

Abstract
Pantothenate kinase-associated neurodegeneration (PKAN is a neurodegenerative disease with unresolved pathophysiology. Previously, we observed reduced Coenzyme A levels in a Drosophila model for PKAN. Coenzyme A is required for acetyl-Coenzyme A synthesis and acyl groups from the latter are transferred to lysine residues of proteins, in a reaction regulated by acetyltransferases. The tight balance between acetyltransferases and their antagonistic counterparts histone deacetylases is a well-known determining factor for the acetylation status of proteins. However, the influence of Coenzyme A levels on protein acetylation is unknown. Here we investigate whether decreased levels of the central metabolite Coenzyme A induce alterations in protein acetylation and whether this correlates with specific phenotypes of PKAN models. We show that in various organisms proper Coenzyme A metabolism is required for maintenance of histone- and tubulin acetylation, and decreased acetylation of these proteins is associated with an impaired DNA damage response, decreased locomotor function and decreased survival. Decreased protein acetylation and the concurrent phenotypes are partly rescued by pantethine and HDAC inhibitors, suggesting possible directions for future PKAN therapy development.
Keywords: DNA damage, HDAC inhibitors, NBIA, PKAN, protein acetylation
INTRODUCTION
Recently a large body of evidence has emerged to suggest that protein acetylation plays an important role in key cellular processes (Choudhary et al, 2009). The balance between Histone or K-acetyltransferases (HATS or KATS) and histone deacetylases (HDACs) is well documented as influential in the homeostasis of protein acetylation (Allis et al, 2007; Lee and Workman, 2007; Shahbazian and Grunstein, 2007). Acetyl-Coenzyme A (Acetyl-CoA) is the source for the acyl group that is transferred to lysine residues, and it was demonstrated that down-regulation of enzymes required for the synthesis of acetyl-CoA induce reduction in acetylation of specific proteins (Starai et al, 2004; Takahashi et al, 2006; Wellen et al, 2009). However, it is completely unknown whether levels of metabolites themselves influence protein acetylation. Considering that CoA is a central metabolic cofactor involved in over 100 metabolic reactions (Leonardi et al, 2005) and is also required to synthesize acetyl-CoA from citrate or acetate, CoA is an interesting candidate-metabolite with potential influence over protein acetylation. Remarkably, a possible role of CoA metabolism on protein acetylation has never been directly investigated and it is unclear whether or not protein acetylation levels respond to decreasing concentrations of cellular CoA or whether increased activity of HATS/KATS compensate for decreased levels of Coenzyme A. The de novo biosynthesis route of CoA is a well-conserved enzymatic pathway. The first and rate-limiting step, the phosphorylation of vitamin B5, is catalysed by the enzyme pantothenate kinase (PANK; Leonardi et al, 2005; Fig 1A). The CoA biosynthesis pathway has received renewed attention after the discovery that mutations in the human PANK2 gene are associated with the severe neurodegenerative disease PANK-associated neurodegeneration (PKAN; Zhou et al, 2001). The pathology of PKAN is complex, and exactly how impaired de novo biosynthesis of CoA is linked to neurodegeneration as in PKAN is largely unknown (Gregory et al, 2009). In Drosophila melanogaster a PANK ortholog is present and referred to as dPANK/fumble (Afshar et al, 2001; Bosveld et al, 2008). dPANK/Fbl Drosophila mutants possess a neurodegenerative phenotype and a greatly reduced life span. We recently showed that down-regulation of the enzyme PANK (dPANK/Fbl) in flies and cultured cells results in decreased levels of total CoA, and further that addition of the compound pantethine to the food restored CoA levels and rescued the mutant phenotype (Bosveld et al, 2008; Rana et al, 2010). This model can now be used to further manipulate and measure CoA levels and to study directly the biological consequences of decreased CoA levels and to understand the molecular mechanisms underlying PKAN. The neurodegenerative Drosophila PKAN model is further characterized by increased sensitivity to DNA damage, an explanation for which is currently lacking. Here we exploited this Drosophila model to investigate (1) whether or not decreased CoA levels affect the acetylation levels of specific proteins and (2) if so, whether this abnormal acetylation status of specific proteins coincide with the pleiotropic phenotype of the Drosophila model for PKAN and (3) if so, whether restoration of acetylation levels of specific proteins can rescue apparent PKAN-related phenotypes in Drosophila.
Figure 1. Acetylation levels of specific proteins are decreased when CoA levels are reduced.

- The de novo biosynthesis route of Coenzyme A: Vitamin B5 is converted in several steps into CoA. PANK (referred to as dPANK/Fbl in Drosophila) is required for the first conversion step. HoPan is a potent chemical inhibitor of PANK enzymatic activity.
- Whole cell lysates of control S2 cells and dPANK/Fbl-depleted cells (by RNAi) were used to analyse acetylated protein levels using an antibody specifically recognizing acetylated-lysine residues. Control cells and dPANK/Fbl-depleted cells were left untreated or treated with pantethine (0.1 mM), with TSA or with TSA and pantethine.
- Quantification of the relative levels of acetylation compared to control cells for the indicated band of 55 kD in size and quantification of the relative levels of acetylation for the indicated bands around 17–11 kD.
- S2 cells were incubated with 0.5 mM HoPan, and/or with 0.1 mM Pantethine. Whole cell extracts were probed with anti-acetyl-lysine antibody.
- dPANK/Fbl depleted cells were left untreated or increasing concentrations of CoA were added to the cell culture medium. Whole cell extracts were probed with an acetyl-lysine antibody.
- Quantification of the relative levels of acetylation compared to control cells after addition of various concentrations of CoA for the indicated band of 55 kD in size and quantification of the relative levels of acetylation for the indicated bands around 17–11 kD. Asterisks indicate bands that show a decreased signal in the dPANK/Fbl-depleted or HoPan treated cells. Efficiency of the RNAi treatment was controlled by using an antibody specifically recognizing dPANK/Fbl (Bosveld et al, 2008). As a loading control tubulin was used.
