

Abstract
Acute and chronic inflammatory disorders are characterized by detrimental cytokine and chemokine expression. Frequently, the chemotactic activity of cytokines depends on a modified N-terminus of the polypeptide. Among those, the N-terminus of monocyte chemoattractant protein 1 (CCL2 and MCP-1) is modified to a pyroglutamate (pE-) residue protecting against degradation in vivo. Here, we show that the N-terminal pE-formation depends on glutaminyl cyclase activity. The pE-residue increases stability against N-terminal degradation by aminopeptidases and improves receptor activation and signal transduction in vitro. Genetic ablation of the glutaminyl cyclase iso-enzymes QC (QPCT) or isoQC (QPCTL) revealed a major role of isoQC for pE1-CCL2 formation and monocyte infiltration. Consistently, administration of QC-inhibitors in inflammatory models, such as thioglycollate-induced peritonitis reduced monocyte infiltration. The pharmacologic efficacy of QC/isoQC-inhibition was assessed in accelerated atherosclerosis in ApoE3*Leiden mice, showing attenuated atherosclerotic pathology following chronic oral treatment. Current strategies targeting CCL2 are mainly based on antibodies or spiegelmers. The application of small, orally available inhibitors of glutaminyl cyclases represents an alternative therapeutic strategy to treat CCL2-driven disorders such as atherosclerosis/restenosis and fibrosis.
Keywords: CCL2, drug development, glutaminyl cyclases, inflammation, pyroglutamate
→See accompanying article http://dx.doi.org/10.1002/emmm.201100161
INTRODUCTION
CCL2 plays a pivotal role in different inflammatory disorders such as atherosclerosis (Charo & Taubman, 2004), fibrosis (Inoshima et al, 2004) and Alzheimer's disease (Galimberti et al, 2006). Upon stimulation, CCL2 is secreted by a variety of different cell types and binds to glycosaminoglycanes of the extracellular matrix building up a chemokine gradient, which is important for the attraction of immune cells (Lau et al, 2004; Proudfoot et al, 2003). Importantly, structure/activity studies revealed, that an intact N-terminus is crucial for the ability of CCL2 to attract monocytes. Artificial elongation or truncation of CCL2 leads to a loss of function, albeit receptor interaction is still observed (Hemmerich et al, 1999; Masure et al, 1995; Proost et al, 1998; Zhang et al, 1994). On the basis of these observations, receptor antagonists were developed, demonstrating disease modification in different animal experiments (Usui et al, 2002; Wada et al, 2004).
A pyroglutamate (pE-) residue constitutes the N-terminus of all human MCPs. N-terminal pE-residues have been described for a number of hormones and secreted proteins, such as thyreotropin-releasing hormone (TRH), gonadotropin-releasing hormone (GnRH) and fibronectin (Awade et al, 1994; Blomback, 1967). Here, the pE-residue confers resistance against degradation by aminopeptidases and—as has been already shown for TRH and GnRH—is important for the receptor interaction (Abraham & Podell, 1981; Morty et al, 2006). An N-terminal blockage by pE has been also described for beta-amyloid (Aβ) peptides deposited in Alzheimer's disease. Also within Aβ, the modification has been linked to increased resistance against degradation and enhanced aggregation propensity (Schilling et al, 2008). Likewise, also for the MCPs a protecting function has been suggested (Van Damme et al, 1999), which, in turn, stabilizes chemotactic activity.
The N-terminal pE is post-translationally formed from a glutaminyl precursor by glutaminyl cyclase (E.C. 2.3.2.5, QPCT, QC; Fischer & Spiess, 1987; Schilling et al, 2003) or iso-glutaminyl cyclase (E.C. 2.3.2.5, QPCTL, isoQC; Cynis et al, 2008). QC and isoQC are closely related single-zinc metalloenzymes, which exhibit nearly identical substrate specificity in vitro (Cynis et al, 2008; Schilling et al, 2003; Stephan et al, 2009). The major difference refers to subcellular localization: QC is secreted from expressing cells; isoQC is a resident enzyme of the golgi complex (Cynis et al, 2008). Inhibition of QC-activity has been shown to prevent pE-peptide formation in mammalian cells (Buchholz et al, 2006; Cynis et al, 2006).
Immature CCL8 (MCP-2), possessing an N-terminal glutamine-proline (Gln-Pro)-motif underwent cleavage by dipeptidylpeptidase 4 (DP4/CD26; Van Coillie et al, 1998). Interestingly, all known human MCPs (CCL2, 7, 8, 13) reveal such a Gln-Pro-motif, rendering the peptides potentially prone to cleavage upon suppression of N-terminal pE-formation.
Consequently, the role of QC-activity for N-terminal pE-formation has been investigated in the present study, applying a combinatorial approach of QC or isoQC knockout as well as the pharmacological inhibition of QC/isoQC to modulate CCL2. By prevention of pE-formation, CCL2 is destabilized and alternative, additional degradation pathways are enabled. The importance of the findings is supported by the impact on pE1-CCL2, monocyte infiltration and alleviation of pathology in several analyzed models. The results might open a field of novel, specific small-molecule anti-inflammatory drugs for the treatment of CCL2-related disorders.
RESULTS
QC and isoQC catalyze the formation of pE1-CCL2 in vitro
According to their sequence, all members of the human MCP-subfamily (CCL2, 7, 8, 13) are putative substrates for QC/isoQC and possess an N-terminal pE-residue in their mature state (Fig 1A). Due to the cyclization of an N-terminal glutaminyl precursor into pE, the positive charge of the N-terminus is lost, which confers resistance against cleavage by aminopeptidases such as DP4. In contrast, the immature variants display cleavage sites for aminopeptidase P (ApP), DP4 or in case of CCL2 also for matrixmetalloproteinase-1 (MMP-1; McQuibban et al, 2002; Fig 1A). To determine the catalytic parameters for QC- and isoQC-catalyzed conversion of Q1-CCL2 into pE1-CCL2, we used a spectrophotometric assay and an evaluation, which relies on the integrated form of the Michaelis–Menten equation. This allows the calculation of KM and kcat from a single reaction. QC and isoQC converted Q1-CCL2 into pE1-CCL2. Thus, these small proteins represent the largest substrates subjected to a quantitative analysis so far. Compared to other substrates, however, CCL2 is rather inefficiently cyclized by QCs showing specificity constants (kcat/KM) of 20 mM−1 s−1 for human QC and 28 mM−1 s−1 for human isoQC (Table 1).
Figure 1. Pyroglutamic acid mediates activity of CCL2.
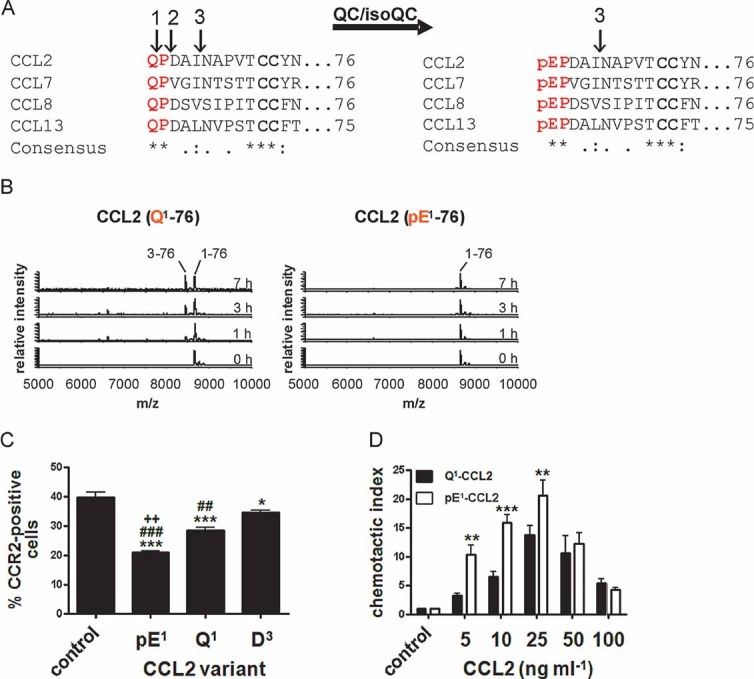
- Immature forms of human MCPs, e.g. Q1-CCL2 display cleavage sites for ApP (1) and DP4 (2). CCL2 can also be processed by MMP-1 (3). All human MCPs are putative substrates of QC/isoQC and they contain in their mature state an N-terminal pE-residue and are protected against aminopeptidase cleavage but not against MMP-1 cleavage.
- N-terminal degradation of human Q1-CCL2 and pE1-CCL2 in human plasma monitored using MALDI-TOF mass spectrometry.
- Internalization of CCR2 from the surface of THP-1 monocytes triggered by CCL2 variants and determined by FACS analysis. (***p < 0.001 and *p < 0.05 vs. control; ###p < 0.001 and ##p < 0.01 vs. D3, ++p < 0.01 vs. Q1, one-way ANOVA followed by Tukey post hoc test, n = 3–8, mean ± SEM).
- Dependence of THP-1 monocyte migration on the concentration of CCL2(Q1-76) and CCL2(pE1-76), assessed using a chamber assay and quantification of cells by FACS analysis (***p < 0.001, **p < 0.01, Q1-CCL2 vs. pE1-CCL2, two-way ANOVA followed by Bonferroni post-test, n = 3–7, mean ± SEM).
Table 1.
Specificity constants for the conversion of human and murine CCL2 by human QC/isoQC and murine QC/isoQC
| Human CCL2 | Murine CCL2 | |
|---|---|---|
| kcat/KM (mM−1 s−1) | kcat/KM (mM−1 s−1) | |
| Human QC | 20 ± 3 | n.d. |
| Human isoQC | 28 ± 3 | n.d. |
| Murine QC | n.d. | 42 ± 8 |
| Murine isoQC | n.d. | 11 ± 0 |
n.d., not determined.
