

Abstract
Obesity is a well-known risk factor for the development of secondary complications such as type 2 diabetes. However, only a part of the obese population develops secondary metabolic disorders. Here, we identify the transcription factor retinoid-related orphan receptor gamma (RORγ) as a negative regulator of adipocyte differentiation through expression of its newly identified target gene matrix metalloproteinase 3. In vivo differentiation of adipocyte progenitor cells from Rorγ-deficient mice is enhanced and obese Rorγ−/− mice show decreased adipocyte sizes. These small adipocytes are highly insulin sensitive, leading to an improved control of circulating free fatty acids. Ultimately, Rorγ−/− mice are protected from hyperglycemia and insulin resistance in the state of obesity. In adipose stromal-vascular fraction from obese human subjects, Rorγ expression is correlated with adipocyte size and negatively correlated with adipogenesis and insulin sensitivity. Taken together, our findings identify RORγ as a factor, which controls adipogenesis as well as adipocyte size and modulates insulin sensitivity in obesity. RORγ might therefore serve as a novel pharmaceutical target to treat obesity-associated insulin resistance.
Keywords: adipogenesis, matrix metalloproteinase 3, obesity, retinoid-related orphan receptor gamma, type 2 diabetes
INTRODUCTION
Obesity is a major risk factor for the development of type 2 diabetes and the metabolic syndrome. Even though there is a clear link between obesity and insulin sensitivity, the degree of insulin resistance in obese subjects can vary enormously (Desai et al, 2006), one possible reason being the variability of adipose tissue plasticity (Sethi & Vidal-Puig, 2007). Adipose tissue mass increases during obesity via two distinct mechanisms, increasing cell number (hyperplasia) and/or increasing adipocyte size (hypertrophy) (Jo et al, 2009; Spalding et al, 2008). Hyperplasia occurs via de novo differentiation of preadipocytes, which reside in the stromal-vascular fraction (SVF) of adipose tissue (Gesta et al, 2007; Rodeheffer et al, 2008). The balance between hyperplasia and hypertrophy strongly influences the metabolic outcome of obesity. While smaller cells retain insulin sensitivity and normal function, large hypertrophic cells are insulin resistant and change their secretory profile towards pro-inflammatory adipocytokines (Roberts et al, 2009; Tilg & Moschen, 2006). As a result, lipolytic release of free fatty acids is enhanced in large adipocytes, leading to cytotoxic fatty acid accumulation in extra-adipose tissue (Unger, 2003). Shifting the balance towards hyperplasia replenishes the pool of small, functional adipocytes, thus ameliorating the metabolic consequences of obesity. A pharmacological example for this mechanism is the clinical use of glitazones activating PPARγ (Crossno et al, 2006; Johnson et al, 2007), which leads to an increase in adipose tissue mass with a shift towards smaller, insulin sensitive adipocytes.
An important regulator of the adipogenic process is the proteolytic remodelling of the extracellular matrix, as preadipocytes have to undergo cell shape transition during differentiation (Kubo et al, 2000). Several classes of metalloproteinases (MMPs) affect the adipogenic process (Lilla et al, 2002). For example, global inhibition of MMP activity inhibits differentiation of preadipocytes and adipose tissue development (Bouloumie et al, 2001; Lijnen et al, 2002). On the other hand, Mmp3-deficient mice show accelerated adipogenesis during mammary gland involution, suggesting a negative regulatory role for MMP3 in adipocyte differentiation (Alexander et al, 2001).
Retinoid-related orphan receptor gamma (RORγ) is a nuclear receptor with an N-terminal (A/B) domain followed by the DNA-binding domain and a C-terminal ligand-binding domain. RORs bind as monomers to the consensus half-site PuGGTCA (Jetten, 2009). Recently, several ligands have been proposed to regulate RORγ transcriptional activity (Solt et al, 2011; Wang et al, 2009). RORγ has two isoforms: the main isoform is expressed in adipose tissue, liver, muscle and kidney, whereas expression of RORγt is restricted to immune cells. RORγt has been studied in the context of interleukin (IL)-17 producing T-helper cells (Zhang et al, 2008; Zhou et al, 2008); the role of RORγ in other tissues, however, is less clear (Meissburger & Wolfrum, 2008). With regard to adipose tissue, single nucleotide polymorphisms of RORγ have been linked to fat accumulation in cattle (Barendse et al, 2007). During in vitro adipogenesis, Rorγ expression is upregulated in late stages of differentiation (Austin et al, 1998). However, a functional role for RORγ in adipose tissue plasticity has yet to be identified.
In this study, we investigated the mechanistic role of RORγ in adipocyte differentiation and show that RORγ is an important negative regulator of adipogenesis. By controlling fat cell development RORγ influences the progression of obesity-associated insulin resistance in mice and humans.
RESULTS
RORγ regulates adipogenesis
In order to identify candidate genes that regulate adipose tissue plasticity, we analysed transcription profiles of visceral adipose SVF from lean and obese patients. The highest upregulated gene in the state of obesity was the transcription factor Rorγ (Table S1 of Supporting information). Increased expression of Rorγ was confirmed by quantitative mRNA analysis in visceral adipose tissue of obese patients versus lean patients (mean values lean 7.9 ± 1.1 vs. obese 17.6 ± 2.9, Fig S1A of Supporting information). As this factor is highly expressed in the preadipocyte-containing SVF, we analysed whether RORγ plays a role in regulating adipose tissue plasticity.
During differentiation, mRNA and protein expression of Rorγ is high in confluent 3T3-L1 preadipocytes, whereas the isoform RORγt could not be detected (Figs 1A and 2A, data not shown). Upon induction of differentiation, Rorγ mRNA and protein levels exhibit a sharp decline suggesting that RORγ might influence the process of differentiation (Figs 1A and 2A). To follow up on this finding, we transiently expressed RORγ in 3T3-L1 preadipocytes and analysed the effect on adipogenesis. RORγ overexpression in 3T3-L1 cells decreases the amount of differentiated adipocytes as well as expression of the mature adipocyte marker adipocyte fatty acid binding protein (A-FABP; Fig 1B and C and Fig S1B of Supporting information). This diminished adipogenesis is retained, albeit to a lesser extent, when rosiglitazone is added, suggesting that RORγ acts upstream of PPARγ (Fig 1B and C). Conversely, siRNA mediated knockdown of RORγ increases the adipogenic potential of 3T3-L1 preadipocytes (Fig 1D and E and Fig S1B of Supporting information). No changes were found in the mRNA expression of closely related family members Rorα and Rorβ suggesting that the effect of RORγ is not due to compensatory regulation of other family members (Fig S1C of Supporting information).
Figure 1. RORγ inhibits adipogenesis.

- A. mRNA expression profile of Rorγ and Mmp3 during 3T3-L1 adipogenesis. Expression was normalized to 36b4 (n = 4).
- B. Fluorescent staining of 3T3-L1 adipocytes overexpressing RORγ or empty vector at day 7 of differentiation with or without rosiglitazone. Automated image acquisition (blue: nuclei, red: cytosol, green: lipid droplets, 12 pictures per well, scale: 100 µm) was followed by automated image quantification using CellProfiler (n = 4).
- C. Whole cell extracts of differentiated 3T3-L1 adipocytes were blotted against the mature adipocyte marker A-FABP using γ-Tubulin as a loading control.
- D,E. Fluorescent staining (n = 4) and A-FABP Western blot of differentiated 3T3-L1 cells transfected with control siRNA or siRNA against RORγ 3 days before induction of differentiation. The results shown are representative of three independent differentiation experiments. Values represent means ± SD, *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001.
Figure 2. RORγ inhibits expression and activity of differentiation genes.

- A. Western blot of nuclear extracts (RORγ, PPARγ, C/EBPα, C/EBPβ and C/EBPδ) or cytosolic extracts (MMP3, PREF-1, LPL and Adiponectin) during differentiation of 3T3-L1 cells overexpressing RORγ or empty vector. Differentiation without rosiglitazone was induced at time point 0 h. Lamin B and γ-Tubulin were used as loading controls.