We show that when CoA levels are reduced, there is no compensatory mechanism to maintain the normal acetylation levels of histones and tubulin, and decreased acetylation of these proteins is associated with key characteristics of a Drosophila model for PKAN. After feeding pantethine or HDAC inhibitors, acetylation levels of histones and tubulin are restored and this coincides with improved viability, locomotor function and survival after DNA damaging insults. Additionally, we demonstrate that the correlation between PANK activity and acetylation of specific proteins is conserved among species and thus decreased protein acetylation may underlay the pathogenesis of PKAN.
RESULTS
CoA levels can be modified and measured and a decrease in CoA leads to decreased acetylation of specific proteins
To investigate the influence of CoA levels on protein acetylation, RNAi was used to down-regulate dPANK/Fbl protein in Drosophila Schneider's S2 cells (Fig 1B). Levels of total CoA are severely reduced under these circumstances (Rana et al, 2010). To determine the general acetylation status of proteins under these conditions, immunoblots of whole cell extracts (from control and dPANK/Fbl-depleted cells) were incubated with an antibody specifically recognizing acetylated-lysine. In dPANK/Fbl-depleted cells the levels of some specific proteins (indicated by asterisks, Fig 1B), between 17–11 and 55 kD in size, appeared to be reduced as compared to control cells (compare lane 1 and lane 3, for quantification see Fig 1C). Only the acetylation levels of specific proteins and not all proteins recognized by the anti-acetyl-lysine antibody were affected under circumstances of decreased CoA levels. Previously, we demonstrated that addition of the compound pantethine restored CoA levels in a dPANK/Fbl-depleted background via a (yet unresolved) non-canonical CoA de novo biosynthesis pathway (Rana et al, 2010). Addition of pantethine reversed the acetylation levels of the indicated proteins back to wild-type (WT; compare lane 3 and 4, Fig 1B and C), indicating that altered acetylation of the indicated proteins coincides with decreased levels of CoA and not with decreased levels of the dPANK/Fbl enzyme. Moreover, inhibition of PANK activity by the selective chemical PANK inhibitor HoPan (Fig 1A; Zhang et al, 2007)) resulted in decreased acetylation of the same proteins, an effect that was also reversed by pantethine (Fig 1D). HoPan treatment results in a block in CoA production in isolated mouse liver (Zhang et al, 2007) and in decreased CoA levels in S2 cells (Fig S1 of Supporting information). To further prove that observed acetylation defects are indeed sensitive and responsive to CoA levels, we supplemented the growth medium of dPANK/Fbl-depleted cells with increasing concentrations of CoA. Addition of CoA restored acetylation levels of the indicated proteins in a dose-dependent manner (Fig 1E, for quantification see Fig 1F). Our results demonstrate that the acetylation levels of at least two proteins are affected under circumstances of reduced CoA. To test whether the effect of dPANK/Fbl-depletion on protein acetylation of the specific proteins was not due to a general increased activity of deacetylases, we measured HDAC activity in the cell extracts. No significant differences in deacetylation rates between control and dPANK/Fbl depleted cells were observed (Fig S2 of Supporting information). Next we tested whether acetylation of the specific proteins could be restored by inhibition of HDACs. Treatment with the HDAC inhibitor Trichostatin A (TSA) resulted in increased acetylation levels of the affected proteins, although acetylation levels in dPANK/Fbl depleted cells remained lower as compared to control cells (Fig 1B, right panel, compare lane 5 and lane 7). Together, these data indicate that reduced acetylation levels of specific proteins in the dPANK/Fbl-depleted background are not caused by increased activity of HDACs, and are most likely the result of decreased levels of CoA. Independent of the reduced CoA levels; the reduced acetylation levels of these specific proteins can be partly restored by inhibiting HDAC activities. This further suggests that normal levels of CoA are required in addition to the balance between HATs and HDACs activities to maintain the proper status of acetylated lysine residues of specific proteins. The acetyl-lysine antibody used in this study possibly recognizes only a minor subset of acetylated proteins and most likely the indicated proteins are among the ones that are abundantly acetylated. In addition, acetylation levels of the indicated proteins, in contrast to the other proteins recognized by the acetyl-lysine antibody, respond most strongly to treatment with HDAC inhibitors and to impaired CoA biosynthesis, suggesting high acetylation/deacetylation dynamics of these specific proteins. In order to evaluate the complete spectrum of acetylated proteins affected by decreased CoA levels, more sensitive assays are required. Here we will focus on the proteins indicated in Fig 1B.
Decreased levels of CoA coincide with decreased acetylation of histones and tubulin
Next we identified the indicated proteins starting with the protein migrating at 55 kD. Previously it has been shown that tubulin is a protein that can be acetylated (Akella et al, 2010; LeDizet and Piperno, 1987) and its molecular weight matches with the protein indicated by the upper asterisk in Fig 1B. Western blot analysis using antibodies that recognize acetylated-tubulin confirmed that indeed in dPANK/Fbl depleted cells; levels of acetylated-tubulin, but not the total levels of tubulin are decreased (Fig 2A compare lane 1 with lane 3, see Fig 2B for quantification). Acetylation levels of tubulin in dPANK/Fbl depleted cells are restored by addition of pantethine and by addition of the HDAC inhibitor TSA (Fig 2A and B) confirming further the results of Fig 1B and C. Altogether these data demonstrate that a decrease in CoA levels coincides with decreased levels of acetylated-tubulin.
Figure 2. Levels of acetylated tubulin and histones are decreased in dPANK/Fbl-depleted cells.

- Cell extracts of control cells and dPANK/Fbl-depleted cells (by RNAi) were analysed by Western blot using antibodies specifically recognizing acetylated-tubulin. Efficiency of RNAi was determined by using a dPANK/Fbl antibody and tubulin was used as a loading control. Control cells and dPANK/Fbl-depleted cells were left untreated, treated with pantethine, with TSA or with TSA and pantethine.