Suppression of pE-formation opens alternative pathways for CCL2 degradation
In order to show, that human CCL2 and its family members CCL7, CCL8 and CCL13 are protected against N-terminal truncation by aminopeptidases, the immature (glutaminyl-) and mature (pyroglutamyl-) recombinant peptides were incubated with human DP4. CCL2(Q1-76) was susceptible to cleavage by recombinant human DP4 leading to a rapid generation of CCL2(D3-76). Formation of CCL2(pE1-76) by recombinant human QC prevented N-terminal truncation. In addition, human CCL7, 8 and 13, which also bear a pE-residue in their mature state, are protected against truncation by DP4. The unblocked N-termini of these peptides are, however, all susceptible to truncation due to their conserved N-terminal Gln-Pro-motif (Fig 1 of Supporting Information). The results were corroborated using ApP (not shown). In addition, the incubation of CCL2 with human plasma proved the presence of competing QC and protease activities within the circulation. Importantly, CCL2(pE1-76) was completely resistant to cleavage (Fig 1B). Hence, the prevention of pE-formation by QC-inhibition has the potential to enhance chemokine degradation in the circulation by suppression of N-terminal maturation and, thereby, opening of alternative degradation pathways not present under normal conditions.
Alternative degradation impairs the bioactivity of CCL2
The functional consequences of the alternative processing of CCL2 were investigated using a CCR2 receptor internalization assay in human THP-1 monocytes. CCL2(pE1-76) was most potent in eliciting CCR2 internalization compared to CCL2(Q1-76) and the DP4-cleavage product CCL2(D3-76; Fig 1C). Furthermore, truncation of CCL2 increases the concentration needed for full receptor internalization (Fig 2 of Supporting Information). In addition, the N-terminal heterogeneity affects the potency of monocyte attraction. Among the different variants, CCL2(pE1-76) exhibited the highest efficacy closely followed by CCL2(Q1-76). In contrast, truncation by DP4 inactivates CCL2 leading to a shift to higher concentrations needed to elicit chemotaxis (Fig 3 of Supporting Information). A direct comparison of CCL2(Q1-76) and CCL2(pE1-76) emphasized the higher potency of CCL2(pE1-76) to attract monocytes at low concentrations (Fig 1D).
A similar influence of the pE-modified N-terminus was also observed with CCL7, 8 and 13 when compared to the respective DP4 cleavage products (Fig 3 of Supporting Information). Thus, a prevention of pE-formation at the N-termini of members of the MCP-family results in destabilization and suppression of the chemotactic function.
IsoQC but not QC knockout alters pE1-CCL2 formation ex vivo and in vivo
The analysis of pE1-CCL2-formation in vitro revealed similar proficiencies of QC and isoQC for conversion of CCL2(Q1-76) (Table 1). To study the role of QC and isoQC for the formation of CCL2(pE1-76) in vivo, primary monocytes/macrophages, neurons and glia cells from QC (QC−/−) knockout mice (Schilling et al, 2011) and newly generated isoQC (isoQC−/−) knockout mice were isolated and stimulated with lipopolysaccharide (LPS). For analysis of the conditioned media, novel enzyme-linked immunosorbent assays (ELISAs) were used, which permit a discrimination of total-CCL2 and CCL2(pE1-76) from human and murine origin (Fig 4 and Table 1 of Supporting Information).
Primary cells isolated from QC−/− mice secreted equal amounts of CCL2(pE1-76) and total-CCL2 into the conditioned medium after LPS-stimulation (Fig 2A). In contrast, a significantly reduced concentration of CCL2(pE1-76) has been observed in conditioned medium of primary cells isolated from isoQC−/− mice after LPS stimulation (Fig 2B).
Figure 2. IsoQC is the major pE1-CCL2 forming enzyme in vivo.
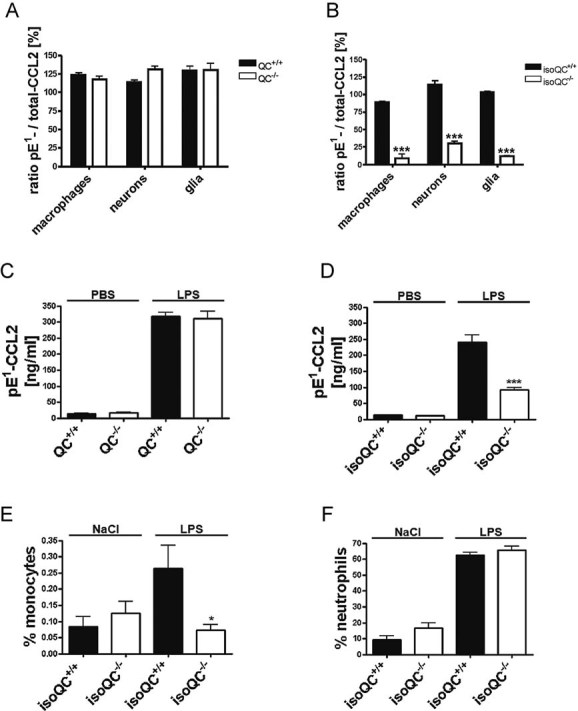
- Analysis of the ratio of total-CCL2 and pE1-CCL2 secreted from LPS-stimulated primary cells isolated from QC−/− mice compared to WT littermates (QC+/+; n = 4–5, mean ± SEM).
- Analysis of the ratio of total-CCL2 and pE1-CCL2 secreted from LPS-stimulated primary cells isolated from isoQC−/− mice compared to WT littermates (isoQC+/+; ***p < 0.001 vs. isoQC+/+, Student's t-test, n = 4, mean ± SEM).
- pE1-CCL2 formation in serum after peripheral injection of LPS in male QC−/− mice compared to WT littermates (QC+/+; n = 6–7, mean ± SEM).
- pE1-CCL2 formation in serum after peripheral injection of LPS in male isoQC−/− mice, compared to WT littermates (QC+/+; ***p < 0.001 vs. isoQC+/+ +LPS, Student's t-test, n = 5–8, mean ± SEM).
- IsoQC-deficiency leads to impaired monocyte recruitment to lungs after intranasal LPS-application in isoQC−/− compared to WT littermates (isoQC+/+; *p < 0.05 vs. isoQC+/+ +LPS, Student's t-test, n = 7–8, mean ± SEM).
- Analysis of neutrophils in bronchoalveolar fluid after intranasal LPS-application in isoQC−/− compared to WT littermates (isoQC+/+; n = 7–8, mean ± SEM).
To study the differential role of QC and isoQC in vivo, we stimulated mice from both knockout lines by a peripheral (intraperitoneal; i.p.) injection of LPS and determined the pE1-CCL2 concentration in serum. As expected, the LPS-injection led to an increase of mature pE1-CCL2 in serum of wild-type (WT) littermates from both lines. While in QC−/− mice alterations of the concentration of CCL2(pE1-76) are similar (Fig 2C), the isoQC−/− mice exhibit significantly less CCL2(pE1-76) compared to isoQC+/+ littermates (Fig 2D), which is in line with the observations from primary cell cultures.
The effect of an isoQC knock out on CCL2 activity was further analyzed in a model of lung inflammation, which is based on an intranasal application of LPS. The differential cell count shows the infiltration of neutrophils and lymphocytes into the alveolar space (Fig 5 of Supporting Information). A difference between isoQC+/+ and isoQC−/− could not be observed. However, when analysing bronchoalveolar lavage fluid (BALF) for infiltrating monocytes using markers 7/4 (Ly6B.2) and Ly6G, isoQC−/− mice showed a significant reduction (Fig 2E), whereas, the number of infiltrating neutrophils remained unaffected (Fig 2F), suggesting a specific effect of isoQC-deficiency on the monocyte infiltration. However, the concentrations of CCL2(pE1-76) within the BALF were below the limit of quantification of the ELISA (not shown).
Therefore, the results were further substantiated in a model of thioglycollate-induced peritonitis, where the CCL2 levels as well as the cell migration could be readily analyzed. The infiltrating monocyte population was determined by fluorescence-activated cell sorting (FACS) analysis using the markers 7/4 (Ly6B.2) and Ly6G. The infiltrating monocyte population shows a high expression of 7/4 (7/4high) and a low expression of Ly6G (Ly6Glow) compared to the infiltrating granulocyte population represented by high expression of 7/4 (7/4high) and Ly6G (Ly6Ghigh; Fig 3A and D). The deficiency in QC did not influence the number of monocytes and granulocytes in the peritoneal lavage fluid after thioglycollate challenge (Fig 3A and B). Accordingly, a difference in total-CCL2 and CCL2(pE1-76) levels could not be detected between QC−/− and QC+/+ mice (Fig 3C). Again, a knockout of isoQC prevented monocyte recruitment (Fig 3D and E), whereas, recruitment of granulocytes remained unaffected (Fig 3E). The specific differences in monocyte recruitment were accompanied by significantly reduced CCL2(pE1-76) concentrations in isoQC−/− mice compared to WT littermates (Fig 3F). Hence, isoQC is apparently the major enzyme for the generation of pE1-CCL2 in vivo.
Figure 3. IsoQC depletion results in impaired monocyte recruitment after thioglycollate challenge.

- Representative FACS analysis showing the infiltrating monocyte population in QC+/+ and QC−/− mice dissected by specific staining.
- Quantification of infiltrating monocytes and granulocytes in QC+/+ mice (black bars) and QC−/− mice (open bars; n = 8–13, mean ± SEM).
- Lavage fluid from (B) was analyzed for total-CCL2 (black bars) and pE1-CCL2 (open bars; n.s., not significant, Student's t-test, mean ± SD).