- B,C,D. qPCR for the adipogenic genes c-Jun, c-Fos and A-Fabp during differentiation of 3T3-L1 cells overexpressing RORγ or empty vector (n = 4).
- E. Firefly luciferase activity of the PPARγ response element (PPRE) normalized to renilla luciferase before, 1 and 2 days after induction of differentiation in 3T3-L1 cells overexpressing RORγ or empty vector (n = 4).
- F. Firefly luciferase activity of PPARγ promoter normalized to renilla luciferase in 3T3-L1 cells overexpressing RORγ or empty vector during differentiation at indicated time points (n = 8). The results shown are representative of three independent experiments. Values represent means ± SD, *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001.
To further pinpoint at which stage RORγ interferes with the differentiation process, we analysed expression of key regulatory adipogenic genes in a time course of differentiation. Transient overexpression of RORγ increased endogenous protein levels approximately 3–4-fold and resulted in a decreased expression of key adipogenic proteins such as C/ebpα, C/ebpβ, C/ebpδ and Pparγ (Fig 2A). In contrast, protein expression of Pref1, a marker for undifferentiated preadipocytes, was not downregulated upon induction of differentiation in cells overexpressing RORγ. These findings were corroborated by the fact that induction of adipogenic genes such as c-Jun, c-Fos and A-Fabp was repressed by RORγ (Fig 2B–D). Taken together, these data demonstrate that RORγ exerts its inhibitory effect during the initial steps of adipogenesis repressing early adipogenic genes.
To confirm that RORγ acts upstream of PPARγ, we measured PPARγ transcriptional activity in differentiating cells transiently overexpressing RORγ or control vector. PPARγ activity in RORγ transfected cells was suppressed in the first 2 days after induction of differentiation (Fig 2E), which is in line with our observation that overexpression of RORγ led to a reduction in Pparγ expression (Fig 2A). On the other hand, activity of the RORγ promoter was not affected by the PPARγ agonist rosiglitazone suggesting that PPARγ does not regulate Rorγ expression (Fig S1D of Supporting information). As REV-ERBα is known to modulate transcription of PPARγ (Wang & Lazar, 2008), we tested whether RORγ could directly influence Pparγ expression or whether the effect was due to reduced C/EBP activity, as would be expected from the lowered expression levels of these transcription factors (Fig 2A). As seen in Fig 2F, RORγ did not influence PPARγ promoter activity at early time points. However, induction of PPARγ during differentiation was inhibited in cells transfected with RORγ. Taken together, these findings demonstrate that RORγ tightly regulates adipocyte differentiation by regulating early phase genes, which in turn leads to a reduction in PPARγ expression and activity.
The effect of RORγ on adipogenesis is mediated by MMP3
To identify new target genes of RORγ that are involved in the repression of adipogenesis, we analysed transcription profiles of 3T3-L1 preadipocytes transiently overexpressing RORγ (Table S2 of Supporting information). Genes upregulated by RORγ and highly expressed in murine SVF were chosen for a functional siRNA screen to test their impact on adipogenesis (Fig 3A). Furthermore, we analysed whether transcription of these genes is regulated by RORγ using a nuclear run-on assay (Fig 3B and Fig S1E of Supporting information). Whereas transcription of several identified genes was upregulated by RORγ, only knockdown of MMP3 led to an increase in adipocyte differentiation (Fig 3A). We confirmed Mmp3 mRNA expression to be upregulated by transient or viral overexpression of RORγ (Fig 3C). Interestingly, expression of Mmp3 mRNA and protein during early 3T3-L1 adipogenesis closely follows the expression profile of Rorγ with high expression in preadipocytes that is downregulated upon induction of differentiation (Figs 1A and 2A). To elucidate whether MMP3 is a direct target gene of RORγ, we screened the MMP3 promoter for potential RREs. The promoter sequence contains two potential RREs: the first RRE (AGGTCA) matches the consensus half-site, whereas the second RRE has the half-core motif AGGTAA. Using different luciferase constructs, we could show that RORγ-induced transcriptional activation of the MMP3 promoter depends on the first, but not on the second RRE and reaches levels similar to the known RORγ target gene IL-17 (Fig 3D). These findings were corroborated by ChIP assays demonstrating binding of RORγ to the first but not to the second RRE in the MMP3 promoter (Fig 3E).
Figure 3. MMP3 is a target gene of RORγ that inhibits adipogenesis.
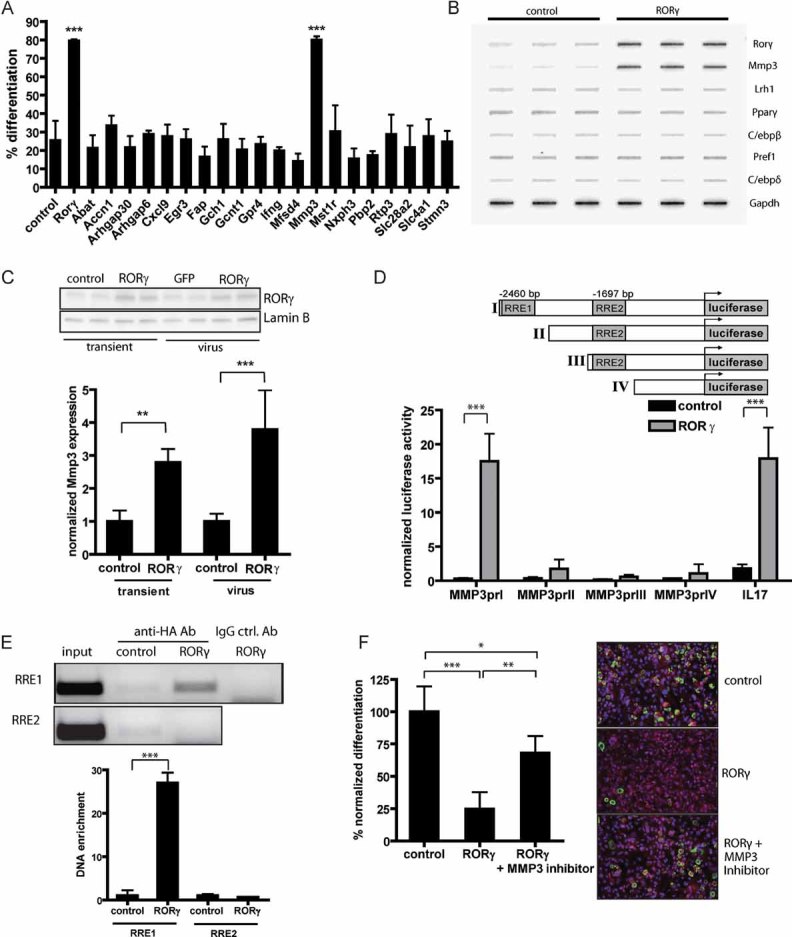
- Functional siRNA screen of potential RORγ target genes identified by microarray analysis of 3T3-L1 cells overexpressing RORγ. 3T3-L1 cells were transfected with siRNA pools, differentiated and fluorescently stained to assess the percentage of differentiated cells (n = 4).
- Nuclear run-on assay for transcriptional activity of genes in undifferentiated 3T3-L1 cells infected with control or RORγ-overexpressing lentivirus. 32P-labelled mRNA was hybridized to cDNA from the indicated genes on a slot blot and analysed by autoradiography.
- Expression of Mmp3 mRNA in 3T3-L1 preadipocytes overexpressing RORγ by transient transfection or viral infection.
- MMP3 promoter constructs with potential ROR response elements (RREs) cloned into a luciferase reporter vector. Luciferase assay in 3T3-L1 preadipocytes overexpressing RORγ or control vector 48 h after transfection normalized to renilla luciferase activity. Activation of IL17 promoter was used as a control (n = 4).
