

See related article in New England Journal of Medicine http://dx.doi.org/10.1056/NEJMc1100859
Typical haemolytic uraemic syndrome (HUS) is a thrombotic microangiopathy, leading to severe renal disease as well as life-threatening extrarenal complications. HUS is primarily caused by infections with enterohaemorrhagic Escherichia coli (EHEC) and 5–20% of EHEC infections result in HUS. EHEC bacteria produce several virulence factors, of which Shiga toxins are believed to play a central role. About 10% of all HUS cases are not caused by E. coli and are thus termed atypical HUS. These are observed as familial or sporadic forms and are most commonly caused by a dysregulation of the alternative pathway of the complement system due to inherited mutations of, or acquired auto-antibodies against, complement regulator proteins. The, thereby, impaired control of complement, which plays a prominent role in the humoral immune system and in immune homeostasis, leads to a hyperactive state, including activation of endothelial cells and platelets, inflammation of small vessel and eventually to the destruction of the kidney and other organs (summarized in Orth et al, 2009).
Lapeyraque et al have published the successful use of the licensed terminal complement C5 inhibitor eculizumab for the treatment of severe Shiga toxin-associated HUS in three 3-year-old children with devastating prognoses (Lapeyraque et al, 2011). The three patients continued to show progression of the HUS despite having received multiple plasma exchanges. Treatment with eculizumab markedly improved their clinical status, in particular their neurological and renal functions, after two to four administrations of this complement inhibitor. Dialysis was reported to be discontinued after 3–16 days in all three patients. Recovery was attributed to eculizumab and absence of mutations of complement proteins or auto-antibodies directed against these corroborated the diagnosis of a severe Shiga toxin-associated HUS and not an atypical HUS in each case (Lapeyraque et al, 2011).
What was the reason for the authors to use this potent complement inhibitor and why a complement inhibitor in the first place? Indeed, extensive complement activation was assumed in one of their patients presenting with low C3 and elevated C3d serum concentrations and both the decrease of the native protein and the increase of the split product are indicative of complement activation. However, this was likely not enough evidence per se to dare a complement inhibitory treatment with eculizumab, a substance never used for this application.
The development of eculizumab goes back more than 20 years when a murine, monoclonal antibody directed against C5 (N19-8) was characterized, which was able to almost completely block the terminal complement cascade by preventing both C5a and terminal complement complex (TCC) formation (Würzner et al, 1991). These properties of the N19-8 prototype were retained in the humanized recombinant single-chain Fv fragment (Evans et al, 1995), which initiated the search for even more potent C5 blockers and eventually led into the generation of the medical drug eculizumab (Soliris®).
Eculizumab has been demonstrated to block complement activation in the treatment of patients with paroxysmal nocturnal haemoglobinuria (PNH), in which red blood cells are lysed by complement due to an inherited lack of inhibitors on their cell surface, and is, therefore, approved for this application. As eculizumab works for PNH, and atypical HUS also shows uncontrolled complement activation, this humanized antibody was also successfully used in several patients with atypical HUS due to mutations in complement regulator protein factor H, both in adults and children (summarized and detailed, respectively, in Zimmerhackl et al, 2010).
A major puzzle piece came when a complement expert became head of the Austrian Reference Laboratory for EHEC and set out to examine whether not only the atypical, but also the typical HUS is associated with unregulated complement activity (Orth et al, 2009), corroborated by similar clinical courses, albeit apparently different causal origins—an inherited mutation (or an acquired auto-antibody) on the one hand and an EHEC infection on the other. Already 30 years ago, Monnens and co-workers have found increased breakdown products of C3 and factor B in the serum of EHEC-infected typical HUS patients, suggesting an activation of the complement system, possibly via the alternative pathway. However, no definite explanation or role in the disease was given for these observations and a concurring reaction of the complement system in systemic infectious disease is not unusual (summarized in Orth et al, 2009). The first definite establishment of a role of complement in typical HUS was achieved by the finding that Shiga toxin 2 activates complement and also binds to its main regulator, factor H, leading to a reduced protective function of this regulator on the cell surface—a stronger activation accompanied by a weaker control (Orth et al, 2009). The involvement of alternative pathway activation in typical HUS was further corroborated by Thurman and co-workers who showed significantly increased plasma levels of complement activation products Bb and SC5b-9 in these patients and thus in a clinical setting (summarized in Lapeyraque et al, 2011). Congress contributions by several other labs have corroborated the role of complement in typical HUS. Taken together, these data provide a rationale for using eculizumab in typical HUS (Fig 1).
Figure 1. Upon infection, EHEC bacteria stay in the gut, but the released Shiga toxin is taken up and circulates in the blood affecting several organs, primarily the kidney.
