKβ | Wada et al (2009) | ||
| Prevents the interaction between IRF3 and PIN1 | Yang et al (2009) | ||
| TRIM21 | Upregulation | Promotes ubiquitination and proteasomal degradation of IRF3 | Higgs et al (2008) |
| Promotes ubiquitination and proteasomal degradation of IRF7 | Higgs et al (2010), Young et al (2011) | ||
| Activates IRF8 via K63-linked polyubiquitination | Kong et al (2007) | ||
| Negatively regulates IRF5 and IRF8 | Espinosa et al (2009) | ||
| TRIM23 | No effect | Promotes IRF3 and NF-κB activation via K27-linked polyubiquitination of NEMO | Arimoto et al (2010) |
| TRIM25 (EEP) | Upregulation | Promotes K63-linked polyubiquitination of RIG-I and triggers antiviral signalling | Gack et al (2007), Zeng et al (2010) |
| TRIM27 | No effect | Binds and inhibits IKKα, IKKβ, TBK1 and IKKi via ubiquitin-independent mechanisms | Zha et al (2006) |
| TRIM30 | Upregulation (mouse) | Promotes lysosome-mediated TAB2 and TAB3 degradation and inhibits TLR4 signalling | Shi et al (2008) |
| TRIM32 | No effect | Activates NF-κB via proteasomal degradation of PIASy | Albor et al (2006) |
| TRIM38 (RoRet) | Upregulation | Causes NF-κB activation when overexpressed | Matsuda et al (2003) |
| TRIM56 | Upregulation | Promotes K63-linked polyubiquitination of STING and activates TBK1 | Tsuchida et al (2010) |
NEGATIVE REGULATION OF TLR4 SIGNALLING BY TRIM30α
Although TLR4 activation is critical for provoking an innate response to ward off pathogens, massive production of inflammatory cytokines and other molecules is harmful and leads to septic shock. Therefore, to avoid inappropriate inflammatory responses, TLR signalling is negatively controlled by multiple mechanisms. Shi et al identified TRIM30α, whose expression is upregulated by stimulation with various TLR agonists, including LPS, as a negative regulator of TLR4 signalling (Shi et al, 2008). They found that TRIM30α interacts with TAK1, TAB2 and TAB3 and induces the degradation of TAB2 and TAB3 (Fig 2). This degradation depends on the lysosomal degradation pathway rather than the conventional ubiquitin–proteasome pathway. TRIM30 knockdown in cell lines leads to enhanced TAB2 expression associated with increased NF-κB activation as well as increased production of TNFα and IL-6 in response to LPS stimulation. Further in vivo studies showed that mice overexpressing TRIM30α are more resistant to LPS-induced septic shock than control mice. Therefore, TRIM30α is involved in a negative feedback regulation of TLR4-induced inflammatory responses. There is no human ortholog of mouse TRIM30, suggesting that other TRIM proteins may participate in TAB2 and TAB3 degradation in humans. In addition, it was demonstrated that human TRIM5α can promote TAB2 degradation and thus contribute to the inhibition of NF-κB (Tareen & Emerman, 2011). TRIM5α also has the ability to induce NF-κB-driven promoters after transient transfection, suggesting that TRIM5α has a dual role in controlling NF-κB activity.
REGULATION OF RIG-I SIGNALLING BY TRIM25
Gack et al reported that the CARD of RIG-I is ubiquitinated upon virus infection and that this modification is essential for the activation of downstream signalling (Gack et al, 2007). They purified TRIM25 as a protein that interacts with RIG-I and demonstrated that it acts as a ubiquitin E3 ligase, which promotes RIG-I polyubiquitination linked to K63 (Fig 3). This modification is required for interaction with the CARD of IPS-1. TRIM25-deficient cells fail to induce cytokines in response to viruses that are sensed by RIG-I. Therefore, TRIM25 is an essential molecule that initiates RIG-I-mediated pathways. Interestingly, non-structural protein 1 (NS1) of influenza A virus, which is known to suppress host antiviral responses, was shown to directly interact with TRIM25 (Gack et al, 2009). This interaction results in suppression of the E3 ligase activity of TRIM25, thereby, preventing RIG-I ubiquitination and the subsequent initiation of antiviral responses.
Zeng et al examined the molecular mechanisms of how RIG-I is activated by TRIM25 in a ubiquitination-dependent manner using an in vitro cell-free system (Zeng et al, 2010). They showed that, in addition to the binding of viral RNA to RIG-I, ubiquitination of RIG-I is required for IRF3 activation. Notably, they found that this ubiquitination is unique in that RIG-I associates with free unanchored K63-linked ubiquitin chains that are catalysed by TRIM25. Therefore, it is possible that unanchored ubiquitin chains act as a signalling platform that allows RIG-I to recruit IPS-1 for robust IRF3 activation.
