

See related article in EMBO Molecular Medicine http://dx.doi.org/10.1002/emmm.201100158
CC chemokine ligand 2 (CCL2) or monocyte chemoattractant protein-1 (MCP-1) is an important chemoattractant for monocytes and macrophages (Deshmane et al, 2009). CCL2 is the main ligand for the chemokine receptor CCR2, which mediates CCL2's biological activity. CCR2 is a seven transmembrane spanning Gαi protein-coupled receptor (GPCR) constitutively expressed at high levels on monocytes and macrophages. CCL2 expression is induced in a variety of tissue resident cells (e.g. endothelial cells) and immune cells (e.g. macrophages) by inflammatory mediators (e.g. TNF, LPS and IFNγ). CCL2 mobilizes monocytes out of the bone marrow and recruits circulating monocytes into sites of infection and inflammation. Perhaps more than any other chemokine system, CCL2 and CCR2 have been shown to play important pathological roles in vivo in murine models of disease. For example, in murine models of atherosclerosis, experimental autoimmune encephalomyelitis and obesity-induced diabetes, mice deficient in CCL2 and/or CCR2 have markedly reduced monocyte accumulation in diseased tissue and are protected from disease (Charo & Ransohoff, 2006). Murine studies have also demonstrated the importance of CCL2 in malignancy and metastasis, including a recent study demonstrating a role for CCL2 in recruiting inflammatory monocytes into tumours, which facilitated breast cancer metastasis (Qian et al, 2011).
»…inhibiting CCL2 and CCR2 is still a very active area for drug development with potential application for a multitude of important chronic human diseases.«
Inhibiting CCL2 and CCR2 activity has thus been thought to be a promising new approach for treating inflammatory diseases. Antibody or small molecule chemical blockade of CCL2–CCR2 signalling is one strategy for therapeutic intervention that has shown promise in animal models of inflammatory diseases. However, this approach has not yet translated to effective human therapies. The reasons for the lack of efficacy in the clinic are not known and may relate to inappropriate disease indication, inherent limitations of the drug being tested or the timing of drug administration (Schall & Proudfoot, 2011). For example, anti-CCR2 antibodies were effective for prophylaxis of inflammatory arthritis in a murine model but actually worsened disease when used in a therapeutic context (Bruhl et al, 2004), highlighting the complexity of targeting chemokines to treat disease. While anti-CCL2 therapies have not yet made their way to the clinic, other effective therapies may derive some of their therapeutic efficacy by indirectly inhibiting CCL2 expression, such as the anti-TNF agents (Taylor et al, 2000). In addition, potential complications of inhibiting CCL2 and CCR2 need to be considered since this chemokine system has been shown to play important roles in monocyte-mediated host defense. For example, inhibition of the CCL2–CCR2 chemokine axis has the potential to impair host defenses against pathogens, as has been demonstrated in murine models of Listeria monocytogenes, Mycobacteria tuberculosis, Toxoplasma gondii and Cryptococcal neoformins infection, or impair the immune response to sterile inflammation, as has been demonstrated in murine models of Alzheimer's disease and macular degeneration (Ambati et al, 2003; El Khoury et al, 2007; Serbina et al, 2008). Nonetheless, inhibiting CCL2 and CCR2 is still a very active area for drug development with potential application for a multitude of important chronic human diseases. New therapies that target CCL2 and CCR2 are on the horizon, but whether they can combine sufficient efficacy with ample safety remains to be seen. The elucidation of new mechanisms of CCL2 regulation, such as the glutaminyl cyclase pathway, presents a new option for chemokine-targeted therapeutics.