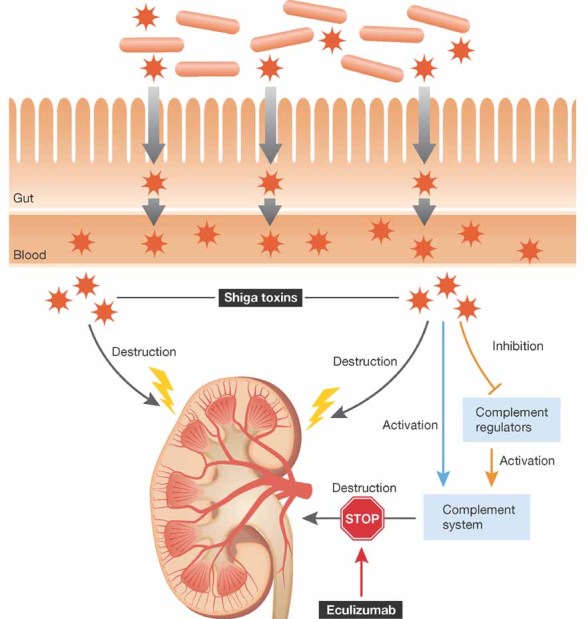
Apart from the direct toxic effect on cells, Shiga toxin activates the alternative pathway of complement (blue arrow) and this activation is enhanced by binding of the toxin to the major fluid phase complement inhibitor, factor H, debilitating its protective function (orange arrow). This enhanced complement activation can be blocked by eculizumab, targeting C5 cleavage (STOP sign).
»…The publication by Lapeyraque et al triggered treatment of more than 300 HUS patients during the 2011 EHEC O104:H4 outbreak in Northern Germany…«
With these indications at hand, Lapeyraque et al had the courage to use the complement inhibitor eculizumab, licensed for a disease with uncontrolled complement activation due to an absent inhibitor (PNH), and successfully used in a handful patients with another disease of uncontrolled complement activation due to a mutated inhibitor (atypical HUS), now in a different, albeit related disease. The timely publication of New England Journal of Medicine right at the peak of the recent EHEC O104:H4 outbreak in Northern Germany on May 25, 2011, then provided the means to treat in particular those patients with poor prognosis and those with cerebral affections as the three children treated by Lapeyraque et al.
More than 300 patients have been treated with eculizumab at present and the initial data are somewhat promising. The German Society for Nephrology has initiated a registry to get an overall view and published advice on the use of eculizumab: http://www.dgfn.eu/index.php?eID=tx_nawsecuredl&u=0&file=uploads/media/Advice_of_DGfN_on_the_use_of_eculizumab_in_EHEC_HUS_outbreak_20110604.pdf&t=1309849615&hash=e01cc3f54554fd1aea9c8d9ffaee8-f45ae16c2b0.
The apparently encouraging data available so far prompted the company distributing eculizumab, Alexion, to initiate a clinical trial in order to evaluate the safety and efficacy of eculizumab in a controlled manner: http://finance.yahoo.com/news/Alexion-Initiates-Clinical-bw-779709396.html?x=0&.v=1.
Thus, several pieces of research and clinical observations, put together well in time, allowed the transfer from bench to bed in record time (2009: HUS is complement-mediated, 2011: HUS is treated by a complement blocker) and initiated the mostly apparently successful treatment of more than 300 patients during the recent O104:H4 outbreak, although detailed analyses of their outcomes will have to follow. The puzzle piece by Lapeyraque et al may be considered as the most important one, as this publication likely represented the final piece of evidence to provide physicians with enough confidence to try to use eculizumab off label for their severe Shiga toxin-associated HUS patients. The apparently successful widespread use of a complement inhibitor in this disease also provided the clinical confirmation that also typical HUS is indeed a complement-mediated disease.
Conflict of interest statement
RW has characterized the prototype of eculizumab, N19-8, but was not involved in the development of the currently commercialized anti-C5. RW is the recipient of an unrestricted educational grant from Alexion, but did not receive any royalties from Alexion. The other authors declare that they have no conflict of interest.
References
- Evans MJ, Rollins SA, Wolff DW, Rother RP, Norin AJ, Therrien DM, Grijalva GA, Mueller JP, Nye SH, Squinto SP, et al. In vitro and in vivo inhibition of complement activity by a single-chain Fv fragment recognizing human C5. Mol Immunol. 1995;32:1183–1195. doi: 10.1016/0161-5890(95)00099-2. [DOI] [PubMed] [Google Scholar]
- Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, Proulx F, Clermont MJ, Le Deist F, Niaudet P, et al. Complement blockade in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364:2561–2563. doi: 10.1056/NEJMc1100859. [DOI] [PubMed] [Google Scholar]
- Orth D, Khan AB, Naim A, Grif K, Brockmeyer J, Karch H, Joannidis M, Clark SJ, Day AJ, Fidanzi S, et al. Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol. 2009;182:6394–6400. doi: 10.4049/jimmunol.0900151. [DOI] [PubMed] [Google Scholar]
- Würzner R, Schulze M, Happe L, Franzke A, Bieber FA, Oppermann M, Götze O. Inhibition of terminal complement complex formation and cell lysis by monoclonal antibodies. Complement Inflamm. 1991;8:328–340. doi: 10.1159/000463204. [DOI] [PubMed] [Google Scholar]
- Zimmerhackl LB, Hofer J, Cortina G, Mark W, Würzner R, Jungraithmayr TC, Khursigara G, Kliche KO, Radauer W. Prophylactic eculizumab after renal transplantation in atypical hemolytic-uremic syndrome. N Engl J Med. 2010;362:1746–1748. doi: 10.1056/NEJMc1001060. [DOI] [PubMed] [Google Scholar]