- Representative FACS analysis showing the infiltrating monocyte population in isoQC+/+ and isoQC−/− mice.
- Quantification of infiltrating monocytes and granulocytes in isoQC+/+ mice (black bars) and isoQC−/− mice (open bars; ***p < 0.001 vs. isoQC+/+ Thio, Student's t-test, n = 10–14, mean ± SEM).
- Lavage fluid from (E) was analyzed for total-CCL2 (black bars) and pE1-CCL2 (open bars; ***p < 0.001 vs. pE1-CCL2 from isoQC+/+ mice, Student's t-test, mean ± SD).
Pharmacological inhibition of QC/isoQC-activity suppresses pE1-CCL2-formation and monocyte infiltration
In order to assess the efficacy of QC/isoQC inhibitors to reduce monocyte infiltration, we selected two compounds, PQ50 and PQ529, from different chemical classes. The compounds are not selective for one isoenzyme and are, therefore, designated as QC/isoQC-inhibitors.
In a first approach, primary murine glia cells were isolated and stimulated using LPS. As expected, the stimulation of the primary cells resulted in a significant increase in CCL2 within the medium. The inhibitor PQ529 decreased the concentration of pE1-CCL2 dose-dependently (Fig 4A). Thereby, the inhibitor led to a slight but significant increase of CCL2 mRNA for the doses of 0.625 and 2.5 µM (Fig 4B), however, PQ529 did not show an effect on CCL2 expression in LPS-stimulated THP-1 cells (Fig 7A of Supporting Information). Similar to treatment of the primary glia cells, PQ529 reduced pE1-CCL2 secreted from murine and human primary macrophages (Fig 6A and B of Supporting Information).
Figure 4. Pharmacological inhibition of QC/isoQC activity in vitro and in vivo reduces pE-CCL2 activity.

- Analysis of total-CCL2 (black bars) and pE1-CCL2 (open bars) after application of varying doses PQ529 to LPS-stimulated primary murine glia cells isolated from C57BL/6J WT mice compared to unstimulated controls (***p < 0.001 vs. pE1-CCL2 (0 µM PQ529), ANOVA followed by Tukey post hoc test, n = 3–4, mean ± SEM).
- Analysis of CCL2 gene expression in LPS-stimulated primary glia cells derived from Fig 4A(*p < 0.05, **p < 0.01 vs. PQ529 0 µM, ANOVA followed by Tukey post hoc test, n = 3–4, mean ± SEM).
- Representative FACS image showing the reduction of infiltrating monocytes after application of PQ529 (30 mg/kg, i.p.).
- Dose-dependent reduction of infiltrating monocytes in absence (black bars) or presence (red bars) of intraperitoneal PQ529 treatment (**p < 0.01 vs. Thio (+), ANOVA followed by Tukey post hoc test, n = 5–6, mean ± SEM, female mice).
- Inhibition of monocyte infiltration after oral application of PQ50 (red bars) and PQ529 (white bar; **p < 0.01 vs. Thio (+), ANOVA followed by Tukey post hoc test, n = 5–6, mean ± SEM, female mice).
In a second approach, we assessed efficacy of the inhibitors PQ50 and PQ529 to suppress monocyte infiltration in vivo, using the thioglycollate-induced peritonitis model. An intraperitoneal application of PQ529 reduced the number of infiltrating monocytes dose-dependently (Fig 4C and D). Moreover, PQ50 and PQ529 were also effective in the model if administered by oral gavage. Both inhibitors reduced the number of monocytes after stimulation with thioglycollate (Fig 4E), without changing the gene expression of CCL2, CCL7, interleukin-6 (IL-6) and tumour-necrosis factor (TNF)-alpha in cells of the peritoneal lavage (Fig 7B–E of Supporting Information). In addition, the level of ICAM1 (Fig 7F of Supporting Information) and CCR2 (not shown) are unchanged suggesting a specific effect on CCL2 maturation. Thus, the N-terminal destabilization of the MCPs by administering small, orally available QC/isoQC-inhibitors has potential to directly affect the recruitment of monocytes in vivo, which might have disease-modifying potential in CCL2-mediated disorders such as atherosclerosis/restenosis.
QC/isoQC-inhibitors attenuate atherosclerotic pathology in a model of accelerated atherosclerosis
Because CCL2 has been implicated in development of atherosclerotic lesions and restenosis, we aimed at testing QC/isoQC-inhibitors in a cuff-model of accelerated atherosclerosis in ApoE3*Leiden mice (Lardenoye et al, 2000). Notably, a characterization of isoQC expression in human tissue revealed a ubiquitous presence of isoQC mRNA (Fig 5). Highest expression was found in lung, liver and thyroid tissue. Likewise, polymerase chain reaction (after reverse transcription) (RT-PCR) proves isoQC expression in human atherosclerotic blood vessels (Arteria femoralis superficialis), which have been isolated during vascular surgery (Fig 8 of Supporting Information). These results were corroborated by Western blot analysis, which identified the isoQC protein in these atherosclerotic vessels (Fig 5 inset). Thus, the expression pattern of isoQC is consistent with the appearance of pathological tissue changes.
Figure 5. IsoQC is ubiquitously expressed in human tissue.
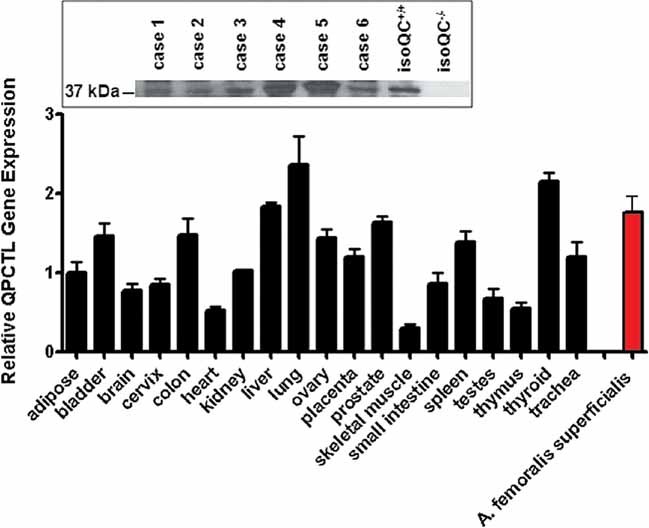
RT-PCR analysis of various human tissues in comparison to isoQC expression in atherosclerotic vessel segments. The inset shows a Western blot analysis of six cases (5 male, 1 female, mean age 53.8 years) revealing the expression of isoQC in atherosclerotic segments of the Arteria femoralis superficialis compared to expression in livers of isoQC+/+ and isoQC−/− mice as internal standard.
Hence, we investigated the therapeutic potential of PQ50 to suppress pE1-CCL2 formation and ameliorate pathological changes in ApoE3*Leiden mice following cuff placement. Two days after surgery, a profound reduction of total adhering cells (45%) and monocytes (67%) at the cuffed vessel segments could be observed in the inhibitor-treated group (Fig 6A). In addition, CCL2 expression was down-regulated in the media and the intima. Analysis of the relative area of the cross-sections positive for CCL2 revealed reductions by 52 and 36% in the media and in the intima, respectively (Fig 6B). Apparently, the reduced adherence of monocytes after vascular surgery resulted in a significant amelioration of lumen stenosis (Fig 6C) and neointimal area (Fig 6D) 2 weeks after surgery. The inhibitor reduced the area of the media (Fig 6D inset) significantly. The overall CCL2 expression is lower compared to the early time point, the CCL2-immunopositive area did not differ between treated and untreated animals (Fig 9F of Supporting Information). Further analysis revealed tendencies to improvements in the intima/media ratio and remaining lumen (Fig 9A and B of Supporting Information). The treatment did not affect the total area within the outer diameter of the vessel segment (Fig 9C of Supporting Information), the area of the interna and the number of foam cells (Fig 9D and E of Supporting Information). Thus, inhibition of QC/isoQC reduced monocyte infiltration has the potential to attenuate downstream atherosclerotic pathology.
Figure 6. QC/isoQC-inhibition alleviates atherosclerotic pathology in a model of cuff-induced accelerated atherosclerosis.
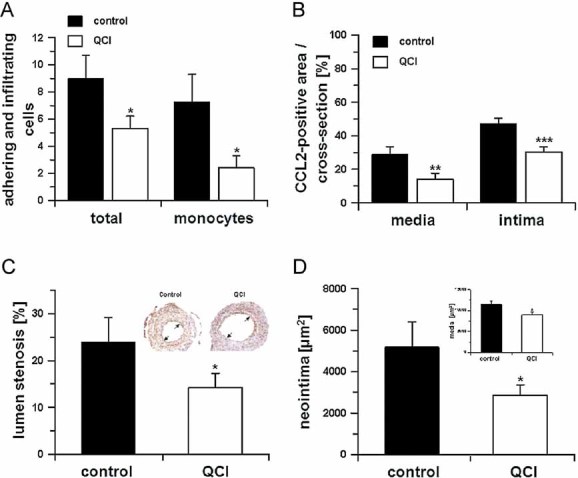
- Monocyte adhesion and total adhering cells 2 days after cuff placement in absence (black bars) or presence (open bars) of PQ50 (QCI) treatment (*p < 0.05 vs. control, Student's t-test, n = 5, mean ± SD).
- In addition, the CCL2-positive area was calculated in cross-sections within the media and neointima in absence (black bars) and presence (open bars) of PQ50 (QCI) treatment (**p < 0.01, ***p < 0.001 vs. control. Student's t-test, n = 5, mean ± SD).
- Morphometric analysis of cuffed vessel segments shows a reduction in the degree of lumen stenosis (*p < 0.05 vs. control, Student's t-test, n = 10, mean ± SD). Inset: Example for lumen stenosis after 2 weeks (black arrows).