- Quantification of the relative levels of tubulin acetylation under the conditions presented in A compared to control cells.
- Cell extracts of control cells and dPANK/Fbl-depleted cells were analysed using Western blot to determine acetylation levels of specific histones. Specific antibodies were used to detect levels of acetylated histone 3 and acetylated histone 4. Control and dPANK/Fbl-depleted cells were left untreated or were treated with pantethine, with TSA or with TSA and pantethine. The efficiency of the RNAi treatment was investigated by the use of an antibody against dPANK/Fbl. H2A was used as a loading control.
- Quantification of the relative levels of histone acetylation under the conditions presented in C compared to control cells.
Next we aimed to investigate the identity of the lower bands (indicated by the lower asterisk in Fig 1B).
The migration pattern of the low molecular weight proteins showing decreased acetylation in CoA-deficient S2 cells correlates well with molecular weights of histone proteins. Histones are among the first and most extensively studied proteins known to be abundantly acetylated (Kurdistani and Grunstein, 2003). We used antibodies that specifically recognize acetylated lysines of histone 3 and 4 to investigate whether the lower candidate bands represent acetylated histones. Indeed, the acetylation of histone 3 and histone 4 is 60% decreased in dPANK/Fbl-depleted cells (Fig 2C, compare lane 1 and lane 3, for quantification see Fig 2D), which is in agreement with the results presented in Fig. .1. Decreased levels of acetylated histones were rescued by addition of pantethine and TSA (Fig 2C and D), demonstrating that CoA levels (and not levels of PANK per se) influence the acetylation status of histones.
Taken together, using our in vitro model we have identified a tight link between cellular CoA levels and acetylation of proteins involved in the architecture of the cytoskeleton (via acetyl-tubulin) and in the integrity of the epigenome (via acetylation of histone tails). Further we aimed to identify the cellular and physiological importance of these specific acetylation defects induced by impairment of PANK activity.
Decreased tubulin acetylation coincides with impaired touch response in Caenorhabditis elegans pantothenate kinase mutants
Although in Drosophila, similar to other species, tubulin undergoes acetylation at the lysine 40 residue, the physiological importance of this modification in flies or Drosophila cell lines has not been demonstrated. On the contrary, in C. elegans reduced acetylation of tubulin has been recently linked with an impaired touch response (Shida et al, 2010). To investigate whether the link between CoA metabolism and tubulin acetylation is evolutionarily conserved, we first tested if impaired function of PANK coincides with reduced acetylation levels of tubulin in C. elegans as well, using a C. elegans mutant carrying a deletion of 773 bp within the pnk-1 gene, an ortholog of human PANK2 (Zhou et al, 2001, Fig S3 of Supporting information and methods of Supporting information). As revealed by Western blot analysis, the pnk-1 mutant animals showed decreased levels of acetylated tubulin, which could be rescued, similarly to the Drosophila model, with the addition of pantethine to the food (Fig 3A). In agreement with the reported link between tubulin acetylation and the function of touch receptor neurons in C. elegans, pnk-1 mutant worms showed a decreased touch response, which was also rescued by pantethine feeding (Fig 3B). Together these data strongly indicate a conserved link between CoA metabolism and tubulin acetylation.
Figure 3. Decreased levels of acetylated tubulin in C. elegans pnk-1 mutants coincide with an abnormal touch response.
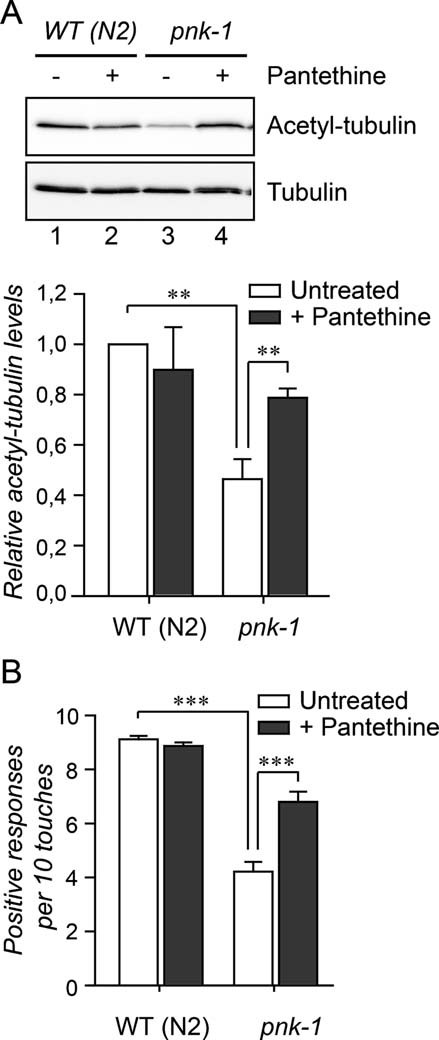
- Extracts of staged L4 + 2 WT and pnk-1 mutant (pnk-1) animals were analysed by Western blot using antibodies specifically recognizing acetylated-tubulin. Tubulin was used as a loading control.
- Touch responses were scored as previously described (Shida et al, 2010) in WT (animals and in pnk-1 mutants under control conditions and after addition of pantethine to the medium. Error bars indicate SEM.