In this issue of EMBO Molecular Medicine, Cynis et al present evidence that enzymes, such as metalloenzyme transferases, specifically glutaminyl cyclase (QPCT, QC) and iso-glutaminyl cyclase (QPCTL, isoQC), play a role in the in vivo biological activity of CCL2 by catalysing the formation of N-terminal pyroglutamate (pE), which confers resistance against aminopeptidase degradation (Cynis et al, 2011). N-terminal pE CCL2 (pE1-CCL2) was as potent a CCR2 ligand and was as active in chemotaxis assays as the mature uncyclized form of CCL2 (Q1-CCL2). The uncyclized form of CCL2, however, was much more susceptible to degradation by aminopeptidases, such as dipeptidyl peptidase-4 (DDP-4). N-terminally degraded forms of CCL2 have markedly diminished biological activity. Both pE1-CCL2 and Q1-CCL2 were secreted in equal amounts from LPS stimulated cells, and isoQC was found to be the enzyme responsible for the N-terminal cyclic conversion of Q1-CCL2 to pE1-CCL2. If formation of pE was prevented by inhibiting isoQC, CCL2 was more prone to N-terminal degradation, which resulted in less functional CCL2 activity. This had the net effect of decreasing monocyte infiltration in vivo, resulting in decreased lung inflammation, thioglycollate-induced peritonitis and atherosclerosis in murine disease models (Fig 1). Thus, the inhibition of the enzyme isoQC resulted in less functional CCL2 activity in vivo and decreased monocyte recruitment into sites of inflammation.
Figure 1. Q1-CCL2, the mature uncyclized form of CCL2, is converted to cyclized pE1-CCL2 by iso-glutaminyl cyclase (isoQC).
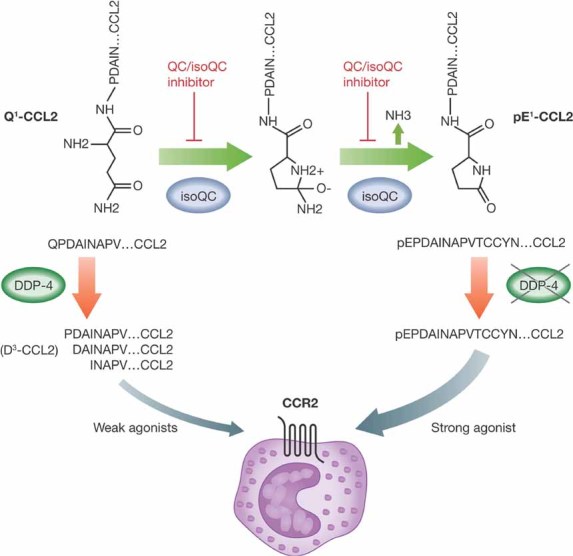
Cyclized pE1-CCL2 is as potent a CCR2 ligand and inducer of moncyte chemotaxis as Q1-CCL2. However, Q1-CCL2 is susceptible to degradation by aminopeptidases, such as DDP-4, which generates products with much weaker biological activity (e.g. D3-CCL2). In contrast, cyclized pE1-CCL2 is resistant to N-terminal proteolytic processing and thus maintains its potency and activity. Inhibition of isoQC enzymatic activity using QC/isoQC inhibitors, results in decreased levels of pE1-CCL2, corresponding increased levels of Q1-CCL2 and subsequent degradation to less active CCL2 variants.
Understanding the structure–function relationship of chemokines has helped to elucidate the importance of the N-terminus. Important structural features of chemokines include a free and flexible N-terminal domain, three antiparallel β-sheets, an N-loop region that follows the first two cysteines and connects the N terminus to the β-sheet region, a C-terminal α-helix and four conserved cysteine residues involved in two intrachain disulphide bonds that stabilize the ‘chemokine structure’ (Fernandez & Lolis, 2002). The importance of the N-terminus for chemokine function is suggested by the ‘two-step’ model of chemokine ligand binding and receptor activation (Fernandez & Lolis, 2002). In this model, the first step is chemokine ligand binding to the chemokine receptor, which leads to a conformational change in the GPCR. The second step is activation of the GPCR by the N-terminus of the chemokine, which interacts with a defined region within the GPCR transmembrane helices. This results in signal transduction through G proteins and a cascade of numerous downstream effector molecules that results in directed cell migration. The strength of signal amplification is determined by the stability of the ligand–receptor complex and receptor–effector protein complexes. Thus, the N-terminus plays a critically important role in initiating this activation and amplification process.