- Neointima formation and media thickness (inset) of the cuffed vessel segments of mice sacrificed after 2 weeks, treated in absence (black bars) and presence (open bars) of QC-inhibitor PQ50 (QCI; *p < 0.05 vs. control, Student's t-test, n = 10, mean ± SD).
DISCUSSION
CCL2 is a key mediator for the infiltration of monocytes to inflamed tissues and plays a pivotal role in several inflammatory disorders, such as atherosclerosis (Boring et al, 1998; Gosling et al, 1999; Mori et al, 2002), cancer (Fridlender et al, 2010), pancreatitis (Marra, 2005), Alzheimer's disease (Galimberti et al, 2006) or multiple sclerosis (Gerard & Rollins, 2001). With regard to therapeutic interventions in those chemokine-mediated processes, it has been suggested, that targeting the ligands rather than the receptor generates effective compounds. Such approaches comprise, among others, the disruption of CCL5–CXCL4 ligand interactions (Koenen et al, 2009) or dominant negative ligand antagonism for CCL2 (Ni et al, 2001) and RANTES (Proudfoot et al, 1996) or the suppression of the generation of chemokine gradients by interference with the glycosaminoglycan binding sites within CCL2.
The results presented here introduce a novel mechanism to reduce the activity of CCL2 under different inflammatory conditions (Fig 7). The principle is to interfere with a post-translational modification, which obviously contributes to efficient receptor interaction and stabilization of chemokines towards degradation by alternative proteolytic pathways. In contrast to approaches such as CCL2-binding by mirror-image RNA oligonucleotides (Eulberg & Klussmann, 2003) or bindarit as inhibitors of CCL2 synthesis (Bhatia et al, 2005), the here presented approach can take advantage of small, orally available molecules which inhibit QC/isoQC-activity to modulate N-terminal pE-formation and, in turn, CCL2-activity. The suppression of the N-terminal pE-formation leads to diminished receptor activation and, importantly, the chemokine is rapidly cleaved by enzymes such as DP4, ApP and MMP-1. Thus, the concept is particularly based on expression of the potential degrading enzymes at the sites of inflammation, e.g. the vessel wall or circulation.
Figure 7. The role of isoQC for regulating monocyte recruitment in CCL2-driven disorders.
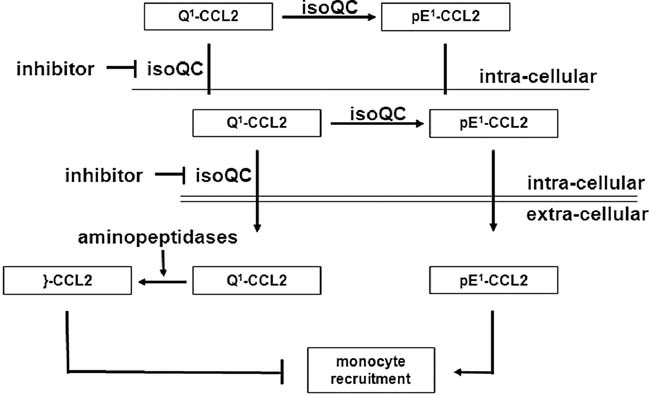
IsoQC is the major enzyme for maturation of CCL2 in vivo ensuring stability and receptor activation leading to monocyte migration. Application of small molecules, which inhibit isoQC provokes the secretion of immature forms (Q1-CCL2) prone to aminopeptidase cleavage generating truncated forms (}-CCL2), devoid of bioactivity under physiological conditions.
The finding that isoQC is the sole enzyme for conversion of Q1-CCL2 into pE1-CCL2 in vivo was unexpected. QC and isoQC are ubiquitously expressed throughout the body and the specificity of both enzymes for various substrates is almost identical (Stephan et al, 2009). However, QC shows a high expression in brain tissues and is secreted with putative substrates, such as TRH (Bockers et al, 1995; Hartlage-Rubsamen et al, 2009) in a regulated fashion (Hartlage-Rubsamen et al, 2011). In contrast, isoQC has been recently described as resident enzyme of the golgi complex and possesses an equal distribution throughout the body (Cynis et al, 2008). A first hint for isoform-specific substrate conversion came from the analysis of the hypothalamic-pituitary-gonadal axis, regulated by pE1-GnRH, and the hypothalamic-pituitary-thyroid axis, regulated by pE1-TRH, in QC knockout mice (Schilling et al, 2011). TRH undergoes extensive prohormone processing within secretory vesicles (Nillni & Sevarino, 1999) and is, therefore, most likely converted by QC. In contrast, the N-terminal glutamine residue of GnRH is already liberated after signal peptidase cleavage in the ER and might be accessed by both, QC and isoQC. Indeed, QC knockouts show a mild hypothyreodism characterized by a reduction in T4, but absence of hypogonadism (Schilling et al, 2011). This suggests a mainly isoQC-mediated conversion of substrates devoid of extensive prohormone processing, like CCL2 and GnRH. Obviously, neither in the golgi complex nor in secretory vesicles does CCL2 come into contact with QC. Therefore, differences in substrate conversion in vivo are mainly based on different sub-cellular localization within expressing cells.
The specificity of CCL2 for monocytes offers the chance to interfere selectively with disease conditions related to monocyte chemotaxis and activation, thereby, avoiding general suppression of immune function. In a first attempt to prove efficacy of the approach, we selected the cuff-induced accelerated atherosclerosis model to show the potency of QC/isoQC-inhibition in alleviation of atherosclerosis since monocytes are fundamental in the development of atherosclerotic pathology (Charo & Taubman, 2004). These mice develop intimal lesions within 2 weeks at a defined site of the arterial tree, making it an attractive model for drug screening (Lardenoye et al, 2000). It has to be considered, however, that the use of a transgenic mouse line and the rapid development of the atherosclerotic pathology after cuff placement do not reflect the long-term mechanisms leading to human pathology completely. For instance, a lack of fibrous caps and necrotic cores has been described for the lesions in the cuff model (Lardenoye et al, 2000). It was suggested, that the cuff placement in ApoE3*Leiden mice mimics early steps in atherosclerotic plaque formation. Because those early events have been linked to CCL2, the model appeared feasible for an initial study to obtain a proof of principle. Likewise, application of PQ50 might represent a novel treatment option, e.g. against restenosis after coronary balloon angioplasty.
It is known that the CCL-induced inflammatory response cascade is biologically redundant, i.e. the CCLs have multiple receptor specificities. While CCL2 binds primarily to CCR2, CCL8 binds to CCRs 1, 2, 3, 5, similarly CCL7 and CCL13 bind to CCRs 1, 2 and 3 (Horuk, 2009; Koenen & Weber, 2010). Blocking the N-terminal pE-formation could suppress receptor activity of not only CCL2 but also the signalling by structurally related MCPs. Since CCL heterodimers of related chemokines have been described, the proposed small molecule therapeutic approach targeting the inhibition of QC/isoQC might be more efficacious than targeting a single CCL by passive immunization or a CCR by a specific receptor antagonist, proposed recently as a ‘multiple receptor approach’ by Koenen and Weber (Koenen & Weber, 2010).
Besides the above-mentioned chemokines, other substrates of QC and isoQC might be affected by inhibition of these enzymes, which might have potential to cause side effects. The here described QC and isoQC ko mice do not have a severe phenotype and even constitutive double (QC and isoQC) ko mice are viable. Likewise, PQ50 has been already applied in high doses in models of Alzheimer's disease for up to 6 months without overt signs of toxicity (Schilling et al, 2008), clearly suggesting presence of a therapeutic window for suppression of both QCs. The absence of a severe phenotype upon QC suppression might be due to a significant spontaneous cyclization of N-terminal glutamine residues (Seifert et al, 2009), leading to a constitutive low-level generation of pE-peptides. Interestingly, our studies also support a background pE-formation even in presence of QC-inhibitor (Fig 2B and Fig 4A). Finally, this is also of importance in the light of the crucial function of CCL2 for various biological processes, such as chemotaxis (Lu et al, 1998), arteriogenesis (Hoefer et al, 2005), neovascularization and tissue repair (Anghelina et al, 2006).
Summarizing, compelling evidence suggests an underlying inflammatory response in the vast majority of acute and chronic diseases. Among others, CCL2 has been shown to be a major player in a number of disease conditions, stimulating monocyte migration, maturation and regulating the expression of other cytokines such as IL-1, IL-6 and TNF-α (Dewald et al, 2005; Jiang et al, 1992). Inhibitors of QC/isoQC are approaching human phase 1 trials, and the findings of the present study support anti-inflammatory strategies to alleviate pE-chemokine driven disorders by those drugs.
MATERIALS AND METHODS
QC-inhibitors
QC-inhibitors (1-(3-(1H-Imidazol-1-yl) propyl)-3-(3,4-dimethoxyphenyl)thiourea; PQ50; Buchholz et al, 2006) and 1-(1H-benzo[d]imidazol-5-yl) -5-(4-propoxyphenyl) imidazolidine-2,4-dione (PQ529; Probiodrug) were applied in all studies. The compounds belong to different chemical classes possessing either imidazole (PQ50) or benzimidazole (PQ529) as warhead. The Ki-values of PQ50 at pH 8 are for human QC: 71 nM, human isoQC: 236 nM, murine QC: 170 nM, murine isoQC: 246 nM. The Ki-values of PQ529 at pH 8 are for human QC: 38 nM, human isoQC: 4 nM, murine QC: 27 nM, murine isoQC: 2 nM.
Catalytic parameters
The generation of pE1-CCL2 was monitored by a spectrophotometric assay essentially as described elsewhere (Schilling et al, 2003). The progress curve was evaluated using GraFit 5 (Erithacus Software Ltd.) by applying the integrated form of the Michaelis–Menten equation {t = (P/v) − [K/v*ln(1 − (P/S)]} allowing the determination of catalytic parameters from single reactions.