Decreased levels of CoA are associated with an impaired DNA damage response in a Drosophila model for PKAN
Next we investigated whether defects in histone acetylation correlate with specific phenotypes observed in the Drosophila PKAN model. Drosophila mutants that carry a mutation in genes coding for various enzymes required for the de novo synthesis of CoA (dPANK/Fbl, dPPCS, and dPPAT-DCPK) demonstrate increased sensitivity to DNA damaging agents (Bosveld et al, 2008). It is currently unknown why mutants that suffer from decreased levels of CoA are hypersensitive to DNA damage. Changes in histone acetylation are tightly linked with a competent DNA damage response (van Attikum and Gasser, 2005) and increased acetylation of specific histone tails has already been reported in yeast, flies and humans after induction of DNA damage (Chen et al, 2008; Das et al, 2009; Vempati et al, 2010; Yuan et al, 2009). We investigated histone acetylation in response to induced DNA damage under circumstances of reduced levels of CoA. Hereto, control cells and dPANK/Fbl-depleted cells were irradiated to induce DNA double strand breaks and the dynamics of histone acetylation were investigated at various time points (Fig 4A). In control cells, a rapid histone acetylation was observed within 15 min after irradiation. The histone acetylation levels returned to those of control conditions during recovery of the cells. Increased histone acetylation was observed for various lysine residues (Lys5 of histone 2, Lys9 of histone 3 and Lys16 of histone 4), indicating the presence of a general histone acetylation induction in response to impaired DNA integrity. In dPANK/Fbl-depleted cells, this response was impaired, for all the lysine residues tested (Fig 4A). dPANK/Fbl-depleted cells showed a markedly smaller increase in histone acetylation (as for H4K16-ac), or a smaller increase combined with delayed increase in histone acetylation (as for H3-ac and H2K5-ac). Next we investigated whether dPANK/Fbl depleted cells, like Drosophila CoA mutants are more sensitive to irradiation. Hereto, dPANK/Fbl-depleted and control cells were irradiated with various doses of ionizing radiation, and 6 days after exposure the number of surviving cells was determined. dPANK/Fbl-depleted cells showed a reduced survival as compared to control cells (Fig 4B). To further investigate the influence of decreased histone acetylation on radiation sensitivity, HDAC inhibitors were used and their influence on the survival of dPANK/Fbl deficient cells was tested. HDAC inhibitors (TSA or sodium butyrate (NaBut) were given 4 h prior to ionizing radiation and the increase in acetylation was confirmed by measuring levels of acetylated histone 3 (Fig S4A of Supporting information). Pretreatment with HDAC inhibitors did not affect survival of the control RNAi cells. However, dPANK/Fbl-depleted cells showed a significantly higher survival when pretreated with HDAC inhibitors as compared to untreated dPANK/Fbl-depleted cells. Increasing histone acetylation by these means improved the cell survival by approximately 20% (Fig 4B).
Figure 4. Decreased levels of acetylated histones and tubulin are associated with increased sensitivity to irradiation of dPANK/Fbl-depleted cells.
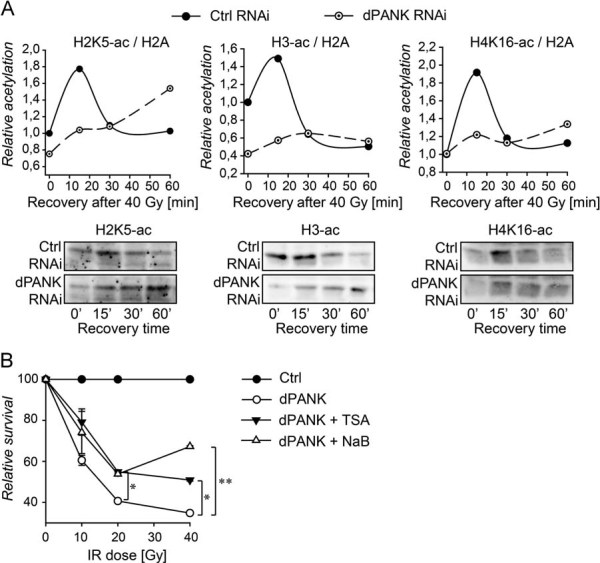
- Control cells and dPANK/Fbl-depleted cells were irradiated (40 Gy) and acetylation levels of specific histone tails were determined after various time points. Levels of H3Ac, H4K16 and H2K5 were determined using specific antibodies for each histone or histone tail. The blots and the quantifications are representative of three-independent experiments.
- Relative cell survival of control cells and dPANK/Fbl-depleted cells was measured after various doses of irradiation (10, 20 and 40 Gy). Cell survival was also determined after treating the cells with HDAC inhibitors (TSA or NaB; See Fig S4A of Supporting information for acetylation levels). Survival of untreated control cells was set to 100% for every dose of irradiation.
Together these data underscore the importance of global histone acetylation in DNA damage responses, as is in agreement with previous results by others (Chen et al, 2008; Das et al, 2009; van Attikum and Gasser, 2005; Vempati et al, 2010; Yuan et al, 2009). Further, our data indicate that hypersensitivity to genotoxic stress of CoA depleted cells may, at least partially, be explained by the impaired acetylation levels of chromatin components.