RIG-I plays a predominant role in initiating innate immune responses to systemic West Nile virus infection. Mice lacking Caspase-12 display a higher viral burden than wild-type (WT) mice, which is associated with defective type I IFN production (Wang et al, 2010). Notably, TRIM25-dependent RIG-I polyubiquitination and the enhancing effects of TRIM25 on RIG-I-mediated IFNβ promoter activity are decreased in cells lacking Caspase-12. Thus, Caspase-12 facilitates TRIM25 function, although the underlying molecular mechanism is still unclear. Although TRIM25-mediated K63-linked polyubiquitination of RIG-I is a prerequisite for triggering antiviral responses, RIG-I can also undergo ISGylation, which leads to the termination of type I IFN production, and it was reported that TRIM25 exhibits E3 ISG15 ligase activity, but it is not clear whether TRIM25 mediates RIG-I ISGylation (Kim et al, 2008). Thus, RIG-I is likely to be regulated by two different modes of modifications.
REGULATION OF STING BY TRIM56
TRIM56 was identified as a molecule whose overexpression enhances dsDNA-induced IFNβ promoter activation (Tsuchida et al, 2010). This enhancement requires the RING domain. TRIM56 associates with STING and overexpression of both proteins together induces remarkable activation of the IFNβ promoter. Notably, TRIM56 is able to target STING lysine 150 for K63-linked polyubiquitination, and this modification is required for STING dimerization. Mutation of this lysine residue in STING abolishes TBK1 recruitment and IFNβ promoter activation. Therefore, TRIM56-mediated STING ubiquitination is required for STING dimerization and subsequent TBK1 activation (Fig 3). Given that TRIM56 is IFN-inducible, it facilitates dsDNA-triggered signalling via modification of STING.
REGULATION OF ANTIVIRAL SIGNALLING PATHWAYS BY TRIM23
Two studies have suggested that TRIM23 positively regulates NF-κB activation. Specifically, it was reported that TRIM23 modulates the function of NEMO and has a unique property in that it can mediate polyubiquitination of NEMO that is linked to K27 (Arimoto et al, 2010). This modification is important for NF-κB activation downstream of RLR and TLR3, which recognizes viral dsRNA (Figs 2 and 3). NEMO was reported to regulate IRF3 activation during virus infection (Zhao et al, 2007), and TRIM23-mediated NEMO ubiquitination is also involved in IRF3 activation. Thus, TRIM23 contributes to virus infection-triggered NF-κB and IRF3 activation by targeting NEMO.
UL144, a gene product of HCMV, was shown to activate NF-κB and induce the expression of the C–C motif chemokine 22 (Ccl22) using a TRAF6-dependent signalling pathway. This induction subverts the host antiviral responses. Poole et al demonstrated that UL144-dependent NF-κB activation requires TRIM23 (Poole et al, 2009). TRIM23 directly associates with UL144 and further enhances the interaction of TRAF6 with UL144, resulting in increased NF-κB activation. However, TRIM23 is unlikely to be involved in RLR signalling pathways for NF-κB activation. Thus, HCMV may specifically provoke TRIM23 activity for the induction of Ccl22.
REGULATION OF IRFS BY TRIM21
TRIM21 is an IFN-inducible TRIM that is present as an autoantigen in the sera of autoimmune patients, such as Sjögren syndrome and systemic lupus erythematosus patients, suggesting its role in autoimmunity. In vitro experiments have indicated that TRIM21 positively or negatively regulates IRF3 activation (Figs 2 and 3). Yang et al demonstrated that TRIM21 interacts with PIN1, a negative regulator of antiviral responses that mediates the ubiquitination and degradation of IRF3 during virus infection (Yang et al, 2009). TRIM21 prevents the interaction between IRF3 and PIN1 and increases the stability of IRF3 to enhance type I IFN induction. It is notable that the RING domain is dispensable for the TRIM21 interaction with PIN1, suggesting that ubiquitination is not involved in TRIM21-mediated IRF3 stabilization. In contrast, Higgs et al showed that TRIM21 has an opposite role (Higgs et al, 2008). They found that TRIM21 interacts with IRF3 and promotes the ubiquitination and degradation of IRF3 to limit IFNβ induction downstream of TLR3, TLR4 and RIG-I (Figs 2 and 3). At present, the reasons underlying the differences between these two studies are unclear.