Degradation of MCPs
Recombinant human CCL2, CCL7, CCL8 and CCL13 (Peprotech) possessing an N-terminal glutaminyl residue were dissolved in 25 mM Tris/HCl pH 7.6 at a final concentration of 10 µg/ml. MCPs were either pre-incubated with recombinant human QC (0.6 µg/ml; Schilling et al, 2002) for 2 h at 30°C and subsequently incubated with human recombinant DP4 (Engel et al, 2003) or incubated with DP4 without QC. Furthermore, CCL2(Q1-76) was either pre-incubated with recombinant human QC and subsequently incubated with human plasma or incubated with human plasma without QC. All samples were analysed using Maldi-TOF-MS.
MALDI-TOF-MS
Matrix-assisted laser desorption/ionization mass spectrometry was performed using the Voyager De Pro (Applied Biosystems) in a linear mode. Five microlitre of the samples from the degradation experiment was mixed and co-crystallized with 5 µl of matrix solution (sinapinic acid 20 mg/ml in acetonitrile/0.1% TFA in water, 50:50 v/v), 1 µl of this solution was spotted on the sample plate and air dried. The analytes were ionized by a nitrogen laser pulse (337 nm) and accelerated under 20 kV with a time-delayed extraction before entering the time-of-flight mass spectrometer. Detector operation was in the positive ion mode ranging from 2000 to 20,000 amu. Insulin and myoglobin solutions (Sigma–Aldrich) were used for calibration in this range according to the instructor's manual from Applied Biosystems. Each spectrum represents the sum of at least 6 × 100 laser pulses.
Cell culture
Human acute monocytic leukaemia cell line THP-1 (DSMZ) was cultured in RPMI160 supplemented with 10% FBS and 50 µg/ml Gentamicin at 37°C and 5% CO2. Cells were checked regularly for mycoplasma contaminations using the VenorGeM-qEP qPCR kit (Minerva).
CCR2 receptor internalization assay
6 × 105 THP-1 cells were incubated in 24-well suspension plates in 500 µl serum-free RPMI 1640 containing the respective ligand [CCL2(pE1-76), CCL2(Q1-76), CCL2(D3-76) and CCL2(I5-76)] for 45 min at 37°C. Afterwards, cells were harvested, resuspended in 80 µl cold PBS and incubated with Fc-blocking reagent (Miltenyi) for 10 min at 4°C. Thereafter, cells were stained with phycoerythrin-conjugated anti-CCR2 antibody (R&D Systems) or IgG2B isotype control for 30 min at 4°C. After washing with PBS, cells were analysed using a BD FACSCalibur (BD, Heidelberg).
TransWell chemotaxis assay
Twenty four-well TransWell plates with a pore size of 5 µm (Corning) were used. Six hundred microlitre of chemoattractant solution were applied to the lower chamber. Serum-free RPMI was applied as negative control. THP-1 cells were harvested and resuspended in RPMI1640 in a concentration of 1 × 106 cells/100 µl and applied in 100 µl aliquots to the upper chamber. Cells were allowed to migrate for 2 h at 37°C. Subsequently, cells from the upper chamber were discarded and the lower chamber was mixed with 50 µl 70 mM EDTA in PBS and incubated for 15 min at 37°C to release cells attached to the surfaces. Afterwards, migrated cells were counted using a cell counter system (Schärfe System) or by cytometric analysis using FACS Calibur based on 5000 beads per sample in BD Trucount tubes (BD) as reference standard.
Generation and genotyping of the isoQC knockout model
IsoQC knockout mice were generated by an ENU induced point mutation. An ENU mutant library (Augustin et al, 2005) was screened for mutations in the isoQC gene and a nonsense mutant with a stop codon in the third exon was identified. In homozygous knockout mice, isoQC-RNA translation stops at amino acid residue 144 and results in a truncated and non-functional isoQC protein.
Genotyping of isoQC ko animals was done by PCR amplification of isoQC exon 3 in a standard PCR reaction applying primers 5′-CGTGGCTCCAGTCACAAG-3′ (sense) and 5′-TCAAGGCTAGCTTGGGCTAC-3′ (antisense). The T → A transversion of the nonsense mutation introduces an AvrII restriction site and AvrII digestion of the PCR fragment results in fragmentation patterns which are indicative for the respective genotype of the tested animals.
Isolation of primary monocytes/macrophages
For isolation of peripheral blood mononuclear cells (PBMCs), animals were anaesthetized using 2% isoflurane and heparinized blood was collected by cardiac puncture. For isolation of human and murine PBMCs, a density gradient was used in Leucosep tubes (50 ml, Greiner) filled with 15 ml of LSM 1077 Lymphocyte Separation Medium (PAA) following manufacturer's recommendation. The cells were resuspended in culture medium, plated in a 25 cm2 tissue culture flask and grown over night at 37°C and 5% CO2. The next day, adhered human or murine monocytes/macrophages were washed once with PBS and subsequently dislodged using accutase (PAA). After centrifugation, cells were counted using a Neubauer counting chamber and plated in a density of 1 × 105 cells per well in a 96-well plate. Cells were stimulated using 1 µg/ml LPS from Escherichia coli strain O55:B5 (Sigma) for 24 h. Afterwards, medium was collected and analysed using total-CCL2 and pE1-CCL2 specific ELISA.
Preparation of primary neuronal and glial cells
Primary neurons were prepared from mouse embryos (E16) and primary glial cells were prepared from newborn pubs at postnatal day 1. Briefly, the brains were removed and afterwards each brain was triturated with a 10 ml pipette. The brain solutions passed 100 µm cell strainers followed by the same step with 40 µm cell strainers (BD). Twenty microlitre of each cell solution was stained with Trypan blue (Sigma) and the vital cells were counted. 6 × 105 cells were spread on poly-l-lysine coated 12-well plate and cultured at 37°C in a humidified atmosphere with 95% air and 5% CO2. To prepare neurons, the medium was exchanged and cells were cultured in DMEM/F12, 5% FBS, 0.05% Gentamicin containing 25% conditioned medium from astrocyte cultures and N-2 (Invitrogen, Karlsruhe) on the next day. Cytosinarabinosid (Sigma) was added (1 µM f.c.) 3 days after preparation. For glial cell isolation, the brains were removed, separated in tubes with DMEM/F12 (Invitrogen), 5% FBS (Invitrogen) containing 0.05% Gentamicin (PAA) and triturated using a 10 ml pipette. The brain solutions passed 100 and 40 µm cell strainers (BD). Cells were stained with Trypan blue (Sigma) and 6 × 105 cells were seeded on poly-l-lysine 12-well plate. The cells were cultured at 37°C in a humidified atmosphere with 95% air and 5% CO2. Seven days later, the medium was renewed. The glia cells consisted of approx. 85% astrocytes and 15% microglia cells and were subjected to the analyses 12 days after seeding.
Dot Blot analysis for characterization of pE1-CCL2 specific antibody
Human CCL2(pE1-38) and human CCL2(D3-38; Probiodrug) in descending concentrations (1000–20 ng) were spotted onto nitrocellulose membranes. Membranes were blocked for 2 h with TBST-M (=TBST (Tris buffered saline + 0.05% Tween-20) +5% skimmed milk) at room temperature with gentle shaking. Membranes were incubated over night at 4°C on a rocking platform with pE1-CCL2 specific antibody clone 2C9 diluted to 1 µg/ml in equal volumes of TBST-M. Secondary anti-mouse antibody conjugated with alkaline phosphatase was used for signal detection, following standard procedures.
Surface plasmon resonance (SPR) analysis
Antibody 2C9 was characterized by SPR. The analyses were performed on a Biacore 3000. Briefly, a CM5 chip was covalently conjugated with 100 response units (RU) CCL2(pE1-38) peptide on flow cell (Fc) 2. Fc 3 and Fc4 was conjugated with 100 RU of differing pE1-peptides. Fc1 was prepared for blank subtraction. 2C9 was diluted in running buffer HBS-EP (Hepes buffered saline + 3 mM EDTA + 0.005% v/v surfactant P20, Biacore) at concentrations ranging from 20 to 1 µg/ml. First, a basal signal was determined with HBS-EP, followed by 180 s of application of antibody dilution, to determine association of antibody to antigen. Then, pure HBS-EP was injected again for another 180 s to observe the dissociation of the corresponding antibody.
pE1-CCL2 specific ELISAs
For determination of murine and human total-CCL2 and pE1-CCL2, specific ELISAs were developed. For murine ELISA, 50 ng of capture antibody rabbit-anti mJE (Peprotech) was coated per well of a 96-well plate in coating buffer (PBS, pH 7.4). Plates were incubated for 4–7 days at 4°C. For human ELISA, 25 ng/well of capture antibody goat-anti hMCP-1 AF (R&D Systems) was coated over night at 4°C. Afterwards, wells were blocked for 2 h by addition of 200 µl blocking buffer [protein free (TBS) blocking buffer (Perbio)] and then washed three times using 300 µl of wash buffer [protein free T20 (TBS) blocking buffer (Perbio)]. Standard peptides (Peprotech) and samples were diluted using dilution buffer [protein free T20 (TBS) blocking buffer)] and 100 µl were applied onto the test plate. The incubation of test samples and standard peptides was for 2 h at room temperature and afterwards the plate was washed three times using wash buffer. For detection of human and mouse pE1-CCL2, anti-pE1-CCL2 specific monoclonal antibody clone 2C9 (Probiodrug) was applied in a concentration of 0.5 µg/ml in combination with anti-mouse-HRP conjugate (KPL). For the detection of murine total-CCL2, goat-anti mouse CCL2 (Santa Cruz) was applied in combination with anti-goat-IgG-HRP conjugate (R&D Systems). For the detection of human total-CCL2, mouse-anti human MCP-1 antibody (Biolegends) was used in combination with anti-mouse-IgG-HRP (KPL). Antibodies were diluted in dilution buffer, applied in a volume of 100 µl to each well and incubated for 2 h at room temperature. Thereafter, wells were washed three times with 300 µl of wash buffer and the chromogen SureBlue (KPL) was applied in a volume of 100 µl to each well and incubated in the dark. After 30 min, the reaction was abrogated using 50 µl Stop Solution (1.2 N H2SO4) and absorption was determined at 450 nm. Reference wavelength of 550 nm was subtracted from sample absorption at 450 nm.