Decreased levels of CoA are associated with impaired locomotor function in a Drosophila model for PKAN
The Drosophila model for PKAN is further characterized by a decreased survival rate, neurodegeneration and by impaired locomotor function (Afshar et al, 2001; Bosveld et al, 2008; Rana et al, 2010; Wu et al, 2009). The impaired locomotor function is most likely (at least partly) caused by the neurodegeneration. We next investigated whether these phenotypes also correlate with decreased acetylation levels. Especially acetylation of tubulin and histones as we report here for dPANK/Fbl depleted cells have previously been shown by others to be associated with neurodegeneration and with abnormal neuronal functioning (Akella et al, 2010; Creppe et al, 2009; Dompierre et al, 2007; Fischer et al, 2010; Kontopoulos et al, 2006; Monti et al, 2009; Saha and Pahan, 2006; Shida et al, 2010). In the experiments as described above Drosophila S2 cultured cells were used and first we tested whether in Drosophila dPANK/fbl mutant whole organisms (Drosophila PKAN model) levels of acetylated tubulin and histones were also decreased. Western blot analysis using extracts of third instar larvae indeed demonstrated that levels of acetylated tubulin and histones were decreased in dPANK/fbl homozygous larvae as compared to WT larvae (Fig 5A compare lane 1 and lane 3, for quantification see Fig 5B). Homozygous dPANK/fbl flies show a reduced eclosion rate, evidenced by the relative low number of homozygous adults compared to heterozygous adults (the ratio heterozygous:homozygous adult survivors is 16, whereas, based on genetic inheritance this is expected to be 2). First we investigated whether addition of various HDAC inhibitors (valproic acid (VPA), sodium phenylbutyrate (PBA) or TSA to the larval food increased the eclosion rate of homozygous dPANK/fbl flies. VPA and PBA did not result in a significant rescue (Fig S5 of Supporting information) however; TSA addition increased the survival rate of the homozygous mutant progeny in a concentration-dependent manner (Fig 5D). VPA and PBA could only be used in relatively low concentrations, because the concentrations commonly used for an efficient HDAC inhibition (above 1 mM) induced lethality when fed during larval development. TSA, on the other hand, is less toxic. Moreover, TSA is a potent and broad-spectrum inhibitor acting on all Drosophila HDACs (Cho et al, 2005; Foglietti et al, 2006). The most effective concentration of TSA (0.2 µM) was used for further studies and we demonstrated that this induces a partial restoration of the decreased levels of acetylated tubulin and histones in dPANK/fbl mutant larvae (Fig 5A and B). This coincided with an increase in locomotor function assessed by larval crawling as a read-out assay (Fig 5C). These data suggest a correlation between tubulin- and histone-acetylation levels and dPANK/fbl mutant phenotypes. These data are however not conclusive as to whether restoration of only these specific proteins is sufficient for improvement of locomotor function and survival, because acetylation levels of other proteins may also restore upon TSA feeding. Nonetheless, the tight correlation between CoA levels, acetylation of tubulins and histones and the specific phenotypes in dPANK/Fbl depleted cells and flies suggests that an altered status of acetylation of specific proteins may explain the pleiotropic mutant phenotype.
Figure 5. Decreased levels of acetylated histones and tubulin are associated with decreased survival and decreased locomotor function of dPANK/Fbl mutant larvae.
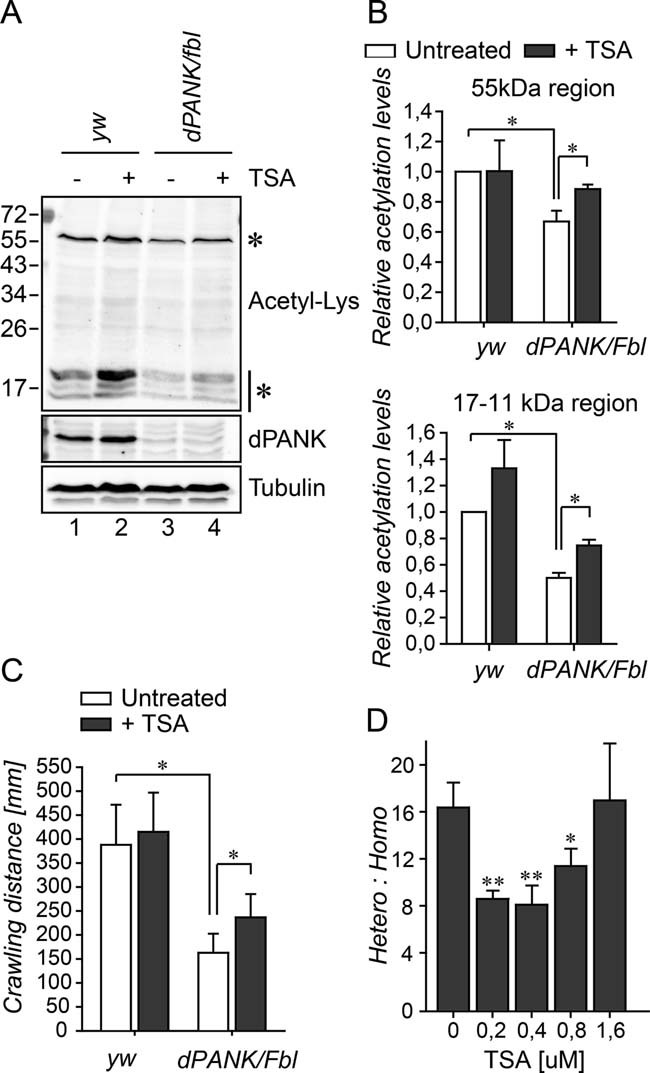
- Extracts of WT and dPANK/Fbl homozygous third instar larvae were analysed for their levels of acetylated proteins using acetyl-Lys antibody. Tubulin was used as a loading control and the dPANK antibody was used to demonstrate the reduced expression of dPANK/Fbl in the mutant larvae. Addition of 0.2 µM TSA to the larval food resulted in increased levels of acetylated histones and tubulin.
- Quantification of the relative intensity of the 55 kDa band (corresponding to acetyl-tubulin) and <17 kDa bands (corresponding to acetyl-histones) in larvae extracts.
- Ability of larvae to crawl a certain distance in 9 min was measured as previously described (Rana et al, 2010). Larval crawling assay was performed in WT larvae and in dPANK/Fbl homozygous mutant larvae untreated or fed with the HDAC inhibitor TSA (0.2 µM).
- dPANK/Fbl/TM3 males and females were crossed and various concentrations of TSA were added to the food. The number of homozygous (dPANK/Fbl/dPANK/Fbl) versus heterozygous dPANK/Fbl/TM3 adults which eclosed was counted.