- Chromatin immunoprecipitation of RORγ in 3T3-L1 cells using anti-HA antibody 6 days after infection with RORγ or control lentivirus. DNA was quantified by qPCR using specific primers for RRE1 and RRE2 and normalized to input (n = 3).
- Fluorescent staining of differentiated 3T3-L1 adipocytes expressing RORγ or control vector during differentiation treated with or without 20 µM of MMP3 inhibitor 2 days before and after induction of differentiation (scale: 100 µm). The results shown are representative of three independent experiments. Values represent means ± SD, *p ≤ 0.05 and **p ≤ 0.01, ***p ≤ 0.001.
To assess whether the effect of RORγ on adipogenesis is mediated by MMP3, we added a MMP3 inhibitor during adipocyte differentiation. Similar to siRNA-mediated knockdown of MMP3 (Fig 3A), pharmacological inhibition of MMP3 enhances 3T3-L1 adipogenesis (Fig S2A of Supporting information). Moreover, inhibition of differentiation by RORγ can be partially rescued with the MMP3 inhibitor (Fig 3F). Additionally, we could show that overexpression of MMP3 prevents the increased differentiation capacity caused by siRNA-mediated knockdown of RORγ (Fig S2B of Supporting information). Conversely, siRNA-mediated knockdown of MMP3 rescues the inhibition of differentiation caused by RORγ, whereas combined knockdown of MMP3 and RORγ does not enhance differentiation compared to knockdown of RORγ alone (Fig S2C of Supporting information). To analyse whether the effect of RORγ on adipogenesis and Mmp3 expression is specific, we measured the effect of RORα and a DNA binding-deficient mutant of RORγ (RORγm) on Mmp3 expression and adipogenesis. We could show that RORα inhibited differentiation as described previously (Ohoka et al, 2009) but did not modulate Mmp3 expression. A DNA binding-deficient RORγm, which does not bind to DNA because of the mutations C31A and C49A in the DNA binding domain (data not shown), influenced neither adipogenesis nor Mmp3 expression (Fig S2D and E of Supporting information). Taken together, these results show that MMP3 is a direct target of RORγ and that inhibition of adipogenesis by RORγ is at least partly mediated through induction of MMP3.
RORγ is a key factor regulating preadipocyte differentiation in vivo
To assess whether the effect of RORγ on adipogenesis could be recapitulated in vivo, we implanted murine adipose SVF containing adipocyte precursor cells into mice (Kawaguchi et al, 1998, 1999). SVF from GFP-expressing mice resuspended in Matrigel and injected into the subcutaneous layer of the neck could be retrieved as a distinct pad 4–6 weeks after transplantation. Cells in these pads stained positive for GFP as well as for the adipocyte marker A-FABP and had a distinct adipocyte structure when acceptor mice were fed a high-fed diet (Fig 4A). Taken together, these results demonstrate that preadipocytes derived from a donor mouse can undergo adipogenesis in vivo without major host cell infiltration. To determine the effect of RORγ on the process of adipogenesis in vivo, we injected GFP positive SVF of wild-type and Rorγ knockout animals into C57Bl/6 acceptor mice. After 4 weeks of high-fat diet excised pads with Rorγ−/− cells contained more mature adipocytes than pads with cells from control animals, demonstrating that the absence of RORγ potentiates adipocyte differentiation (Fig 4A, right panel). Quantification of adipocyte numbers by automated image analysis demonstrated a 2.5-fold increase in de novo formed adipocytes in the absence of RORγ (Fig 4B). In contrast, differentiation of SVF implanted into mice fed with chow diet was lower compared to high-fat diet feeding and did not differ between Rorγ−/− and wild-type SVF cells (Fig S3A of Supporting information).
Figure 4. Loss of RORγ leads to increased adipocyte formation in vivo.

- A. Injection of SVF from different donor mice (as indicated) into the scapular subcutaneous region of C57Bl/6 mice. Fat pads were excised after 6 or 4 weeks of HFD feeding and stained as indicated.
- B. Quantification of adipocyte differentiation in SVF transplants from wild-type and Rorγ−/− mice fed a high-fat diet for 4 weeks (n = 5).
- C. Mean adipocyte size of Rorγ−/− and wild-type mice crossed into the ob/ob background or fed a high-fat diet for 8 weeks, insets are fat sections from representative samples of female DIO wild-type and Rorγ−/− mice (n = 7–10).
- D,E. Adipocyte size distributions from subcutaneous and visceral adipose tissue of female DIO wild-type and Rorγ−/− mice (n = 7).
- F. Mmp3 mRNA expression in visceral SVF of Rorγ−/− and wild-type mice on chow diet (n = 5). Values represent means ± SD, *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001.
Loss of RORγ leads to increased formation of adipocytes in mouse models of obesity
We next examined whether the altered differentiation capacity of Rorγ−/− preadipocytes affects fat cell size within the adipose tissue. Fat cell sizes from adipose tissue sections were determined in obese Rorγ−/− and wild-type animals by automated image analysis. Whereas Rorγ−/− animals on a chow diet showed mildly decreased mean adipocyte sizes compared to their wild-type littermates (Fig S3B of Supporting information), this effect was potentiated in obese Rorγ−/− animals fed a high-fat diet for 8 weeks (DIO) or crossed into the leptin-deficient ob/ob background (Fig 4C). Analysis of the adipocyte size distribution revealed that in Rorγ−/− animals especially the number of small adipocytes (4000–8000 µm2 in size) is increased (Fig 4D and E). To estimate the number of adipocytes, we also quantified whole fat volume using CT scanning and fat pad excision. Fat pad weights did not differ in mice fed a chow diet and were mildly decreased in obese Rorγ−/− mice (data not shown, Fig S3C and D of Supporting information). Based on the average adipocyte volume and the size of the fat pad, we quantified the increase in adipocyte number in the different models. Such calculations will only yield estimations since the cells are not perfectly round and sections are not cut in the middle of each cell. Nevertheless, we observed a slight increase in adipocyte numbers in male DIO subcutaneous and visceral adipose tissue in Rorγ−/− animals compared to wild-type littermates (6 and 11%). In female DIO and in male ob/ob subcutaneous and visceral adipose tissue, the calculated increase in adipocyte numbers was much more pronounced (average of 40%). To confirm that the observed changes in adipocyte size were caused by altered differentiation capacity rather than altered adipocyte function, functional assays were performed in 3T3-L1 adipocytes with siRNA-mediated knockdown or overexpression of RORγ. Neither glucose uptake nor lipolysis were altered by RORγ (Fig S3E–G of Supporting information, data not shown). Differences in adipocyte size were not due to altered energy balance as Rorγ−/− mice did not exhibit altered food intake or oxygen consumption in metabolic cage experiments after 7 weeks of high-fat diet (Fig S3H and I of Supporting information). As MMP3 is a direct target gene of RORγ and at least partially responsible for the alteration of adipogenesis, we compared mRNA expression of Mmp3 in visceral SVF of 12-week-old Rorγ−/− and wild-type animals on a normal chow diet. Mmp3 expression was reduced 2.4-fold in the knockout animals (Fig 4F) suggesting that MMP3 is regulated by RORγ in vivo.