LPS-induced inflammation in mice
Experiments were performed under approval number LALLF MV/TSD/7221.3-1.1-017/09 granted to the Institute of Diabetes, Karlsburg, Germany. Mice (C57BL/6J, age 8–12 weeks) were injected with LPS (2 mg/kg b.w. i.p.) or with vehicle (PBS). Five hours post-injection, blood was collected by cardiac puncture and serum was prepared and snap-frozen until analysis using a CCL2-specific ELISA.
LPS-induced lung inflammation
The study was conducted at Fraunhofer ITEM, Department of Immunology, Allergology and Immunotoxicology under the approval number 33.12-42502-04-07/1241. Young adult (8–12 weeks old) female mice (isoQC−/− and isoQC+/+) were used for the study. Animals were age-matched and randomized by weight. For the induction of respiratory inflammation, the animals were intranasally exposed to 800 ng LPS (E. coli O111:B4) in 50 µl NaCl or to 0.9% NaCl (control) on four consecutive days. Animals were sacrificed 24 h after last intranasal challenge and a necropsy including bronchoalveolar lavage (BAL) was performed. After BAL-fluid (BALF) collection (2 × 800 µl in 0.9% NaCl) flow cytometric investigation of the cells was performed. Therefore, 0.1 ml of the cell suspensions of every animal were transferred into FACS-tubes and stained for the expression of 7/4 (Ly6B.2)-FITC (Serotec)/Ly6G-PE (Miltenyi). To block unspecific binding, the cells were resuspended using 100 µl of 1:100 diluted CD16/32 in FACS-buffer (PBS, 5% FCS; Fc-Block). In addition, cytospots were prepared and stained according to Pappenheim to evaluate differential cell counts.
Thioglycollate-induced peritonitis
Experiments were approved by LVwA Sachsen-Anhalt (authorization: 203.h-42502-2-856probio). C57BL/6J mice (Charles River) were injected (i.p.) with 25 ml/kgbody weight of sterile 8% w/v thioglycollate (Sigma–Aldrich). In addition, QC inhibitors were applied i.p. or p.o. Four hours after stimulation, mice were anaesthetized by isoflurane and peritoneal lavage was performed with 8 ml pre-warmed PBS. Cells were harvested from 1 ml lavage fluid and used for staining of surface expression markers. Cells were blocked with CD16/32 (Invitrogen) and stained with 7/4 (Ly6B.2)-FITC (Serotec)/Ly6G-PE (Miltenyi) and isotype controls (eBioscience). Flow cytometric analysis was performed using FACSCalibur based on 5000 beads per sample in BD Trucount tubes (BD) as reference standard.
Human tissue
Pieces of Arteria femoralis superficialis were collected from patients undergoing vascular surgery under the approval of the ethics commission of the Martin-Luther-University Halle (Approval: September 21, 2009). A written informed consent, documented by the Department of Vascular Surgery of the St. Elisabeth and St. Barbara Hospital in Halle, was received from every patient prior to inclusion in the study. Tissue was harvested by an experienced surgeon and subsequently washed with cold PBS, cut into pieces and either snap-frozen or stored in fixative.
IsoQC-specific antibodies and immunoblotting
Western blot analysis was performed essentially as described elsewhere (Cynis et al, 2008). IsoQCs were detected using newly generated polyclonal antibodies against the human (anti-isoQC 5406) and murine (anti-isoQC 5407) enzyme. Anti-isoQC 5406 was generated in rabbit by immunization with the peptide MRSGGRGRPRLRLGERGLMEPLLPPKRRLLPRVRC and anti-isoQC 5407 was generated by immunizing a rabbit using the peptide MSPGSRGRPRQRLEDRGLMKPPSLSKRRLLPRVQC. Both peptides were coupled via a C-terminal cystein to Limulus Polyphemus Hemocyanin (LPH). After final bleeding the antibodies were affinity purified using the immunization peptides coupled to an activated CNBr sepharose column (Sigma). For immunoblotting, the antibodies were used in a dilution of 1:500 at 4°C over night followed by incubation with HRP-conjugated goat anti-rabbit IgG (1:2000, 4°C, 1 h; Cell Signaling) and detection using SuperSignal West Femto (Perbio).
RT-PCR
RNA was isolated using the NucleoSpin RNA II kit or the XS kit for gene expression analysis in cells from peritoneal lavage (both Macherey Nagel) according to the manufacturer's instructions. RNA concentration was measured using a NanoDrop 2000 spectrophotometer (Peqlab). Gene expression in different normal human tissues was analysed applying the FirstChoice Human Total RNA Survey Panel (Ambion). RNA (0.1–1 µg) was reversely transcribed into complementary DNA (cDNA) using random primers (Roche) and Superscript III (Invitrogen). Quantitative PCR was performed in a Rotorgene3000 (Corbett Research) using the Rotor-Gene SYBR Green PCR kit and the Quantitect primer assay HsQPCTL (Qiagen) or specific primers for human CCL2 (NM_002982.3), mouse TNF-alpha (NM_013693.1), mouse ICAM-1 (NM_010493.2), mouse CCL2 (NM_011333.1), mouse CCL7 (NM_013654.2) and mouse IL-6 (NM_031168.1) all synthesized by Metabion. The primer sequences are depicted in Supporting Information Table 2. Relative amounts of gene expression were determined with the Rotorgene software version 4.6 in comparative quantitation mode. RPII (NM_000937.2), GAPDH (NM_002046.3) and YWHAZ (NM_003406.2) were used as reference genes (RefG) for the human tissue gene expression analysis, whereas, mouse YWHAZ (NM_011740.2) and mouse PHKA1 (NM_008832.2) for gene expression analysis in cells of mouse peritoneal lavage. The PCR was verified by product melting curves and single amplicons were confirmed by agarose gel electrophoresis.
Immunohistochemistry
Tissue was fixed using zinc fixative (BD Pharmingen), embedded in low-melting-point paraffin (DCS Innovative Diagnostic Systems), and sectioned at 5 µm on a rotating microtome. After de-paraffinization, immunostainings were performed by incubation (overnight, 4°C) with primary antisera (anti-CD68; anti-sma, Dako). For immunodetection, biotinylated IgGs (Vector Laboratories) were used, followed by ABC-detection and diaminobenzidine (DAB) staining (Vector Laboratories). For staining of blood vessels, slides were incubated (overnight, 4°C) with biotinylated UEA1 (Vector Laboratories) followed by ABC-detection and DAB-staining. Elastin-staining was performed according to the manufacturer's protocol (Elastika van Gieson staining, Merck 1.15974).
The paper explained
PROBLEM
Inflammation is an underlying symptom described for the vast majority of disorders. Thereby, the chemokine CCL2 plays a pivotal role for the formation of atherosclerotic plaques and restenotic changes after coronary angioplasty and correlates also with neurodegeneration. Multiple receptor specificities of chemokines point to the need of approaches targeting the chemokine ligands rather than a single chemokine receptor. There are no small molecule inhibitors available inhibiting directly CCL2 function.
RESULTS
The authors used a combined biochemical and cell biological approach to investigate pyroglutamate formation, an important post-translational step in CCL2 maturation. The study provides insights into the function of N-terminal pyroglutamate residues for stability and bioactivity of CCL2. In addition, alternative pathways for CCL2-inactivation were identified by inhibition of glutaminyl cyclase activity. The approach was validated in different models in vitro and in vivo showing potential of small molecule enzyme inhibitors of glutaminyl cyclase. In addition, studies in newly generated gene knock outs for two described glutaminyl cyclases in mice points to a pivotal role of the golgi-resident isoQC for CCL2 maturation.
IMPACT
This study provides insights into a novel mechanism, which is useful for the generation of drugs targeting stability and bioactivity of a small subset of chemokines, however, showing a pivotal role in a number of inflammatory conditions. Inhibitors of glutaminyl cyclases are currently approaching clinical phase I trials.
Cuff-induced accelerated atherosclerosis in ApoE3*Leiden mice
After approval by the animal welfare committee, 30 male ApoE3*Leiden mice (age 12 weeks) were fed a hypercholesterolemic (1% w/w cholesterol, 0.05% cholate) semi-synthetic western type of diet consisting of 15% cacao butter, 1% corn oil, 40.5% sucrose, 10% starch, 20% casein, 5.95% cellulose, 2% choline, 0.2% methionine, 5.35% vitamin and mineral mix for 3 weeks prior to surgical cuff placement aiming at plasma cholesterol levels of 7.5–12 mM. Thereafter, the mice underwent surgical non-constricting cuff placement (day 0) and were divided into two groups, matched for plasma cholesterol levels. The mice either received control (acidified) drinking water or drinking water containing the QC inhibitor (1-(3-(1H-imidazol-1-yl)propyl)-3-(3,4-dimethoxyphenyl)thiourea hydrochloride (PQ50) in a concentration of 2.4 mg/ml. Seven days after start of treatment, the inhibitor concentration was reduced to 1.2 mg/ml. Five mice of each group were sacrificed after 2 days for analysis of monocyte adhesion and infiltration, and 10 mice were sacrificed after 2 weeks for histomorphometric analysis to quantify the inhibition of accelerated atherosclerotic lesions and neointima formation.