IRF7 has a similar structure to IRF3, and its activation is also controlled by phosphorylation. IRF7 plays an essential role in type I IFN induction by pDCs, which predominantly recognize viruses through TLR7 and TLR9. These TLRs utilize MyD88 to recruit IRF7 (Kawai et al, 2004). Higgs et al reported that TRIM21 negatively regulates IRF7 function (Higgs et al, 2010). It interacts with IRF7 and promotes ubiquitin–proteasome-mediated degradation of IRF7 to limit IFNα production in response to TLR7 and TLR9 ligands (Fig 2). Young et al also reported that TRIM21 negatively regulates IRF7 function via proteasome-dependent degradation (Young et al, 2011). They found that TRIM21 interacts with Fas-associated protein with death domain (FADD), which was reported to participate in IRF7 activation during RLR signalling, and that this interaction enhances the TRIM21 ubiquitin ligase activity to promote the ubiquitination and subsequent degradation of IRF7 (Fig 3). Thus, TRIM21 negatively regulates IRF7 in TLR7, TLR9 and RLR signalling pathways.
In addition to the regulation of type I IFN, TRIM21 was reported to regulate IRF8 function (Figs 2 and 3). IRF8 plays an essential role in the development and function of macrophages, DCs and B cells, and is required for the induction of cytokines such as IL-12p40 and type I IFNs during TLR signalling and virus infection. Kong et al purified TRIM21 as a protein that associates with IRF8 in IFNγ- and TLR9-activated macrophages (Kong et al, 2007). TRIM21 promotes K63-linked polyubiquitination of IRF8 and enhances the induction of IL-12p40.
The murine TRIM21 gene was disrupted by two independent groups, producing markedly different phenotypes. Espinosa et al reported that TRIM21-deficient mice display no overt abnormal phenotypes (Espinosa et al, 2009). However, these mice spontaneously exhibit severe dermatitis in response to ear tag injuries. Moreover, these mice develop an autoimmune phenotype characterized by hypergammaglobulinemia and autoantibodies to DNA, associated with upregulation of the inflammatory cytokines IL-6, IL-12p40 and IL-17, which are involved in Th17 cell differentiation by draining lymph node cells. TRIM21-deficient mice consistently show decreased ubiquitination of IRF3, IRF5 and IRF8 (Figs 2 and 3). Therefore, TRIM21 prevents the development of local injury-initiated autoimmunity by negatively regulating IRF activity in vivo. In contrast, Yoshimi et al showed that the cytokine production by B cells, T cells and DCs derived from TRIM21-deficient mice is comparable to that by the corresponding cells from WT mice (Yoshimi et al, 2009). However, only embryonic fibroblast cells derived from TRIM21-deficient mice produce higher cytokine levels than WT cells, and this is caused by increased NF-κB DNA-binding activity. Although the reasons why these two studies produced different phenotypes remain unclear, it is possible that differences in the knockout strategies or the environmental conditions for the mouse colonies affected the phenotypes.
REGULATION OF ACTIVATION OF CANONICAL AND NON-CANONICAL IKKS BY TRIM27 AND TRIM21
Overexpression of TRIM27/RFP1 suppresses IL-1-, virus infection- and TNF-induced cytokine production. The protein binds IKKα and IKKβ to suppress their catalytic activities (Zha et al, 2006). In addition, TRIM27 binds IKKi and TBK1 to suppress IRF3 activation after virus infection. Therefore, TRIM27 negatively regulates antiviral and inflammatory responses by targeting all the IKKs (Figs 2 and 3). Since the RING domain of TRIM27 is dispensable for the inhibition of NF-κB and IRF3, the E3 ligase activity is unlikely to be required in its function. Wada et al reported that TRIM21 conjugates a monoubiquitin to IKKβ. Monoubiquitinated IKKβ is then delivered to the autophagosome for degradation (Wada et al, 2009; Figs 2 and 3). Thus, TRIM21 may negatively regulate IKKβ-mediated NF-κB activation through autophagy.
ACTIVATION OF NF-κB BY TRIM20, TRIM13, TRIM38 AND TRIM32
FMF is an autoinflammatory disease caused by mutations in the MEFV (Mediterranean fever) gene encoding TRIM20 (pyrin). It is speculated that FMF-associated mutations of TRIM20 may lead to activation of NF-κB as well as caspase-1-dependent IL-1β production pathways. In this regard, it was reported that TRIM20 is cleaved by caspase-1 and that the cleaved fragment of TRIM20 interacts with IκBα and the RelA subunit of NF-κB (Chae et al, 2008). The interaction of cleaved TRIM20 with RelA enhances RelA nuclear translocation, whereas, the interaction with IκBα supports IκBα degradation, resulting in increased NF-κB activation. Notably, TRIM20 variants with FMF-associated mutations are cleaved more efficiently than WT TRIM20 by caspase-1, and FMF patients display increased FMF fragments and IκBα degradation.