Loss of RORγ protects against obesity-induced insulin resistance
As Rorγ−/− mice show alterations in adipocyte size distribution, we examined the metabolic consequences of altered fat cell size in obese mouse models. Mean body weight was slightly reduced for male and female knockout animals on a high-fat diet (Fig 5A and Fig S4A of Supporting information), whereas chow fed animals did not show any changes in body weight (Fig S4B of Supporting information). In all models, Rorγ−/− mice were protected from obesity-induced hyperglycemia (Fig 5B), whereas no effect on glucose levels was observed in chow diet fed animals (Fig S4C of Supporting information). Decreased blood glucose levels in obese Rorγ−/− mice were accompanied by lower insulin levels in the fasted and in the fed state (Fig 5C and Fig S4D of Supporting information). In an insulin tolerance test, male Rorγ−/− animals showed improved insulin sensitivity compared to control mice after 8 weeks of high-fat diet (Fig 5D). Besides whole body insulin sensitivity, we also tested insulin sensitivity of isolated primary adipocytes from obese Rorγ−/− mice and control littermates. As expected from the observed differences in adipocyte size, insulin-mediated repression of lipolysis in obese Rorγ−/− animals was similar to wild-type chow fed animals. In contrast, adipocytes from obese wild-type mice were completely insulin-resistant (Fig 5E). In line with this, concentrations of circulating free fatty acids were significantly decreased in obese Rorγ−/− mice both in fasted and fed state (Fig 5F and Fig S4E of Supporting information). Triglyceride concentrations were mildly altered only in male high-fat diet animals (Fig S4F of Supporting information).
Figure 5. Loss of RORγ protects against diet-induced insulin resistance in mice.

- A. Body weight of Rorγ−/− and wild-type mice (12 weeks) crossed into the ob/ob background or fed a high-fat diet (DIO) for 8 weeks (n = 7–10).
- B,C. Fasting blood glucose and serum insulin of ob/ob and high-fat diet Rorγ−/− and wild-type mice (n = 7–10).
- D. Insulin tolerance test with male Rorγ−/− and wild-type mice after 8 weeks of high-fat diet injected intraperitoneal (i.p.) with 0.75 U/kg insulin (n = 7).
- E. In vitro lipolysis of isolated adipocytes from 12-week-old wild-type mice on a chow diet, ob/ob wild-type and ob/ob Rorγ−/− mice treated with or without insulin normalized to cell number and set to 100% for basal lipolysis (n = 4).
- F. Fasting free fatty acid serum concentrations of ob/ob and high-fat diet Rorγ−/− and wild-type mice (n = 7–10).
- G. Expression of Rorγ, Mmp3 and A-Fabp in visceral adipose tissue of male C57Bl/6 mice during a time course of high-fat diet feeding. Body weight and mean adipocyte size of the animals was monitored.
To analyse how Rorγ and Mmp3 expression is regulated in the course of obesity development and how this regulation is linked to fat cell size, we monitored body weight, adipocyte size and mRNA expression in visceral adipose tissue of mice in a time course of high-fat diet feeding for 24 weeks (Fig 5G). Interestingly, Rorγ expression initially dropped within the first weeks of high-fat diet feeding before expression increased during later stages of diet-induced obesity. Expression of the RORγ target gene Mmp3 also increased at later stages of obesity. When looking at mean adipocyte size during the course of high-fat diet feeding, increased Rorγ expression at later stages paralleled accelerated adipocyte hypertrophy.
As RORγ is expressed in muscle tissue as well as in fat, we analysed the ability of RORγ to modulate glucose uptake in muscle cells. Neither overexpression by viral infection with Ad-RORγ, nor siRNA-mediated knockdown of RORγ in differentiated C2C12 cells influenced basal or insulin-stimulated glucose uptake, whereas knockdown of insulin receptor (IR) completely abolished insulin-induced glucose uptake. These results suggest that the metabolic changes observed in Rorγ−/− mice are not due to altered muscle function (Fig S4G–J of Supporting information).
As RORγ is implicated in immunological processes, we quantified different markers of inflammation associated with insulin resistance including TNFα, INFγ, IL-1β, IL-2, IL-12 and IL-6 in serum of Rorγ−/− mice. To further address the inflammatory state of adipose tissue in obese wild-type and Rorγ−/− mice, we also measured fat-specific expression of the pro-inflammatory cytokines Tnfα and Il-6 as well as Cd68 and Emr1 as markers of macrophage infiltration. None of the inflammation markers linked to adipose tissue inflammation and macrophage infiltration was significantly altered in these mice (Fig S5A and B of Supporting information). Furthermore, we analysed expression of RORγ in different subfractions of adipose SVF. As can be seen in Fig S5C of Supporting information, RORγ is expressed both in CD4+ immune cells as well as in preadipocytes (Rodeheffer et al, 2008), albeit with a approximately fourfold higher expression in the latter population.
Taken together, our results show that adipose tissue of obese Rorγ−/− mice is composed of small, insulin-sensitive adipocytes with intact lipolytic control, which prevents systemic free fatty acid overload and diet-induced insulin resistance.
RORγ expression in obese subjects predicts adipocyte size and insulin sensitivity
As we have shown that ablation of RORγ serves as a protective mechanism against the development of obesity-associated insulin resistance in mice, we next analysed the link between Rorγ expression and insulin resistance in human obesity. To this end, we obtained subcutaneous fat biopsies from 21 obese patients (11 female and 10 male) with body mass indices (BMI) of 32–56 kg/m2. As shown in previous studies, we observed a significant link between mean fat cell size and whole body insulin resistance determined by HOMA-index (Roberts et al, 2009; Fig S6A of Supporting information). To quantify the hyperplastic response of adipose tissue, we measured the differentiation potential of human SVF from subcutaneous adipose tissue biopsies using ex vivo differentiation assays, in which adipogenesis was quantified by high-throughput image analysis (Fig S6B of Supporting information). Interestingly, we found an inverse correlation between mean fat cell size and the differentiation potential of the corresponding SVF, which suggests that fat cell size is also partly determined by the ability to produce new fat cells (Fig S6C of Supporting information). Rorγ mRNA expression in SVF showed a positive correlation with adipocyte size and an inverse correlation with adipocyte differentiation potential (Fig 6A and B), demonstrating that Rorγ expression predicts adipocyte cell number in fat of obese patients similar to mice. Furthermore, expression of the RORγ target gene Mmp3 in the SVF is correlated both with RORγ expression and with adipocyte size (Fig 6C and Fig S6D of Supporting information). Rorγ mRNA expression in the SVF of obese subjects correlates with insulin sensitivity as measured by HOMA index (Fig 6D); however, Rorγ mRNA expression does not show a significant correlation with BMI suggesting that RORγ is a predictor of insulin resistance independent of body weight in obese patients (Fig S6E of Supporting information).
Figure 6. Expression of RORγ correlates with adipocyte size and insulin resistance in humans.
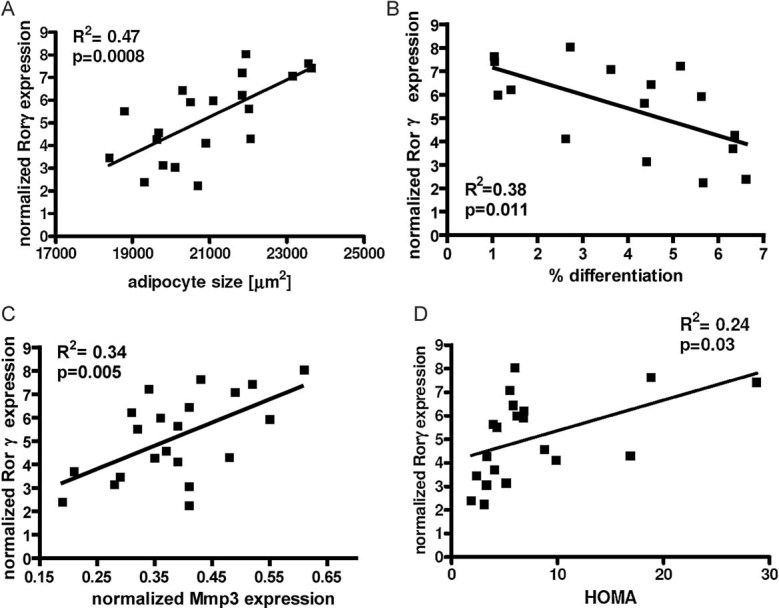
- Rorγ mRNA expression normalized to Gapdh in human subcutaneous SVF of obese subjects plotted against mean adipocyte cell size (n = 20).
- Rorγ mRNA expression in human subcutaneous SVF of obese subjects plotted against percent in vitro differentiated adipocytes of human subcutaneous SVF (n = 16).