Impairment of pantothenate kinase function correlates with decreased acetylation of tubulin and histones in various mammalian cell models for PKAN
Finally, we aimed to test whether inhibition or down-regulation of PANK in a human cell line also results in decreased levels of acetylated tubulin and acetylated histones. Firstly, human PANK activity was inhibited using HoPan in HEK293 cells (Fig 6A). Western blot analysis using the acetyl-lysin, the acetyl-tubulin, the acetyl-H3 and the acetyl-H4 antibodies demonstrated that levels of acetylated tubulin and levels of acetylated histones (Fig 6A) were decreased. These effects were rescued by addition of pantethine or TSA to the medium (Fig 6A). In light of the above data, it is highly relevant to investigate levels of histone and tubulin acetylation in material derived from PKAN patients. We investigated acetylation levels in available patient-derived lymphoblasts. In these cells no significant difference in histone acetylation could be observed (Fig S6 of Supporting information). However, it should be noted that lymphoblast cells of patients do not show any phenotype and therefore it will be of higher relevance to investigate protein acetylation levels in tissues (when available in the future) that are affected in PKAN patients, such as the globus pallidus (Kruer et al, 2011). In order to test whether neuronal cells show the same response to impaired PANK function, SHSY-5Y cells (a human derived neuroblastoma cell line; Biedler et al, 1978) were used and HoPan treatment induced a decrease in tubulin and histone acetylation, an effect reverted by pantethine or TSA treatment (Fig 6B). Finally, we aimed to test whether down-regulation of specifically PANK2 (the causative gene of the neurodegenerative disease PKAN) in a human cell line also resulted in decreased levels of acetylated tubulin and acetylated histones. Human PANK2 was down-regulated using siRNAs in HEK293 cells (Fig 6C). Western blot analysis using the acetyl-lysin antibody demonstrated that levels of acetylated tubulin (Fig 6C, high exposure) and levels of acetylated histones (Fig 6C low exposure) were decreased. These results were further confirmed using specific antibodies against acetyl H3 and acetyl H4 and hPANK2 depletion resulted in a 50% decrease in histone 3 as well as histone 4 acetylation as compared to cells treated with control siRNA (Fig 6D, for quantification see Fig 6E). Addition of pantethine to the cell culture medium increased the acetylation in siPANK2 treated cells to the level indistinguishable from that of control cells (Fig 6D). These results demonstrate that in human cells, impaired de novo biosynthesis of CoA is also associated with decreased levels of acetylation of a specific set of proteins. Additionally, we tested VPA for its potential to increase acetylation in PANK2-depleted background. VPA is an HDAC inhibitor of a high interest for mammalian systems because it is able to cross the blood–brain barrier and has been proposed as a possible treatment for neurodegeneration (reviewed in Monti et al, 2009). Treatment with VPA increased acetylation levels of histones in hPANK2-depleted cells (Fig 6D and E).
Figure 6. Impairment of human PANK activity in vitro results in decreased levels of tubulin and histone acetylation, an effect reverted by pantethine and by HDAC inhibitors.

- HEK293 cells were left untreated or were treated with HoPan. Control and HoPan treated cells were co-treated with pantethine or TSA. Acetyl-lysine, acetyl-tubulin, acetyl-H3 and acetyl-H4 antibodies were used to analyse acetylation levels of tubulin and histones. Histone H2A and tubulin were used as loading controls.
- As in A but human SHSY-5Y cells were used.
- HEK293 cells transfected with a control siRNA or with an siRNA against human PANK2 were analysed for the levels of acetylated proteins. Acetyl-lysine antibody was used. Low exposure (left panel) was shown to determine levels of histone acetylation (lowest asterisk), high exposure (right panel) was shown to determine levels of acetyl-tubulin (highest asterisk). Efficiency of RNAi was confirmed using an antibody specifically recognizing human PANK2. Tubulin was used as a loading control.
- Control cells and human PANK2 depleted cells were investigated for their levels of acetyl H3 and acetyl H4. Cells were additionally left untreated or were treated with pantethine or VPA. Specific antibodies were used to determine levels of histone H3 and histone H4. The efficiency of human PANK2 RNAi was determined by a specific human PANK2 antibody. H3 and GAPDH were used as loading controls.
- Quantification of the relative levels of acetylated histone 3 and acetylated histone 4 compared to control cells.
DISCUSSION
In this study we show that in addition to H/KATs and HDACs, which are the key proteins controlling protein acetylation, levels of the metabolite CoA affect the status of protein acetylation as well. We demonstrate that under conditions of reduced levels of CoA, there is no compensatory mechanism able to maintain normal histone and tubulin acetylation. Numerous reports exist about the role of CoA in metabolic processes (reviewed in Leonardi et al, 2005), however, an influence of levels of this cofactor on protein acetylation as we describe here has never been reported. Recent results of other studies are in line with our observations. It has been demonstrated that enzymes required for the synthesis of acetyl-CoA from acetate or citrate also influence the acetylation status of histones (Starai et al, 2004; Takahashi et al, 2006; Wellen et al, 2009). Although neither CoA nor acetyl-CoA levels were directly measured in these studies, these results are in agreement with our observations and with the model presented in Fig 7. Our results reveal that levels of tubulin and histone acetylation are decreased but still detectable under conditions of CoA reduction. In addition, acetylation levels of other proteins recognized by the acetyl-lysin antibody seem to be unaffected. It will be of interest to investigate how much residual CoA is required to maintain acetylation levels of specific proteins. Until now no literature exists addressing these issues. Previously we demonstrated that in adult dPANK/fbl (hypomorph) mutant flies levels of CoA are undetectable with the method used (Rana et al, 2010), however, we cannot conclude that CoA levels are actually zero. In dPANK/Fbl RNAi depleted Drosophila S2 cells, levels of CoA decrease to 30% (Rana et al, 2010) and after HoPan treatment CoA levels drop to 50% (this manuscript). Under all these circumstances, acetylation of tubulin and histones is still detectable but clearly decreased. Most likely under these conditions there is a residual CoA and acetyl-CoA pool left, sufficient for the detected protein acetylation or alternatively there exist an additional source other than acetyl-CoA for protein acetylation. It will be of interest to investigate in a more detailed way, how levels of CoA in specific subcellular compartments influence the acetylation of specific proteins over time and whether this is tissue specific and how this affects specific cellular processes.