TRIM13/RFP2 contains a transmembrane domain and is localized in the ER and nucleus (Lerner et al, 2007). Transfection studies indicated that TRIM13 overexpression activates the NF-κB promoter (Matsuda et al, 2003). TRIM38/RoRet overexpression was also reported to activate NF-κB-driven transcription (Matsuda et al, 2003). However, the physiological roles of these TRIMs in innate immunity remain unclear.
TRIM32 is overexpressed in skin cancer cells. TRIM32 was found to target PIASy, an inhibitor of NF-κB, for ubiquitination and degradation, thereby, allowing NF-κB activation (Albor et al, 2006). This pathway is linked to the NF-κB-dependent upregulation of anti-apoptotic genes induced by UV-B radiation or TNFα. Consequently, in cells overexpressing TRIM32, PIASy is degraded and NF-κB is enhanced, thus promoting cancer formation by preventing apoptosis. A recent report showed that PIASy has the ability to suppress virus infection-induced type I IFN production through an unknown mechanism, suggesting the possibility that TRIM32 facilitates virus infection-induced type I IFN production pathways by targeting PIASy for degradation (Kubota et al, 2011).
SENSING RETROVIRUS AND ACTIVATION OF INNATE IMMUNE SIGNALLING BY TRIM5
TRIM5 is known to restrict infection of retroviruses such as HIV-1 by binding to the viral capsid and inducing premature uncoating. Petrel et al recently showed that TRIM5 has a role in eliciting inflammatory responses to retroviral capsids (Fig 4). Overexpression of human TRIM5 activates signalling pathways that lead to the activation of both NF-κB and AP-1, and conversely, TRIM5 knockdown decreases the expression of NF-κB- and AP1-dependent genes involved in inflammatory responses (Pertel et al, 2011). TRIM5 was found to bind to the TAK1 complex (TAK1, TAB2 and TAB3) and E2 ubiquitin conjugation enzymes UBC13 and UEV1A, which promote the synthesis of unattached ubiquitin chains linked to K63. This results in the activation of TAK1 and subsequent induction of the expression of NF-κB- and AP1-dependent genes. Interestingly, TRIM5 binding to the HIV-1 capsid lattice enhances TRIM5 E3 ubiquitin ligase activity. Thus, TRIM5 acts as a PRR that recognizes the retrovirus capsid and activates the TAK-1-dependent inflammatory signalling pathways.
Figure 4. Recognition of retrovirus and activation of NF-κB by TRIM5.
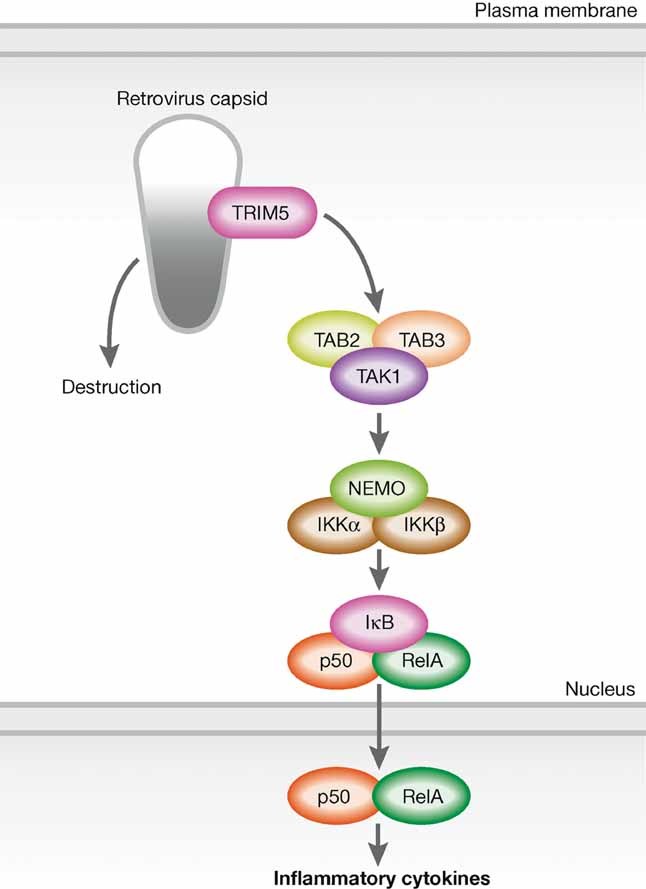
TRIM5 recognizes retrovirus capsid and promotes the synthesis of unanchored ubiquitin chains linked to K63. These chains trigger a proinflammatory response by activating the TAK1 kinase complex. Binding TRIM5 to the capsid causes activation of TRIM5 E3 ubiquitin ligase activity as well as destruction of capsid to prevent infections.