- Rorγ mRNA expression plotted against Mmp3 expression in subcutaneous SVF of obese patients (n = 21).
- Rorγ expression in subcutaneous SVF plotted against HOMA index of obese subjects (n = 20). Values represent means ± SD, *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001.
DISCUSSION
Our results identify RORγ as a negative regulator of adipogenesis through expression of its target gene MMP3. The adipocyte size profile in obese Rorγ knockout mice is shifted towards smaller cells and these mice remain insulin sensitive in adipose tissue as well as in the whole body. Since adipocyte precursor cells from Rorγ knockout mice show a higher differentiation capacity in a physiological environment, it is intriguing to speculate that Rorγ knockout mice respond to nutrient overload with an increased hyperpastic response in adipose tissue, generating small, insulin-sensitive adipocytes by de novo differentiation. Indeed, it has been proposed that stimulation of adipogenesis improves whole body insulin sensitivity through reduced systemic free fatty acid overload and that this is one reason for the differences between metabolically healthy and metabolically unhealthy obese subjects (Lonn et al, 2010; O'Connell et al, 2010).
During high-fat diet feeding of mice, adipocyte size is only mildly affected in the early phase of weight gain, whereas adipocyte hypertrophy mainly occurs in late adiposity. This mirrors fat cell development in humans proposed by Spalding et al who suggested that adipocyte hyperplasia decreases with age (Spalding et al, 2008). Interestingly, Rorγ expression in adipose tissue drops at the onset of high-fat diet feeding and increases later on concomitant with pronounced adipocyte hypertrophy. These results further hint at a role for Rorγ in adipogenesis in response to nutritional overload.
To establish a direct link between RORγ, adipocyte hyperplasia and insulin sensitivity, a conditional knockout of RORγ in adipocyte precursor cells would be necessary. Unfortunately, no specific preadipocyte marker has been described to date that would allow recombination of RORγ solely in adipocyte precursor cells. Furthermore, it is possible that other tissues and cell types also contribute to the observed phenotype. To this end, we tested the role of RORγ in mature adipocytes and metabolic muscle function, as RORγ is expressed in these cell types (Austin et al, 1998; Raichur et al, 2007). Neither adipocytes nor muscle cells showed altered glucose metabolism dependent on RORγ. Moreover, improved insulin sensitivity in Rorγ knockout mice apparently does not depend on decreased expression of classical inflammatory cytokines. However, it is possible that secretion of the RORγ target gene MMP3 from adipocytes and other cell types influences adipogenesis in a paracrine manner and/or exerts additional functions. Therefore, it would be interesting to explore whether expression of MMP3 is regulated by RORγ in other cell types apart from adipocytes and preadipocytes and whether lack of MMP3 mirrors the metabolic phenotype of Rorγ knockout animals.
In obese, nondiabetic patients, Mmp3 expression is decreased in subcutaneous adipose tissue compared to lean subjects (Traurig et al, 2006), whereas we observe an increase of Rorγ and Mmp3 in the SVF of visceral adipose tissue from obese, diabetic patients. These discrepancies could be due to a fat depot-specific effect, as visceral adipose tissue is an independent determinant for the development of obesity-associated metabolic complications (Heilbronn et al, 2004; Ravussin & Smith, 2002), or due to differences in the degree of insulin resistance and further studies are needed to address this point. Within a cohort of obese patients, however, our data suggests that Rorγ expression is mainly associated with changes in insulin sensitivity.
The link between RORγ and MMP3 regulating adipogenesis is intriguing as extracellular matrix remodelling by MMPs is an important aspect of adipocyte differentiation (Lilla et al, 2002). Whereas global inhibition of MMPs reduces differentiation (Chavey et al, 2003), Mmp3−/− mice show an approximately 35% accelerated adipocyte differentiation during mammary gland involution (Alexander et al, 2001). Given the low specificity of MMPs for distinct extracellular matrix proteins, it will be a challenge to explore the exact nature of matrix reorganization by MMP3 leading to altered adipocyte differentiation in future studies.
In a time course of adipocyte differentiation, RORγ and MMP3 are immediately repressed upon induction paralleled by downregulation of other inhibitory markers such as Pref1 (Smas & Sul, 1993), which suggests an inhibitory role for RORγ in early differentiation events. This is corroborated by the fact that RORγ inhibits expression of early transcription factors such as C/ebpβ and C/ebpδ leading to decreased expression of C/ebpα and Pparγ and thus to reduced transcriptional activity of PPARγ. In contrast to REV-ERBα, which directly modulates activity of the PPARγ promoter (Wang & Lazar, 2008), RORγ did not alter PPARγ promoter activity in undifferentiated cells. As RORγ-mediated repression of adipogenesis cannot be reversed by the PPARγ activator rosiglitazone, it can be presumed that RORγ inhibits adipogenesis at an early stage through a PPARγ-independent mechanism, which ultimately leads to a less pronounced induction of PPARγ. RORα has been previously described to inhibit adipogenesis by blocking the transcriptional activity of C/EBPβ, thus reducing the induction of Pparγ and C/ebpα expression (Ohoka et al, 2009). As RORγ regulates genes preceding induction of C/EBPβ and RORα does not regulate Mmp3 expression it is likely that RORγ and RORα regulate adipogenesis through different pathways.
Mice with decreased RORα (sg/sg) expression are resistant towards the development of diet-induced obesity (Lau et al, 2008). The mechanism for this phenotype is tightly linked to lipid mobilization as well as increased energy expenditure and also involves central regulation of food intake. These findings are quite different from RORγ-deficient mice, which do not show major changes in triglyceride or cholesterol serum levels nor altered energy expenditure suggesting that RORα and RORγ control different metabolic aspects under high-fat diet feeding.
Loss of RORγ leads to an increase in total adipocyte numbers in female DIO mice and in ob/ob mice, whereas this effect is less pronounced in male DIO mice. One possibility could be a sexual dimorphism phenotype, as regulation of adipogenesis includes gender specific aspects (Dieudonne et al, 2000). Further studies will be needed to address this point.
Given the fact that RORγ in adipose tissue is linked to insulin sensitivity and that activity can be regulated by ligands, that is by hydroxylated cholesterols or the sulfonamide T0901317 (Kumar et al, 2009; Solt et al, 2011; Wang et al, 2009) RORγ might be a novel target for the treatment of obesity-associated insulin resistance. Inhibition of RORγ would mirror the effect of glitazones in clinical use, where activation of PPARγ leads to improved insulin sensitivity partly through de novo adipocyte differentiation (Czoski-Murray et al, 2004; Zanchi et al, 2007).
Taken together, our studies reveal RORγ as a novel factor in adipocyte differentiation through regulation of its target gene MMP3. RORγ expression in adipose tissue is linked to adipocyte size, thereby influencing lipolytic control and insulin resistance in obesity.
MATERIALS AND METHODS
Animals
All mouse models were maintained in the C57Bl/6J background and kept on a 12-h/12-h light/dark cycle in a pathogen-free animal facility. Groups of mice were fed a high-fat diet (Provimi Kliba AG) containing 60% fat for 8 weeks. RORγ global knockout mice (Sun et al, 2000) were a kind gift from D. Littmann.
Plasmids
RORγ cDNA amplified from murine adipocyte cDNA was subcloned into pCR2.1 (Invitrogen) and subsequently into pcDNA3 (Invitrogen) for transfection or pCCL.sin.PPT.hPGK.IRES.GFP.PRE for lentiviral production. For adenoviral production HA-tagged RORγ was cloned into pENTRcmv and recombined to pAdDEST (Invitrogen). MMP3 was amplified from murine adipose tissue cDNA and cloned into the pcDNA3-V5/his-TOPO vector (Invitrogen). Different parts of the MMP3promoter (Fig 3C) or 3 kb of the RORγ promoter or 700 bp of the PPARγ promoter (Wang & Lazar, 2008) were cloned from murine C57Bl/6J genomic DNA into pGL3. Single siRNAs or siRNA pools were purchased from Dharmacon. Primer sequences are available upon request.