Surgical procedure of cuff placement
At the time of surgery, mice were anaesthetized with an intraperitoneal injection of 5 mg/kg Dormicum, 0.5 mg/kg Domitor and 0.05 mg/kg Fentanyl. This cocktail gives complete narcosis for at least 1 h and can be quickly antagonized with Antisedan 2.5 mg/kg and Anexate 0.5 mg/kg. A longitudinal 1 cm incision was made in the internal side of the leg and the femoral artery is dissected for 3 mm length from the femoral nerve and femoral vein. The femoral artery is looped with a ligature and a non-constrictive fine bore polyethylene tubing (0.4 mm inner diameter, 0.8 mm outer diameter and length 2 mm) is longitudinally opened and sleeved loosely around the femoral artery. The cuff is closed up with two ligature knots. The skin is closed with a continued suture. After surgery, the animals were antagonized and placed in a clean cage on top of a heating pad for a few hours.
Sacrifice of the animals
For histological analysis, animals were sacrificed either 2 or 14 days after cuff placement. The thorax of the anaesthetized animals was opened and a mild pressure-perfusion (100 mmHg) with 4% formaldehyde was performed for 3 min by cardiac puncture. After perfusion, a longitudinal 2 cm incision was made in the internal side of the leg and the cuffed femoral artery was harvested as a whole and fixed overnight in 4% formaldehyde and processed to paraffin.
Analysis of monocyte adhesion and CCL2 expression
Adhesion of leukocytes in general and monocytes/macrophages in particular to the activated endothelium of the cuffed vessel wall was investigated by microscopic analysis of cross-sections harvested 2 days after cuff placement. The number of adhering and/or infiltrating leukocytes in general, identified as adhering cells at the luminal side of the vessel segment, and monocytes/macrophages in particular was counted and illustrated as cells per cross-section or as defined areas per cross-section. Monocytes were identified by specific immunohistochemical staining using AIA31240 antibody (Accurate Chemicals), recognizing monocytes and macrophages. In addition on these sections a specific immunohistochemical staining for CCL2 was performed.
Analysis of vascular remodelling and accelerated atherosclerosis
Vessel wall remodelling accelerated atherosclerosis and neointima formation were analysed morphometrically in all mice sacrificed after 14 days. A full comparison between the two groups was performed for all relevant vessel wall parameters (neointima formation, vascular circumference (i.e. outward remodelling), media thickness and lumen stenosis). Accelerated atherosclerosis was analysed by immunohistochemical staining for macrophages in the lesion area by AIA31240 antibody. The sections were also stained for CCL2.
Statistical analysis
Statistical analyses comprise two-tailed, unpaired Student's t-test, one-way ANOVA followed by Tukey post hoc analysis or two-way ANOVA followed by Bonferroni post-test. Excel (Microsoft) or Graphpad prism 4 (Graphpad Sofware Inc.) was used.
Acknowledgments
The authors gratefully acknowledge the technical assistance of Kathrin Klemm, Sandra Torkler, Katja Menge, Anja Fothe, Olga Sharma, Anke Ryll, Nadine Taudte, Beate Kohlstedt, Claudia Göttlich and Stephan Reich. This work was supported by the Investment Bank of Saxony-Anhalt, grant #6003373000.
Supporting information is available at EMBO Molecular Medicine online.
Conflict of interest statement: HC, TH, DF, AK, KG, MK, JUR, RW, MW, AS, MH, RE, UH and SS are employees of Probiodrug. RS, SG, WJ and AM are employees of Ingenium Pharmaceuticals. HUD serves as CSO of Probiodrug and Managing Director of Ingenium Pharmaceuticals GmbH, a daughter company of Probiodrug, and holds stocks of the Probiodrug group.
Author contributions
HC and SS planned most of the experiments; SS, HC, AK, FS, RW, RE, AS, MH and TH performed the biochemical and cell biological experiments; MW generated and purified the applied antibodies; JUR, KG and MK established the CCL2(pE1-76)-specific ELISA; UW, DT and IH are vascular surgeons and isolated the human atherosclerotic vessels; SG and RS generated the QC and isoQC knock out animals; AK, TH, AM and DF conducted the thioglycollate-induced peritonitis assay; SB and EJF performed the intraperitoneal LPS-treatment in mice; PHAQ and MRdV performed the cuff-induced atherosclerosis study; WJ analysed the human tissues; UH supervised medicinal chemistry developing QC-specific inhibitors; HC, SS and HUD designed the studies and wrote the manuscript; SS and HUD initiated the research and supervised the program.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Abraham GN, Podell DN. Pyroglutamic acid. Non-metabolic formation, function in proteins and peptides, and characteristics of the enzymes effecting its removal. Mol Cell Biochem. 1981;38:181–190. doi: 10.1007/BF00235695. [DOI] [PubMed] [Google Scholar]
- Anghelina M, Krishnan P, Moldovan L, Moldovan NI. Monocyte/macrophages cooperate with progenitor cells during neovascularization and tissue repair. Am J Pathol. 2006;168:529–541. doi: 10.2353/ajpath.2006.050255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin M, Sedlmeier R, Peters T, Huffstadt U, Kochmann E, Simon D, Schöniger M, Garke-Mayerthaler S, Laufs J, Mayhaus M, et al. Efficient and fast targeted production of murine models based on ENU mutagenesis. Mamm Genome. 2005;16:405–413. doi: 10.1007/s00335-004-3028-2. [DOI] [PubMed] [Google Scholar]
- Awade AC, Cleuziat P, Gonzales T, Robert-Baudouy J. Pyrrolidone carboxyl peptidase (Pcp): an enzyme that removes pyroglutamic acid (pGlu) from pGlu-peptides and pGlu-proteins. Proteins. 1994;20:34–51. doi: 10.1002/prot.340200106. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Ramnath RD, Chevali L, Guglielmotti A. Treatment with bindarit, a blocker of MCP-1 synthesis, protects mice against acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1259–G1265. doi: 10.1152/ajpgi.00435.2004. [DOI] [PubMed] [Google Scholar]
- Blomback B. Derivatives of glutamine in peptides. Methods Enzymol. 1967;11:398–411. [Google Scholar]
- Bockers TM, Kreutz MR, Pohl T. Glutaminyl-cyclase expression in the bovine/porcine hypothalamus and pituitary. J Neuroendocrinol. 1995;7:445–453. doi: 10.1111/j.1365-2826.1995.tb00780.x. [DOI] [PubMed] [Google Scholar]
- Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- Buchholz M, Heiser U, Schilling S, Niestroj AJ, Zunkel K, Demuth HU. The first potent inhibitors for human glutaminyl cyclase: synthesis and structure–activity relationship. J Med Chem. 2006;49:664–677. doi: 10.1021/jm050756e. [DOI] [PubMed] [Google Scholar]
- Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004;95:858–866. doi: 10.1161/01.RES.0000146672.10582.17. [DOI] [PubMed] [Google Scholar]
- Cynis H, Schilling S, Bodnar M, Hoffmann T, Heiser U, Saido TC, Demuth HU. Inhibition of glutaminyl cyclase alters pyroglutamate formation in mammalian cells. Biochim Biophys Acta. 2006;1764:1618–1625. doi: 10.1016/j.bbapap.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Cynis H, Rahfeld JU, Stephan A, Kehlen A, Koch B, Wermann M, Demuth HU, Schilling S. Isolation of an isoenzyme of human glutaminyl cyclase: retention in the golgi complex suggests involvement in the protein maturation machinery. J Mol Biol. 2008;379:966–980. doi: 10.1016/j.jmb.2008.03.078. [DOI] [PubMed] [Google Scholar]
- Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- Engel M, Hoffmann T, Wagner L, Wermann M, Heiser U, Kiefersauer R, Huber R, Bode W, Demuth HU, Brandstetter H. The crystal structure of dipeptidyl peptidase IV (CD26) reveals its functional regulation and enzymatic mechanism. Proc Natl Acad Sci USA. 2003;100:5063–5068. doi: 10.1073/pnas.0230620100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulberg D, Klussmann S. Spiegelmers: biostable aptamers. Chembiochem. 2003;4:979–983. doi: 10.1002/cbic.200300663. [DOI] [PubMed] [Google Scholar]