Figure 7. CoA metabolism and protein acetylation are tightly linked.
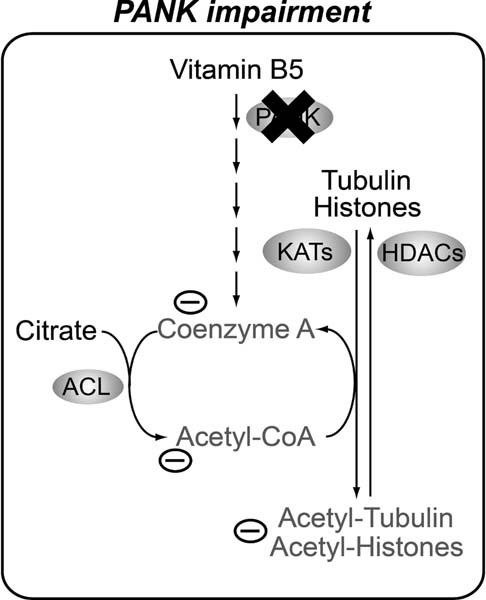
Under WT conditions, CoA is synthesized de novo from Vitamin B5 at normal levels. CoA is incorporated in acetyl-CoA and the latter serves as the acetyl source for various protein acetylation reactions. A tight interplay between KATs and HDACs determine the acetylation levels of specific proteins resulting in normal homeostasis of cells and tissues. In cells and organisms suffering from impaired function of PANK, CoA de novo biosynthesis is disturbed, and levels of CoA are decreased. Reduced levels of CoA result in decreased levels of acetyl-CoA and reduced acetylation of tubulin and histones. The latter is associated with decreased survival, impaired locomotor function and an impaired DNA damage response. We suggest that decreased acetylation levels of specific proteins may explain part of the pleiotropic phenotype of the Drosophila PKAN model and possibly part of the pathogenesis of PKAN.
The importance of normal CoA metabolism is underscored by the fact that PKAN patients who carry a mutation in the PANK 2 gene suffer from an early onset and a severe form of neurodegeneration (Zhou et al, 2001). The pathophysiology of this disease is not understood. Recently, we showed using a Drosophila PKAN model that levels of CoA are severely reduced when PANK is affected (Rana et al, 2010). Here we show that reduced levels of CoA coincide with reduced acetylation levels of histones and tubulin and it is of interest that these ‘marker’ proteins are associated with neurodegenerative conditions reviewed in (Fischer et al, 2010; Saha and Pahan, 2006). Our observations suggest that at least part of the impaired locomotor abnormalities of the Drosophila PKAN model can be explained by decreased acetylation levels of specific proteins and this may also explain the complex pathophysiology of PKAN patients. Although the above-mentioned studies and our results together demonstrate a strong link between histone and tubulin acetylation and neurodegeneration, it should be stressed that neither the published studies nor our data exclude that acetylation levels of other proteins may also explain part of the observed effects.
Another feature that correlates with decreased levels of CoA and impaired histone- and tubulin acetylation in Drosophila dPANK/fbl mutants and dPANK/Fbl-depleted cells is their increased sensitivity to DNA damaging agents. This correlation is in line with other reports, suggesting that normal acetylation of histones is required for a functional DNA damage response (van Attikum and Gasser, 2005). Therefore, we propose that the DNA damage sensitive phenotype of the Drosophila PKAN model may be explained by decreased acetylation levels of a specific set of proteins. It is yet unclear whether or not the presence of DNA damage as a result of an impaired DNA damage response is somehow linked to neurodegeneration or impaired locomotor function, however, previously we demonstrated that inducing DNA damage in WT fruitflies results in reduced climbing activities (Bosveld et al, 2008) and there are multiple examples of neurodegenerative diseases associated with an impaired DNA damage response (reviewed in Katyal and McKinnon, 2008).
Our data demonstrate that defects in PANK function in various cells and organisms are associated with a decrease in acetylated tubulin. This is of high interest because there is extensive literature in which abnormal tubulin acetylation is linked to impaired neuronal functioning. Amongst others, acetylated tubulin is associated with stable tubulin filaments and defects in tubulin acetylation are associated with abnormal transport in neuronal cells (Dompierre et al, 2007), abnormal branching of projection neurons (Creppe et al, 2009), neuromuscular defects (Akella et al, 2010) and impaired function of touch receptor neurons (Shida et al, 2010; for a recent review see Perdiz et al (Perdiz et al, 2011)).
Although it remains to be proven it is attractive to speculate that the neurological defects observed in PKAN affected individuals may be partly explained by abnormal tubulin acetylation.
All together our results indicate a conserved link between CoA metabolism and protein acetylation and when disturbed this coincides with a pleiotropic phenotype as observed in the Drosophila model for PKAN (Afshar et al, 2001; Bosveld et al, 2008; Rana et al, 2010; Zhou et al, 2001). These results provide a novel explanation for how the disturbed synthesis of CoA as in PKAN may lead to neurodegeneration. We furthermore show that the Drosophila PKAN phenotype is partly rescued by adding specific compounds to the food that either replenish CoA levels (pantethine) or restore protein acetylation levels (TSA). This knowledge can be of use to develop possible future therapies for PKAN.