The paper explained
PROBLEM
Obesity is a prevalent disease in western societies and poses a well-known risk for patients to develop secondary malignancies such as type 2 diabetes, hypertension and cardiovascular complications. Interestingly, only a part of obese subjects develops these problems, which is still largely unexplained, but could be due to differences in the development of adipose tissue. One hypothesis in this regard is that patients with small adipocytes are better at controlling the release of fatty acids from adipose tissue, thereby, protecting other tissues from toxic or harmful lipids.
RESULTS
In a screen of obese and lean patients we identified the transcription factor RORγ to be highly expressed in obesity. In addition, we could show that this factor controls adipose tissue development through a gene, which is responsible for reshaping the extracellular environment, namely matrix metalloproteinase 3. If RORγ is depleted from adipocyte precursor cells, these cells differentiate much more readily into adipocytes, both in vitro as well as in vivo. This enhanced potential to develop fat cells can lead to the buildup of small adipocytes that are highly insulin sensitive, and, thereby, much better at controlling lipid release. In mice, loss of RORγ protects from the development of hyperglycemia, insulin resistance and ultimately type 2 diabetes. In obese human subjects, expression of this factor is inversely correlated with the ability to develop adipocytes and also with general insulin sensitivity, suggesting that RORγ might indeed be an important factor for adipose tissue development in humans.
IMPACT
Thus, RORγ might serve as a new pharmaceutical target for the treatment of obesity-associated insulin resistance.
Cell culture
3T3-L1 preadipocytes were cultured on collagen-coated plates in high-glucose DMEM (Invitrogen) supplemented with 10% foetal bovine serum and penicillin/streptomycin. For differentiation experiments, 3T3-L1 cells were seeded in 96-well plates at a density of 1.5 × 104/cm2 and transfected the next day using Fugene (Roche) and overexpression vectors or siRNA according to the manufacturer's protocol. Adipogenesis was induced 3 days after transfection with 1 µg/ml insulin, 1 µM dexamethasone and 0.115 mg/ml isobutylmethylxanthine, followed by 2 days of insulin medium (1 µg/ml) and 2 days of normal medium. Where indicated, the MMP3 inhibitor II (Merck) was added 2 days before and after induction of differentiation. For stable transfection, cells were kept on geneticin (500 µg/ml) for 2 weeks. C2C12 was cultured in high-glucose DMEM (Invitrogen) supplemented with 10% foetal bovine serum and penicillin/streptomycin. For differentiation experiments serum concentration was reduced to 1%.
Automated analysis of adipocyte differentiation
Differentiated cells were fixed with 5% formaldehyde prior to staining with BODIPY for lipid droplets, Hoechst for nuclei and Syto60 for cytosolic staining (Invitrogen). Twenty-five pictures per well were taken with an automated microscope imaging system (CellWorx/Operetta). Pictures were analysed using Cell Profiler or the Harmony software as described previously (Meissburger et al, 2011; Trajkovski et al, 2011).
Viral infection
Lentiviral stocks were prepared as described (Naldini et al, 1996). For functional assays differentiated 3T3-L1 cells were infected for 24 h with virus containing medium supplemented with polybrene (8 µg/ml) 2 days after induction of differentiation. Infection at this late time point did not alter differentiation capacity of the cells. Differentiation was continued until day 8. For adenoviral production HEK293A cells were infected with crude pAd-RORγ virus lysate. Adenovirus was purified using a column system (Virapur). C2C12 cells were infected in media supplemented with 0.0001% poly-lysine for 24 h and glucose uptake was performed 3 days after infection.
Western blotting
Whole cell lysates, cytosolic extracts or nuclear extracts were prepared as described previously (Wolfrum et al, 2003). A-FABP was detected with an antibody raised against recombinant mouse A-FABP (N. Haunerland). C/EBPα,β,δ and PPARγ (Cell Signaling) as well as RORγ (Abcam) and RORγt (eBioscience) were detected according to the manufacturer's instructions. γ-Tubulin (Sigma) or Lamin B (Cell Signaling) were used as loading control.
Luciferase assay, chromatin immunoprecipitation
Luciferase assays were performed as described before (Wolfrum et al, 2003). For ChIP assays 3T3-L1 cells were infected with RORγ or control lentivirus at 60% confluence. Days 6–8 after transfection chromatin immunoprecipitation was performed using the chromatin IP Assay Kit (LucernaChem) according to the manufacturer's protocol. Two putative RREs in the MMP3 promoter were tested: RRE1 (−2940 to −2928) TTCCCAAGGTCA and RRE2 (−1727 to −1715) TTAAGTAGGTAA.
Nuclear run-on assay
The nuclear run on assay was performed in undifferentiated 3T3-L1 preadipocytes according to the method described by Reeder and Roeder (Reeder & Roeder, 1972). In brief, nuclei were extracted 5 days after virus infection and transcription was allowed to finish in the presence of 32P-ATP. cDNA for the genes of interest was immobilized on a slot blot, labelled mRNA was hybridized to the blot and analysed by autoradiography. Primer sequences are available upon request.
Real-time PCR
mRNA was isolated and transcribed into cDNA using the Multi-MACS cDNA kit (Miltenyi). mRNA expression was assessed by real-time PCR using SybrGreen (Invitrogen) according to the manufacturer's protocol. mRNA expression was normalized to 36b4 or Gapdh. Primer sequences are available upon request. For expression of RORγ and MMP3 in human samples TaqMan gene expression assays (Ambion) were used according to the manufacturer's protocol.
Microarrays
Total RNA was extracted using Trizol (Invitrogen). Genomic DNA was digested using the DNA-free kit (Ambion) and RNA was labelled with either Affimetrix labelling system (human samples) or with One-Colour RNA Spike-In Kit (Agilent) for mouse samples. Labelled cRNA was hybridized to human genome U133 arrays (Affimetrix) or 44k whole mouse genome arrays (Agilent). For the human analysis six patients per group were analysed. For identification of RORγ target genes two independent experiments using transient overexpression of RORγ were performed. Genes that were upregulated >twofold and show a high expression also in primary mouse SVF (internal microarray data) were selected for a functional siRNA differentiation screen. Data were submitted to the EMBL-EBI database and the accession numbers are E-MEXP-3313 for the human SVF array and E-MEXP-3312 for the RORg 3T3L1 cell array.
Glucose uptake and lipolysis
Glucose uptake and lipolysis was determined essentially as described previously (Wolfrum et al, 2003). Briefly, cells were preincubated with 10 nM (C2C12), 20 nM (primary adipocytes) or 50 nM (3T3-L1 adipocytes) insulin for 10 min. Primary adipocytes were spun through dinoyl-phthalate oil, washed and the aqueous phase was collected. Osmium-tetroxide fixed primary adipocytes (Mersmann & Macneil, 1986) were counted for normalization. Glycerol release in the supernatant was measured to assess lipolysis using glycerol reagent (Sigma) according to the manufacturer's protocol.
In vivo differentiation of SVF
SVF from subcutaneous and visceral fat was prepared as described before (Hansen et al, 1998). Briefly, fat tissue was dissected, minced and incubated in collagenase type II for 1 h at 37 °C. Pelleted stomal-vascular fraction was strained through a 40 µm net, erythrocytes were lysed and 106 cells were resuspended in 100 µl of Matrigel (BD). Cells were injected subcutaneously into a skin fold of the neck. The protocol for injection and differentiation was adapted from Kawaguchi et al (Kawaguchi et al, 1998, 1999). After 4–6 weeks Matrigel pads were excised and treated as described below for the determination of adipocyte size. From each pad pictures of three full sections were taken and adipocyte numbers as well as the number of nuclei were determined automatically with Cell Profiler.