CONCLUSIONS
It is now clear that members of the TRIM family of proteins exhibit antiviral activity by modulating not only viral proteins but also cellular proteins involved in innate immune signalling pathways. Individual TRIM proteins regulate innate immune signalling pathways through distinct mechanisms. TRIM21 influences multiple IRF activities while TRIM27 suppresses IRF3 and NF-κB by targeting all the IKKs. TRIM25 specifically targets RIG-I for activation of antiviral signalling pathways, and TRIM56 facilitates DNA-triggered signalling pathways. TRIM30α dampens TLR4 signalling by targeting the TAK1 complex. It is noteworthy that these TRIMs exhibit distinct E3 ligase activities. K63-linked ubiquitination is promoted by TRIM25 and TRIM56 while K48-linked ubiquitination is triggered by TRIM21. TRIM27 induces monoubiquitination, and TRIM23 promotes K27-linked ubiquitination. TRIM5 mediates ubiquitination of TAK1 during retrovirus infection. Notably, the E3 ligase activity is not always linked to the antiviral activity of TRIMs. The C-terminal SPRY domain of TRIM5a is a prerequisite for restricting HIV-1 replication. TRIM27-mediated suppression of NF-κB and IRF3 activities does not require the RING domain. TRIM30α induces TAB2 and TAB3 degradation in a lysosome-dependent manner rather than through conventional proteasome pathways. At present, however, our knowledge about the functions of TRIMs is limited. Future characterization of all the TRIM proteins, that is, the identification of associated molecules and the generation of knockout mice, will be required to elucidate the whole picture of the TRIM protein functions with regard to innate immune responses. Moreover, the development of drugs that can control the activities of TRIMs will lead to new strategies for infectious, inflammatory and autoimmune diseases in which aberrant PRR signalling is involved.
Pending issues
Studies on the in vivo function of each TRIM protein in antiviral immune responses.
Analysis of genetic mutations or polymorphisms of each TRIM gene in patients of autoimmunity or immunodeficiency.
Studies to identify substrates for each TRIM protein.
Acknowledgments
The authors declare that they have no conflict of interest.
REFERENCES
- Albor A, El-Hizawi S, Horn EJ, Laederich M, Frosk P, Wrogemann K, Kulesz-Martin M. The interaction of Piasy with Trim32, an E3-ubiquitin ligase mutated in limb-girdle muscular dystrophy type 2H, promotes Piasy degradation and regulates UVB-induced keratinocyte apoptosis through NFkappaB. J Biol Chem. 2006;281:25850–25866. doi: 10.1074/jbc.M601655200. [DOI] [PubMed] [Google Scholar]
- Arimoto K, Funami K, Saeki Y, Tanaka K, Okawa K, Takeuchi O, Akira S, Murakami Y, Shimotohno K. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc Natl Acad Sci USA. 2010;107:15856–15861. doi: 10.1073/pnas.1004621107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avela K, Lipsanen-Nyman M, Idanheimo N, Seemanova E, Rosengren S, Makela TP, Perheentupa J, Chapelle AD, Lehesjoki AE. Gene encoding a new RING-B-box-coiled-coil protein is mutated in mulibrey nanism. Nat Genet. 2000;25:298–301. doi: 10.1038/77053. [DOI] [PubMed] [Google Scholar]
- Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- Barber GN. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol. 2011;23:10–20. doi: 10.1016/j.coi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind F, Ablasser A, Bartok E, Kim S, Schmid-Burgk J, Cavlar T, Hornung V. Inflammasomes: current understanding and open questions. Cell Mol Life Sci. 2011;68:765–783. doi: 10.1007/s00018-010-0567-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Chetrit E, Chan EK, Sullivan KF, Tan EM. A 52-kD protein is a novel component of the SS-A/Ro antigenic particle. J Exp Med. 1988;167:1560–1571. doi: 10.1084/jem.167.5.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- Billaut-Mulot O, Cocude C, Kolesnitchenko V, Truong MJ, Chan EK, Hachula E, de la Tribonniere X, Capron A, Bahr GM. SS-56, a novel cellular target of autoantibody responses in Sjogren syndrome and systemic lupus erythematosus. J Clin Invest. 2001;108:861–869. doi: 10.1172/JCI13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius AL, Beutler B. Intracellular Toll-like receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- Bullrich F, Fujii H, Calin G, Mabuchi H, Negrini M, Pekarsky Y, Rassenti L, Alder H, Reed JC, Keating MJ, et al. Characterization of the 13q14 tumor suppressor locus in CLL: identification of ALT1, an alternative splice variant of the LEU2 gene. Cancer Res. 2001;61:6640–6648. [PubMed] [Google Scholar]