- Fischer WH, Spiess J. Identification of a mammalian glutaminyl cyclase converting glutaminyl into pyroglutamyl peptides. Proc Natl Acad Sci USA. 1987;84:3628–3632. doi: 10.1073/pnas.84.11.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlender ZG, Buchlis G, Kapoor V, Cheng G, Sun J, Singhal S, Crisanti C, Wang LC, Heitjan D, Snyder LA, et al. CCL2 blockade augments cancer immunotherapy. Cancer Res. 2010;70:109–118. doi: 10.1158/0008-5472.CAN-09-2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti D, Fenoglio C, Lovati C, Venturelli E, Guidi I, Corra B, Scalabrini D, Clerici F, Mariani C, Bresolin N, et al. Serum MCP-1 levels are increased in mild cognitive impairment and mild Alzheimer's disease. Neurobiol Aging. 2006;27:1763–1768. doi: 10.1016/j.neurobiolaging.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2:108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- Gosling J, Slaymaker S, Gu L, Tseng S, Zlot CH, Young SG, Rollins BJ, Charo IF. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J Clin Invest. 1999;103:773–778. doi: 10.1172/JCI5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlage-Rubsamen M, Staffa K, Waniek A, Wermann M, Hoffmann T, Cynis H, Schilling S, Demuth HU, Rossner S. Developmental expression and subcellular localization of glutaminyl cyclase in mouse brain. Int. J Dev Neurosci. 2009;27:825–835. doi: 10.1016/j.ijdevneu.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Hartlage-Rubsamen M, Morawski M, Waniek A, Jager C, Zeitschel U, Koch B, Cynis H, Schilling S, Schliebs R, Demuth HU, et al. Glutaminyl cyclase contributes to the formation of focal and diffuse pyroglutamate (pGlu)-Abeta deposits in hippocampus via distinct cellular mechanisms. Acta Neuropathol. 2011;121:705–719. doi: 10.1007/s00401-011-0806-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmerich S, Paavola C, Bloom A, Bhakta S, Freedman R, Grunberger D, Krstenansky J, Lee S, McCarley D, Mulkins M, et al. Identification of residues in the monocyte chemotactic protein-1 that contact the MCP-1 receptor CCR2. Biochemistry. 1999;38:13013–13025. doi: 10.1021/bi991029m. [DOI] [PubMed] [Google Scholar]
- Hoefer IE, Grundmann S, van Royen N, Voskuil M, Schirmer SH, Ulusans S, Bode C, Buschmann IR, Piek JJ. Leukocyte subpopulations and arteriogenesis: specific role of monocytes, lymphocytes and granulocytes. Atherosclerosis. 2005;181:285–293. doi: 10.1016/j.atherosclerosis.2005.01.047. [DOI] [PubMed] [Google Scholar]
- Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov. 2009;8:23–33. doi: 10.1038/nrd2734. [DOI] [PubMed] [Google Scholar]
- Inoshima I, Kuwano K, Hamada N, Hagimoto N, Yoshimi M, Maeyama T, Takeshita A, Kitamoto S, Egashira K, Hara N. Anti-monocyte chemoattractant protein-1 gene therapy attenuates pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1038–L1044. doi: 10.1152/ajplung.00167.2003. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Beller DI, Frendl G, Graves DT. Monocyte chemoattractant protein-1 regulates adhesion molecule expression and cytokine production in human monocytes. J Immunol. 1992;148:2423–2428. [PubMed] [Google Scholar]
- Koenen RR, Weber C. Therapeutic targeting of chemokine interactions in atherosclerosis. Nat Rev Drug Discov. 2010;9:141–153. doi: 10.1038/nrd3048. [DOI] [PubMed] [Google Scholar]
- Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15:97–103. doi: 10.1038/nm.1898. [DOI] [PubMed] [Google Scholar]
- Lardenoye JH, Delsing DJ, de Vries MR, Deckers MM, Princen HM, Havekes LM, van Hinsbergh VW, van Bockel JH, Quax PH. Accelerated atherosclerosis by placement of a perivascular cuff and a cholesterol-rich diet in ApoE*3Leiden transgenic mice. Circ Res. 2000;87:248–253. doi: 10.1161/01.res.87.3.248. [DOI] [PubMed] [Google Scholar]
- Lau EK, Paavola CD, Johnson Z, Gaudry JP, Geretti E, Borlat F, Kungl AJ, Proudfoot AE, Handel TM. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: implications for structure and function in vivo. J Biol Chem. 2004;279:22294–22305. doi: 10.1074/jbc.M311224200. [DOI] [PubMed] [Google Scholar]
- Lu BB, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra F. Renaming cytokines: MCP-1, major chemokine in pancreatitis. Gut. 2005;54:1679–1681. doi: 10.1136/gut.2005.068593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masure S, Paemen L, Proost P, Van Damme J, Opdenakker G. Expression of a human mutant monocyte chemotactic protein 3 in Pichia pastoris and characterization as an MCP-3 receptor antagonist. J Interferon Cytokine Res. 1995;15:955–963. doi: 10.1089/jir.1995.15.955. [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Gong JH, Wong JP, Wallace JL, Clark-Lewis I, Overall CM. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood. 2002;100:1160–1167. [PubMed] [Google Scholar]
- Mori E, Komori K, Yamaoka T, Tanii M, Kataoka C, Takeshita A, Usui M, Egashira K, Sugimachi K. Essential role of monocyte chemoattractant protein-1 in development of restenotic changes (neointimal hyperplasia and constrictive remodeling) after balloon angioplasty in hypercholesterolemic rabbits. Circulation. 2002;105:2905–2910. doi: 10.1161/01.cir.0000018603.67989.71. [DOI] [PubMed] [Google Scholar]
- Morty RE, Bulau P, Pelle R, Wilk S, Abe K. Pyroglutamyl peptidase type I from Trypanosoma brucei: a new virulence factor from African trypanosomes that de-blocks regulatory peptides in the plasma of infected hosts. Biochem J. 2006;394:635–645. doi: 10.1042/BJ20051593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni W, Egashira K, Kitamoto S, Kataoka C, Koyanagi M, Inoue S, Imaizumi K, Akiyama C, Nishida KI, Takeshita A. New anti-monocyte chemoattractant protein-1 gene therapy attenuates atherosclerosis in apolipoprotein E-knockout mice. Circulation. 2001;103:2096–2101. doi: 10.1161/01.cir.103.16.2096. [DOI] [PubMed] [Google Scholar]
- Nillni EA, Sevarino KA. The biology of pro-thyrotropin-releasing hormone-derived peptides. Endocr Rev. 1999;20:599–648. doi: 10.1210/edrv.20.5.0379. [DOI] [PubMed] [Google Scholar]
- Proost P, Struyf S, Couvreur M, Lenaerts JP, Conings R, Menten P, Verhaert P, Wuyts A, Van Damme J. Posttranslational modifications affect the activity of the human monocyte chemotactic proteins MCP-1 and MCP-2: identification of MCP-2(6-76) as a natural chemokine inhibitor. J Immunol. 1998;160:4034–4041. [PubMed] [Google Scholar]
- Proudfoot AE, Power CA, Hoogewerf AJ, Montjovent MO, Borlat F, Offord RE, Wells TN. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem. 1996;271:2599–2603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA. 2003;100:1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling S, Hoffmann T, Rosche F, Manhart S, Wasternack C, Demuth HU. Heterologous expression and characterization of human glutaminyl cyclase: evidence for a disulfide bond with importance for catalytic activity. Biochemistry. 2002;41:10849–10857. doi: 10.1021/bi0260381. [DOI] [PubMed] [Google Scholar]
- Schilling S, Niestroj AJ, Rahfeld JU, Hoffmann T, Wermann M, Zunkel K, Wasternack C, Demuth HU. Identification of human glutaminyl cyclase as a metalloenzyme. Potent inhibition by imidazole derivatives and heterocyclic chelators. J Biol Chem. 2003;278:49773–49779. doi: 10.1074/jbc.M309077200. [DOI] [PubMed] [Google Scholar]
- Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, Holzer M, Hutter-Paier B, Prokesch M, Windisch M, et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Aβ and Alzheimer's disease-like pathology. Nat Med. 2008;14:1106–1111. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- Schilling S, Kohlmann S, Baeuscher C, Sedlmeier R, Koch B, Eichentopf R, Becker A, Cynis H, Hoffmann T, Berg S, et al. Glutaminyl cyclase (QC) knock out mice show mild hypothyreodism but absence of hypogonadism: implications for enzyme function and drug development. J Biol Chem. 2011;286:14199–14208. doi: 10.1074/jbc.M111.229385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert F, Schulz K, Koch B, Manhart S, Demuth HU, Schilling S. Glutaminyl cyclases display significant catalytic proficiency for glutamyl substrates. Biochemistry. 2009;48:11831–11833. doi: 10.1021/bi9018835. [DOI] [PubMed] [Google Scholar]
- Stephan A, Wermann M, von Bohlen A, Koch B, Cynis H, Demuth HU, Schilling S. Mammalian glutaminyl cyclases and their isoenzymes have identical enzymatic characteristics. FEBS J. 2009;276:6522–6536. doi: 10.1111/j.1742-4658.2009.07337.x. [DOI] [PubMed] [Google Scholar]
- Usui M, Egashira K, Ohtani K, Kataoka C, Ishibashi M, Hiasa K, Katoh M, Zhao Q, Kitamoto S, Takeshita A. Anti-monocyte chemoattractant protein-1 gene therapy inhibits restenotic changes (neointimal hyperplasia) after balloon injury in rats and monkeys. FASEB J. 2002;16:1838–1840. doi: 10.1096/fj.02-0094fje. [DOI] [PubMed] [Google Scholar]
- Van Coillie E, Proost P, Van Aelst I, Struyf S, Polfliet M, De Meester I, Harvey DJ, Van Damme J, Opdenakker G. Functional comparison of two human monocyte chemotactic protein-2 isoforms, role of the amino-terminal pyroglutamic acid and processing by CD26/dipeptidyl peptidase IV. Biochemistry. 1998;37:12672–12680. doi: 10.1021/bi980497d. [DOI] [PubMed] [Google Scholar]
- Van Damme J, Struyf S, Wuyts A, Van Coillie E, Menten P, Schols D, Sozzani S, De Meester I, Proost P. The role of CD26/DPP IV in chemokine processing. Chem Immunol. 1999;72:42–56. [PubMed] [Google Scholar]
- Wada T, Furuichi K, Sakai N, Iwata Y, Kitagawa K, Ishida Y, Kondo T, Hashimoto H, Ishiwata Y, Mukaida N, et al. Gene therapy via blockade of monocyte chemoattractant protein-1 for renal fibrosis. J Am Soc Nephrol. 2004;15:940–948. doi: 10.1097/01.asn.0000120371.09769.80. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Rutledge BJ, Rollins BJ. Structure/activity analysis of human monocyte chemoattractant protein-1 (MCP-1) by mutagenesis. Identification of a mutated protein that inhibits MCP-1-mediated monocyte chemotaxis. J Biol Chem. 1994;269:15918–15924. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.