Adipocyte size
Adipose tissue was fixed in 5% paraformaldehyde over night before paraffin embedding and sectioning to 10 µm slices. H&E staining of sections was performed according to standard procedures (Chen & Farese, 2002). Microscopic pictures were taken and cell size was analysed using Cell Profiler. Fat pads were either weighted after excision or measured with an animal CT-Scanner (LaTheta). At least 5000 adipocytes were measured per animal to determine adipocyte size.
Serum measurements
Blood glucose was measured with a glycometer (Contour). Insulin was determined with the Insulin ELISA kit (Crystal Chem). Triglycerides were measured with the Trig/GB Kit using C.f.a.s. standard (Roche/Hitachi). Free fatty acids were measured with the NEAFA-HR kit (Wako). For serum ILs a micro-bead based assay system (Luminex) was used.
Ex vivo differentiation of human SVF
Differentiation of the preadipocytes was induced as described previously with minor modifications (Isakson et al, 2008). Complete SVF fraction was obtained from subcutaneous biopsies of obese patients and 5 × 104 cells were plated on collagen coated 96-well plates. Cells were induced with a differentiation cocktail consisting of 1 µM insulin, 10 µM dexamethasone, 0.5 mM IBMX, 100 nM pioglitazone, 33 µM biotin and 17 µM pathenonate in DMEM/F12 supplemented with 3% FBS, 2 mM glutamine and antibiotics. Two days post induction medium was changed to a medium containing only insulin and, dexamethasone in DMEM/F12, 10% FBS with glutamine and antibiotics. Cells were analysed as described above at day 8 of differentiation. Differentiated cells were co-stained with adiponection to verify differentiation.
Bioethics
All mouse work was carried out in Zürich and was approved by the Kantonale Veterinäramt Zürich. The human work on obese versus lean patients was performed in Bratislava and approved by the ethics committee of the Slovak Academy of Science. Informed consent was obtained from all patients, studied. The work on obese subjects was carried out at the University Hospital of Heidelberg, with approval from the ethics committee of the University Hospital. Informed consent was obtained from all patients, studied.
Statistical analysis
Results are given as mean ± standard deviation. Statistical analyses were performed using a two-tailed Student's t-test. Linear regression was calculated using Graph Pad Prism.
Acknowledgments
A-FABP antibodies were provided by N. Haunerland. Rory knockout mice were provided by D. Littman. IL-17 promoter construct was a kind gift of Takashi Kobayashi. These studies were supported by ERC-Adipodif (CW), SNF-3100A0-117979 (CW), FP7-LipidomicNet (CW, DG), EASD (GR), BMBF (PPN) and the Roche Research Foundation (BM).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
NB and MG performed the in vivo differentiation experiments; DT performed the analysis on circulating inflammation markers; WL provided help with the CT scanning of mice; BC helped with the ChIP assays; BM and CW performed all other experiments (except for obtaining patient samples); JU and DG obtained the patient material of the obese versus lean subjects; ER, GR and PPN obtained patient material and characterized the obese patient cohort; CW, BM, PPN and GR wrote and edited the manuscript.
For more information
Ensembl:
Rorc [ENSMUSG00000028150] http://www.ensembl.org/Mus_musculus/Gene/Summary?g=ENSMUSG00000028150
Mmp3 [ENSMUSG00000043613]
http://www.ensembl.org/Mus_musculus/Gene/Summary?g=ENSMUSG00000043613
Uniprot:
P51450 (RORG_MOUSE)
http://www.uniprot.org/uniprot/P51450
P28862 (MMP3_MOUSE)
http://www.uniprot.org/uniprot/P28862
MGI:
RORc MGI:104856
http://www.informatics.jax.org/searches/accession_report.cgi?id=MGI:104856
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Alexander CM, Selvarajan S, Mudgett J, Werb Z. Stromelysin-1 regulates adipogenesis during mammary gland involution. J Cell Biol. 2001;152:693–703. doi: 10.1083/jcb.152.4.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin S, Medvedev A, Yan ZH, Adachi H, Hirose T, Jetten AM. Induction of the nuclear orphan receptor ROR gamma during adipocyte differentiation of D1 and 3T3-L1cells. Cell Growth Differ. 1998;9:267–276. [PubMed] [Google Scholar]
- Barendse W, Bunch RJ, Kijas JW, Thomas MB. The effect of genetic variation of the retinoic acid receptor-related orphan receptor C gene on fatness in cattle. Genetics. 2007;175:843–853. doi: 10.1534/genetics.106.064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouloumie A, Sengenes C, Portolan G, Galitzky J, Lafontan M. Adipocyte produces matrix metalloproteinases 2 and 9: involvement in adipose differentiation. Diabetes. 2001;50:2080–2086. doi: 10.2337/diabetes.50.9.2080. [DOI] [PubMed] [Google Scholar]
- Chavey C, Mari B, Monthouel MN, Bonnafous S, Anglard P, Van Obberghen E, Tartare-Deckert S. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. J Biol Chem. 2003;278:11888–11896. doi: 10.1074/jbc.M209196200. [DOI] [PubMed] [Google Scholar]
- Chen HC, Farese RV. Determination of adipocyte size by computer image analysis. J Lipid Res. 2002;43:986–989. [PubMed] [Google Scholar]
- Crossno ,JT, Jr, Majka SM, Grazia T, Gill RG, Klemm DJ. Rosiglitazone promotes development of a novel adipocyte population from bone marrow-derived circulating progenitor cells. J Clin Invest. 2006;116:3220–3228. doi: 10.1172/JCI28510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czoski-Murray C, Warren E, Chilcott J, Beverley C, Psyllaki MA, Cowan J. Clinical effectiveness and cost-effectiveness of pioglitazone and rosiglitazone in the treatment of type 2 diabetes: a systematic review and economic evaluation. Health Technol Assess. 2004;8:iii. doi: 10.3310/hta8130. ix–x, 1–91. [DOI] [PubMed] [Google Scholar]
- Desai MY, Dalal D, Santos RD, Carvalho JAM, Nasir K, Blumenthal RS. Association of body mass index, metabolic syndrome, and leukocyte count. Am J Cardiol. 2006;97:835–838. doi: 10.1016/j.amjcard.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Dieudonne MN, Pecquery R, Leneveu MC, Giudicelli Y. Opposite effects of androgens and estrogens on adipogenesis in rat preadipocytes: evidence for sex and site-related specificities and possible involvement of insulin-like growth factor 1 receptor and peroxisome proliferator-activated receptor {gamma}2. Endocrinology. 2000;141:649–656. doi: 10.1210/endo.141.2.7293. [DOI] [PubMed] [Google Scholar]
- Gesta S, Tseng Y, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Hansen LH, Madsen B, Teisner B, Nielsen JH, Billestrup N. Characterization of the inhibitory effect of growth hormone on primary preadipocyte differentiation. Mol Endocrinol. 1998;12:1140–1149. doi: 10.1210/mend.12.8.0154. [DOI] [PubMed] [Google Scholar]
- Heilbronn L, Smith SR, Ravussin E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int J Obes Relat Metab Disord. 2004;28:S12–S21. doi: 10.1038/sj.ijo.0802853. [DOI] [PubMed] [Google Scholar]
- Isakson P, Hammarstedt A, Gustafson B, Smith U. Impaired preadipocyte differentiation in human abdominal obesity—role of Wnt, TNFα and inflammation. Diabetes. 2008;58:1550–1557. doi: 10.2337/db08-1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7:e003. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J, Gavrilova O, Pack S, Jou W, Mullen S, Sumner AE, Cushman SW, Periwal V. Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PLoS Comput Biol. 2009;5:e1000324. doi: 10.1371/journal.pcbi.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Trasino SE, Ferrante AW, Jr, Vasselli JR. Prolonged decrease of adipocyte size after rosiglitazone treatment in high- and low-fat-fed rats. Obesity (Silver Spring, Md) 2007;15:2653–2663. doi: 10.1038/oby.2007.317. [DOI] [PubMed] [Google Scholar]