- Campbell EM, Dodding MP, Yap MW, Wu X, Gallois-Montbrun S, Malim MH, Stoye JP, Hope TJ. TRIM5 alpha cytoplasmic bodies are highly dynamic structures. Mol Biol Cell. 2007;18:2102–2111. doi: 10.1091/mbc.E06-12-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthagena L, Bergamaschi A, Luna JM, David A, Uchil PD, Margottin-Goguet F, Mothes W, Hazan U, Transy C, Pancino G, et al. Human TRIM gene expression in response to interferons. PLoS One. 2009;4:e4894. doi: 10.1371/journal.pone.0004894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae JJ, Wood G, Richard K, Jaffe H, Colburn NT, Masters SL, Gumucio DL, Shoham NG, Kastner DL. The familial Mediterranean fever protein, pyrin, is cleaved by caspase-1 and activates NF-kappaB through its N-terminal fragment. Blood. 2008;112:1794–1803. doi: 10.1182/blood-2008-01-134932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang AP, Beck JS, Yen HJ, Tayeh MK, Scheetz TE, Swiderski RE, Nishimura DY, Braun TA, Kim KY, Huang J, et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet–Biedl syndrome gene (BBS11) Proc Natl Acad Sci USA. 2006;103:6287–6292. doi: 10.1073/pnas.0600158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de The H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 1991;66:675–684. doi: 10.1016/0092-8674(91)90113-d. [DOI] [PubMed] [Google Scholar]
- Eldin P, Papon L, Oteiza A, Brocchi E, Lawson TG, Mechti N. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J Gen Virol. 2009;90:536–545. doi: 10.1099/vir.0.006288-0. [DOI] [PubMed] [Google Scholar]
- Espinosa A, Dardalhon V, Brauner S, Ambrosi A, Higgs R, Quintana FJ, Sjostrand M, Eloranta ML, Ni Gabhann J, et al. Loss of the lupus autoantigen Ro52/Trim21 induces tissue inflammation and systemic autoimmunity by disregulating the IL-23-Th17 pathway. J Exp Med. 2009;206:1661–1671. doi: 10.1084/jem.20090585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- Frosk P, Weiler T, Nylen E, Sudha T, Greenberg CR, Morgan K, Fujiwara TM, Wrogemann K. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am J Hum Genet. 2002;70:663–672. doi: 10.1086/339083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- Gack MU, Albrecht RA, Urano T, Inn KS, Huang IC, Carnero E, Farzan M, Inoue S, Jung JU, Garcia-Sastre A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009;5:439–449. doi: 10.1016/j.chom.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Duan Z, Xu W, Xiong S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology. 2009;50:424–433. doi: 10.1002/hep.23011. [DOI] [PubMed] [Google Scholar]
- Geoffroy MC, Chelbi-Alix MK. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res. 2011;31:145–158. doi: 10.1089/jir.2010.0111. [DOI] [PubMed] [Google Scholar]
- Gotoh K, Tanaka Y, Nishikimi A, Nakamura R, Yamada H, Maeda N, Ishikawa T, Hoshino K, Uruno T, Cao Q, et al. Selective control of type I IFN induction by the Rac activator DOCK2 during TLR-mediated plasmacytoid dendritic cell activation. J Exp Med. 2010;207:721–730. doi: 10.1084/jem.20091776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas AL. Linear polyubiquitylation: the missing link in NF-kappaB signalling. Nat Cell Biol. 2009;11:116–118. doi: 10.1038/ncb0209-116. [DOI] [PubMed] [Google Scholar]
- Hasegawa N, Iwashita T, Asai N, Murakami H, Iwata Y, Isomura T, Goto H, Hayakawa T, Takahashi M. A RING finger motif regulates transforming activity of the rfp/ret fusion gene. Biochem Biophys Res Commun. 1996;225:627–631. doi: 10.1006/bbrc.1996.1221. [DOI] [PubMed] [Google Scholar]
- Higgs R, Ní Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol. 2008;181:1780–1786. doi: 10.4049/jimmunol.181.3.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs R, Lazzari E, Wynne C, Ni Gabhann J, Espinosa A, Wahren-Herlenius M, Jefferies CA. Self-protection from anti-viral responses—Ro52 promotes degradation of the transcription factor IRF7 downstream of the viral Toll-like receptors. PLoS One. 2010;5:e11776. doi: 10.1371/journal.pone.0011776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]