Modification of the N-terminus has been shown to abrogate chemokine activity and, in some cases, even generate competitive antagonists. This has been shown to be the case for several CC chemokines, including CCL2, CCL5, CCL11 and CCL26 (Fernandez & Lolis, 2002). Elimination of the N-terminal glutamate of CCL2 resulted in reduced chemokine activity, while truncation of additional N-terminal residues resulted the generation of a CCR2 antagonist (Deshmane et al, 2009). Furthermore, removal of N-terminal residues 2–8 resulted in a dominant negative mutant that formed a heterodimer with full length mature CCL2 and abrogated its activity. This may explain why inhibiting isoQC was so effective at neutralizing CCL2 activity in vivo as uncyclized forms of CCL2, which are susceptible to N-terminal degradation, have the potential to act as dominant negatives in vivo. N-terminal modifications of other CC chemokines, such as CCL5 (RANTES), also modulated its biological activity. For example, addition of an N-terminal methionine, truncation of two N-terminal residues or modification the N-terminal of CCL5 with aminooxypentane generated potent antagonists for CCL5 chemokine receptors. Studies have revealed that the N-terminus of CCL11 (eotaxin-1) and CCL26 (eotaixn-3) are also crucial for signalling through their receptor CCR3. N-terminal truncation of CCL11 or CCL26 generates potent antagonists of their receptor, CCR3.
Manipulation of the glutaminyl cyclase pathway represents a novel approach to CCL2 regulation. However, caution is warranted before considering a broader application of such a strategy for the treatment of inflammatory diseases. Pyroglutamic acid is the N-terminal amino acid of many proteins, hormones and neurotransmitter peptides, including thyrotropin releasing hormone, gonadotropin-releasing hormone and gastrin (Busby et al, 1987). Blockade of pE formation by inhibiting glutaminyl cyclase may therefore have untoward side effects in the nervous and endocrine systems that may not be predictable. The authors suggest that the promiscuity of the glutaminyl cyclase inhibition approach may be a strength of this therapeutic strategy; however, this may actually represent a potential drawback, particularly if one desires only a specific block of CCL2 since all human MCPs may be substrates for QC. Finally, since QC/isoQC is a Golgi-resident enzyme, inhibitors of QC/isoQC need to act intracellularly, introducing an additional level of complexity compared to inhibitors that target extracellular ligands or receptors. With these caveats in mind, as additional mechanisms of chemokine regulation continue to be elucidated, these discoveries will foster a greater understanding of chemokine and chemokine receptor biology, which will hopefully lead to the development of new and exciting agents for the treatment of inflammatory diseases that are both effective and safe.
Acknowledgments
The authors declare that they have no conflict of interest.
References
- Ambati J, Anand A, Fernandez S, et al. An animal model of age-related macular degeneration in senescent Ccl2- or Ccr2-deficient mice. Nat Med. 2003;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- Bruhl H, Cihak J, Schneider MA, Plachy J, Rupp T, Wenzel I, Shakarami M, Milz S, Ellwart JW, Stangassinger M, et al. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol. 2004;172:890–898. doi: 10.4049/jimmunol.172.2.890. [DOI] [PubMed] [Google Scholar]
- Busby WH, Jr, Quackenbush GE, Humm J, Youngblood WW, Kizer JS. An enzyme(s) that converts glutaminyl-peptides into pyroglutamyl-peptides. Presence in pituitary, brain, adrenal medulla, and lymphocytes. J Biol Chem. 1987;262:8532–8536. [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Cynis H, Hoffmann T, Friedrich D, Kahlen A, Gans K, Kleinschmidt M, Rahfeld J-U, Wolf R, Wermann M, Stephan A, et al. The isoenzyme of glutaminyl cyclase is an important regulator of monocyte infiltration under inflammatory conditions. EMBO Mol Med. 2011 doi: 10.1002/emmm.201100158. DOI: 10.1002/emmm.201100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Fernandez EJ, Lolis E. Structure, function, and inhibition of chemokines. Annu Rev Pharmacol Toxicol. 2002;42:469–499. doi: 10.1146/annurev.pharmtox.42.091901.115838. [DOI] [PubMed] [Google Scholar]
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schall TJ, Proudfoot AE. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat Rev Immunol. 2011;11:355–363. doi: 10.1038/nri2972. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PC, Peters AM, Paleolog E, Chapman PT, Elliott MJ, McCloskey R, Feldmann M, Maini RN. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43:38–47. doi: 10.1002/1529-0131(200001)43:1<38::AID-ANR6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]