- Kawaguchi N, Toriyama K, Nicodemou-Lena E, Inou K, Torii S, Kitagawa Y. De novo adipogenesis in mice at the site of injection of basement membrane and basic fibroblast growth factor. Proc Natl Acad Sci USA. 1998;95:1062–1066. doi: 10.1073/pnas.95.3.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi N, Toriyama K, Nicodemou-Lena E, Inou K, Torii S, Kitagawa Y. Reconstituted basement membrane potentiates in vivo adipogenesis of 3T3-F442A cells. Cytotechnology. 1999;31:215–220. doi: 10.1023/A:1008198731341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y, Kaidzu S, Nakajima I, Takenouchi K, Nakamura F. Organization of extracellular matrix components during differentiation of adipocytes in long-term culture. In Vitro Cell Dev Biol Anim. 2000;36:38–44. doi: 10.1290/1071-2690(2000)036<0038:OOEMCD>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Kumar N, Solt LA, Conkright JJ, Wang Y, Istrate MA, Busby SA, Garcia-Ordonez R, Burris TP, Griffin PR. The benzenesulfonamide T0901317 is a novel ROR{alpha}/{gamma} inverse agonist. Mol Pharmacol. 2009;77:228–236. doi: 10.1124/mol.109.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau P, Fitzsimmons RL, Raichur S, Wang SC, Lechtken A, Muscat GE. The orphan nuclear receptor, RORalpha, regulates gene expression that controls lipid metabolism: staggerer (SG/SG) mice are resistant to diet-induced obesity. J Biol Chem. 2008;283:18411–18421. doi: 10.1074/jbc.M710526200. [DOI] [PubMed] [Google Scholar]
- Lijnen HR, Maquoi E, Hansen LB, Van Hoef B, Frederix L, Collen D. Matrix metalloproteinase inhibition impairs adipose tissue development in mice. Arterioscler Thromb Vasc Biol. 2002;22:374–379. doi: 10.1161/hq0302.104522. [DOI] [PubMed] [Google Scholar]
- Lilla J, Stickens D, Werb Z. Metalloproteases and adipogenesis: a weighty subject. Am J Pathol. 2002;160:1551–1554. doi: 10.1016/S0002-9440(10)61100-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonn M, Mehlig K, Bengtsson C, Lissner L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J. 2010;24:326–331. doi: 10.1096/fj.09-133058. [DOI] [PubMed] [Google Scholar]
- Meissburger B, Wolfrum C. The role of retinoids and their receptors in metabolic disorders. Eur J Lipid Sci Technol. 2008;110:191–205. [Google Scholar]
- Meissburger B, Stachorski L, Roder E, Rudofsky G, Wolfrum C. Tissue inhibitor of matrix metalloproteinase 1 (TIMP1) controls adipogenesis in obesity in mice and in humans. Diabetologia. 2011;54:1468–1479. doi: 10.1007/s00125-011-2093-9. [DOI] [PubMed] [Google Scholar]
- Mersmann HJ, Macneil MD. Variables in estimation of adipocyte size and number with a particle counter. J Anim Sci. 1986;62:980–991. doi: 10.2527/jas1986.624980x. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- O'Connell J, Lynch L, Cawood TJ, Kwasnik A, Nolan N, Geoghegan J, McCormick A, O'Farrelly C, O'Shea D. The relationship of omental and subcutaneous adipocyte size to metabolic disease in severe obesity. PLoS One. 2010;5:e9997. doi: 10.1371/journal.pone.0009997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, Kato S, Takahashi Y, Hayashi H, Sato R. The orphan nuclear receptor RORalpha restrains adipocyte differentiation through a reduction of C/EBPbeta activity and perilipin gene expression. Mol Endocrinol. 2009;23:759–771. doi: 10.1210/me.2008-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichur S, Lau P, Staels B, Muscat GEO. Retinoid-related orphan receptor gamma regulates several genes that control metabolism in skeletal muscle cells: links to modulation of reactive oxygen species production. J Mol Endocrinol. 2007;39:29–44. doi: 10.1677/jme.1.00010. [DOI] [PubMed] [Google Scholar]
- Ravussin E, Smith SR. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann N Y Acad Sci. 2002;967:363–378. doi: 10.1111/j.1749-6632.2002.tb04292.x. [DOI] [PubMed] [Google Scholar]
- Reeder RH, Roeder RG. Ribosomal RNA synthesis in isolated nuclei. J Mol Biol. 1972;67:433–441. doi: 10.1016/0022-2836(72)90461-5. [DOI] [PubMed] [Google Scholar]
- Roberts R, Hodson L, Dennis AL, Neville MJ, Humphreys SM, Harnden KE, Micklem KJ, Frayn KN. Markers of de novo lipogenesis in adipose tissue: associations with small adipocytes and insulin sensitivity in humans. Diabetologia. 2009;52:882–890. doi: 10.1007/s00125-009-1300-4. [DOI] [PubMed] [Google Scholar]
- Rodeheffer MS, Birsoy K, Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135:240–249. doi: 10.1016/j.cell.2008.09.036. [DOI] [PubMed] [Google Scholar]
- Sethi JK, Vidal-Puig AJ. Thematic review series: adipocyte biology—adipose tissue function and plasticity orchestrate nutritional adaptation. J Lipid Res. 2007;48:1253–1262. doi: 10.1194/jlr.R700005-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smas CM, Sul HS. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell. 1993;73:725–734. doi: 10.1016/0092-8674(93)90252-l. [DOI] [PubMed] [Google Scholar]
- Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, Liu J, Istrate MA, Kamenecka TM, Roush WR, Vidovic D, et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature. 2011;472:491–494. doi: 10.1038/nature10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, Naslund E, Britton T, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- Sun ZM, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, Littman DR. Requirement for ROR gamma in thymocyte survival and lymphoid organ development. Science. 2000;288:2369–2373. doi: 10.1126/science.288.5475.2369. [DOI] [PubMed] [Google Scholar]
- Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, Heim MH, Stoffel M. MicroRNAs 103 and 107 regulate insulin sensitivity. Nat Adv. 2011;474:649–653. doi: 10.1038/nature10112. [DOI] [PubMed] [Google Scholar]
- Traurig MT, Permana PA, Nair S, Kobes S, Bogardus C, Baier LJ. Differential expression of matrix metalloproteinase 3 (MMP3) in preadipocytes/stromal vascular cells from nonobese nondiabetic versus obese nondiabetic Pima Indians. Diabetes. 2006;55:3160–3165. doi: 10.2337/db06-0373. [DOI] [PubMed] [Google Scholar]
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144:5159–5165. doi: 10.1210/en.2003-0870. [DOI] [PubMed] [Google Scholar]
- Wang J, Lazar MA. Bifunctional role of Rev-erbalpha in adipocyte differentiation. Mol Cell Biol. 2008;28:2213–2220. doi: 10.1128/MCB.01608-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kumar N, Solt LA, Richardson TI, Helvering LM, Crumbley C, Garcia-Ordonez RA, Stayrook KR, Zhang X, Novick S, et al. Modulation of ROR{alpha} and ROR{gamma} activity by 7-oxygenated sterol ligands. J Biol Chem. 2009;285:5013–5025. doi: 10.1074/jbc.M109.080614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum C, Shih DQ, Kuwajima S, Norris AW, Kahn CR, Stoffel M. Role of Foxa-2 in adipocyte metabolism and differentiation. J Clin Invest. 2003;112:345–356. doi: 10.1172/JCI18698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanchi A, Dulloo AG, Perregaux C, Montani JP, Burnier M. Telmisartan prevents the glitazone-induced weight gain without interfering with its insulin-sensitizing properties. Am J Physiol Endocrinol Metab. 2007;293:E91–E95. doi: 10.1152/ajpendo.00024.2007. [DOI] [PubMed] [Google Scholar]
- Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. 2008;9:1297–1306. doi: 10.1038/ni.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.