- Jeon YJ, Yoo HM, Chung CH. ISG15 and immune diseases. Biochim Biophys Acta. 2010;1802:485–496. doi: 10.1016/j.bbadis.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- Keeble AH, Khan Z, Forster A, James LC. TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved. Proc Natl Acad Sci USA. 2008;105:6045–6050. doi: 10.1073/pnas.0800159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Hwang SY, Imaizumi T, Yoo JY. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J Virol. 2008;82:1474–1483. doi: 10.1128/JVI.01650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong HJ, Anderson DE, Lee CH, Jang MK, Tamura T, Tailor P, Cho HK, Cheong J, Xiong H, Morse HCr, et al. Autoantigen Ro52 is an interferon inducible E3 ligase that ubiquitinates IRF-8 and enhances cytokine expression in macrophages. J Immunol. 2007;179:26–30. doi: 10.4049/jimmunol.179.1.26. [DOI] [PubMed] [Google Scholar]
- Kubota T, Matsuoka M, Xu S, Otsuki N, Takeda M, Kato A, Ozato K. Piasy inhibits virus-induced and interferon-stimulated transcription through distinct mechanisms. J Biol Chem. 2011;286:8165–8175. doi: 10.1074/jbc.M110.195255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin B, Zechel C, Garnier JM, Lutz Y, Tora L, Pierrat P, Heery D, Gronemeyer H, Chambon P, Losson R. The N-terminal part of TIF1, a putative mediator of the ligand-dependent activation function (AF-2) of nuclear receptors, is fused to B-raf in the oncogenic protein T18. EMBO J. 1995;14:2020–2033. doi: 10.1002/j.1460-2075.1995.tb07194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner M, Corcoran M, Cepeda D, Nielsen ML, Zubarev R, Ponten F, Uhlen M, Hober S, Grander D, Sangfelt O. The RBCC gene RFP2 (Leu5) encodes a novel transmembrane E3 ubiquitin ligase involved in ERAD. Mol Biol Cell. 2007;18:1670–1682. doi: 10.1091/mbc.E06-03-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chen ZJ. Expanding role of ubiquitination in NF-kappaB signaling. Cell Res. 2011;21:6–21. doi: 10.1038/cr.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21) Proc Natl Acad Sci USA. 2010;107:19985–19990. doi: 10.1073/pnas.1014074107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda A, Suzuki Y, Honda G, Muramatsu S, Matsuzaki O, Nagano Y, Doi T, Shimotohno K, Harada T, Nishida E, et al. Large-scale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene. 2003;22:3307–3318. doi: 10.1038/sj.onc.1206406. [DOI] [PubMed] [Google Scholar]
- Meroni G, Diez-Roux G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays. 2005;27:1147–1157. doi: 10.1002/bies.20304. [DOI] [PubMed] [Google Scholar]
- Nakasato N, Ikeda K, Urano T, Horie-Inoue K, Takeda S, Inoue S. A ubiquitin E3 ligase Efp is up-regulated by interferons and conjugated with ISG15. Biochem Biophys Res Commun. 2006;351:540–546. doi: 10.1016/j.bbrc.2006.10.061. [DOI] [PubMed] [Google Scholar]
- Nisole S, Stoye JP, Saib A. TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. 2005;3:799–808. doi: 10.1038/nrmicro1248. [DOI] [PubMed] [Google Scholar]
- Ozato K, Shin DM, Chang TH, Morse HC., III TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8:849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 2011;472:361–365. doi: 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole E, Groves I, MacDonald A, Pang Y, Alcami A, Sinclair J. Identification of TRIM23 as a cofactor involved in the regulation of NF-kappaB by human cytomegalovirus. J Virol. 2009;83:3581–3590. doi: 10.1128/JVI.02072-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, et al. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet. 1997;17:285–291. doi: 10.1038/ng1197-285. [DOI] [PubMed] [Google Scholar]
- Rajsbaum R, Stoye JP, O'Garra A. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur J Immunol. 2008;38:619–630. doi: 10.1002/eji.200737916. [DOI] [PubMed] [Google Scholar]
- Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, et al. The tripartite motif family identifies cell compartments. EMBO J. 2001;20:2140–2151. doi: 10.1093/emboj/20.9.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone V, Palmieri M, Passamano L, Piluso G, Meroni G, Politano L, Nigro V. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2H. Hum Mutat. 2008;29:240–247. doi: 10.1002/humu.20633. [DOI] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Cairo S, Fontanella B, Ballabio A, Meroni G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol Biol. 2008;8:225. doi: 10.1186/1471-2148-8-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, Takeuchi O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci USA. 2010;107:1512–1517. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Deng W, Bi E, Mao K, Ji Y, Lin G, Wu X, Tao Z, Li Z, Cai X, et al. TRIM30 alpha negatively regulates TLR-mediated NF-kappa B activation by targeting TAB2 and TAB3 for degradation. Nat Immunol. 2008;9:369–377. doi: 10.1038/ni1577. [DOI] [PubMed] [Google Scholar]
- Short KM, Cox TC. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J Biol Chem. 2006;281:8970–8980. doi: 10.1074/jbc.M512755200. [DOI] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Tareen SU, Emerman M. Human Trim5alpha has additional activities that are uncoupled from retroviral capsid recognition. Virology. 2011;409:113–120. doi: 10.1016/j.virol.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity. 2010;33:765–776. doi: 10.1016/j.immuni.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Uchil PD, Quinlan BD, Chan WT, Luna JM, Mothes W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 2008;4:e16. doi: 10.1371/journal.ppat.0040016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada K, Niida M, Tanaka M, Kamitani T. Ro52-mediated monoubiquitination of IKK{beta} down-regulates NF-{kappa}B signalling. J Biochem. 2009;146:821–832. doi: 10.1093/jb/mvp127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Arjona A, Zhang Y, Sultana H, Dai J, Yang L, LeBlanc PM, Doiron K, Saleh M, Fikrig E. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat Immunol. 2010;11:912–919. doi: 10.1038/ni.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Liu B, Wang N, Lee YM, Liu C, Li K. TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J Virol. 2011;85:3733–3745. doi: 10.1128/JVI.02546-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, Goff SP. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell. 2007;131:46–57. doi: 10.1016/j.cell.2007.07.026. [DOI] [PubMed] [Google Scholar]
- Yang K, Shi HX, Liu XY, Shan YF, Wei B, Chen S, Wang C. TRIM21 is essential to sustain IFN regulatory factor 3 activation during antiviral response. J Immunol. 2009;182:3782–3792. doi: 10.4049/jimmunol.0803126. [DOI] [PubMed] [Google Scholar]
- Yap MW, Nisole S, Lynch C, Stoye JP. Trim5 protein restricts both HIV-1 and murine leukemia virus. Proc Natl Acad Sci USA. 2004;101:10786–10791. doi: 10.1073/pnas.0402876101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol. 2010;20:4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- Yoshimi R, Chang TH, Wang H, Atsumi T, Morse HC, III, Ozato K. Gene disruption study reveals a nonredundant role for TRIM21/Ro52 in NF-kappaB-dependent cytokine expression in fibroblasts. J Immunol. 2009;182:7527–7538. doi: 10.4049/jimmunol.0804121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JA, Sermwittayawong D, Kim HJ, Nandu S, An N, Erdjument-Bromage H, Tempst P, Coscoy L, Winoto A. Fas-associated death domain (FADD) and the E3 ubiquitin-protein ligase TRIM21 interact to negatively regulate virus-induced interferon production. J Biol Chem. 2011;286:6521–6531. doi: 10.1074/jbc.M110.172288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J, Han KJ, Xu LG, He W, Zhou Q, Chen D, Zhai Z, Shu HB. The Ret finger protein inhibits signaling mediated by the noncanonical and canonical IkappaB kinase family members. J Immunol. 2006;176:1072–1080. doi: 10.4049/jimmunol.176.2.1072. [DOI] [PubMed] [Google Scholar]
- Zhao T, Yang L, Sun Q, Arguello M, Ballard DW, Hiscott J, Lin R. The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat Immunol. 2007;8:592–600. doi: 10.1038/ni1465. [DOI] [PubMed] [Google Scholar]
- Zou W, Zhang DE. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J Biol Chem. 2006;281:3989–3994. doi: 10.1074/jbc.M510787200. [DOI] [PubMed] [Google Scholar]