MATERIALS AND METHODS
Cell culture and RNAi
Drosophila Schneider's S2 cells were cultured and subjected to RNAi treatment as described previously (Rana et al, 2010). HEK293 and SHSY-5Y cells were cultured according to the standard protocols in dMEM supplemented with 10% FCS and Penicilin/Streptomycin. For RNAi experiments 50 nM PANK2 siGENOM SMARTpool (Dharmacon) or non-silencing control siRNA (Dharmacon) were transfected with Lipofectamine (Invitrogene) for 6 h in serum free medium. Transfection medium was then removed and cells were cultured for 48 h in complete culture medium. Cells were subcultured and transfected again according to the same protocol. Six hours after second transfection complete medium with or without 0.1 mM pantethine was added. Samples were collected 48 h after the second transfection. For HoPan treatment HEK293 and SHSY-5Y cells were cultures in custom made vitamin B5 deficient dMEM (Thermo Scientific) supplemented with dialysed FCS (Thermo Scientific). This medium did not effect normal growth of the cell lines. For all the cell lines studied, pantethine was used in the culture medium at the final concentration of 0.1 mM. HEK 293 and SHSY-5Y cells were treated with HoPan and/or pantethine for 4 and 6 days, respectively.
Inhibitors
PANK inhibitor, HoPan, was previously described (Zhang et al, 2007) and was used in the cell culture medium at a final concentration of 0.5 mM. HDAC inhibitors: TSA, sodium butyrate and VPA were purchased from Sigma. Drosophila S2 cells were treated with 0.5 µM TSA for 24 h, unless mentioned otherwise. Sodium butyrate was used in a final concentration of 5 mM for 5 h. HEK293 and SHSY-5Y cells were treated with 0.25 µM TSA for 7 h or with 2 mM VPA for 48 h.
The paper explained
PROBLEM
PKAN is a severe and mostly early childhood onset disease. The pathophysiology is unclear and currently there is no treatment available for this painful and devastating disease. Affected individuals carry a mutation in the gene coding for PANK 2, an enzyme required for de novo synthesis of Coenzyme A. Coenzyme A is a salient cofactor required for over 100 essential metabolic reactions in living cells. The consequences of impaired Coenzyme A metabolism remain largely unresolved and need to be investigated in order to understand the pathophysiology of PKAN and to design future therapies.
RESULTS
We recently established a Drosophila model for PKAN and showed that levels of Coenzyme A are severely decreased and strongly correlated with a neurodegenerative phenotype. This fruitfly model was used to investigate consequences of Coenzyme A depletion and, thereby, clarify possible mechanisms that underlie neurodegeneration in PKAN. We demonstrate for the first time that decreased levels of Coenzyme A in the fly model and in human cell models of PKAN induce a decrease in levels of tubulin and histone acetylation. Restoration of tubulin and histone acetylation via several means coincides with a partial rescue of the PKAN associated phenotypes at the cellular and at the behavioural level.
IMPACT
Decreased levels of histone and tubulin acetylation are strongly associated with other neurodegenerative diseases and therefore our data suggest that part of the neurodegeneration observed in PKAN models is due to decreased protein acetylation levels. Our data further imply that compounds capable of restoring Coenzyme A levels or protein acetylation levels (or a combination thereof), can be used as starting points for the development of possible future therapies for PKAN.
Immunoblotting
Immunoblotting was performed as previously described (Rana et al, 2010). The following antibodies were used: anti-dPANK/Fbl (previously described; Bosveld et al, 2008), anti-acetyl-lysine (Cell Signaling, #9441), anti-alpha-tubulin (Sigma), anti-acetyl-tubulin (Sigma, clone 6-11B-1), anti-acetyl-H3 (Active Motif, #39139), anti-acetyl-H4 (Millipore, #06-598), anti-H2A (Abcam, #ab13923), anti-acetyl-H4K16 (Active Motif), anti-acetyl-H2K5 (Active Motif), anti-H3 (Millipore), anti-hPANK2 (a gift from J. Gitschier, UCSF) and anti-GAPDH (Fitzgerald Industries). HRP-conjugated secondary antibodies were from Amersham. Western blots from at least three-independent experiments were quantified using Adobe Photoshop SC3.
S2 cells – radiation survival assay
RNAi experiment was performed to knock-down dPANK protein. After 4 days of RNAi cells were subcultured to an equal density and left untreated or treated with HDAC inhibitors. After 4 h cells were washed twice with PBS, resuspended in complete culture medium and plated in the density of 0.4 × 106/ml in 36 mm dishes. Cells were irradiated with Cesium-137 source IBL 637 irradiator (CIS Bio-Internationl). Six days after exposure to IR viable cells were counted with Trypan Blue exclusion test.
Drosophila maintenance and physiological assays
Hypomorphic dPANK/fbl1 strain was used (Afshar et al, 2001; Bosveld et al, 2008), y1w1118 were used as a WT control (Bloomgton Stock Centre). TSA feeding experiments: fly food was prepared by addition of varying concentration of TSA or DMSO (as control) to the standard food, equal numbers of flies (12 females, 4 males) were placed in vials and allowed to lay eggs for 48 h. Late third instar WT and homozygous dPANK/fbl1 larvae were collect for analysis. Larval crawling assays were performed as described before (Rana et al, 2010). To measure the eclosion rates of homozygous dPANK/fbl1 on TSA supplemented or control food, the ratio between heterozygous and homozygous adult survivors in the F1 generation was determined.
C. elegans
C. elegans strains were maintained at 20°C, according to the standard protocols (Brenner, 1974). N2 was used as a WT strain and the PANK deletion mutant, VC927 pnk-1 (ok1435)I/hT2[bli-4(e937) let-? (q782)qIs48](I;III), was obtained from the Caenorhabditis Genetics Center. Touch response assays were performed by blind scoring. L4 larvae were isolated and placed on control NGM agar plates or NGM agar plates supplemented with 0.8 mg/ml pantethine and seeded with E. coli OP50. After 48 h each animal was touched 10 times with an eyelash, altering between the anterior and posterior part of the body. Positive response as movement away behaviour from the stimulation was scored for 60 worms per condition. For Western blotting 40–50 synchronized worms were collected in 25 µl of M9 buffer, frozen in liquid nitrogen and stored in −80°C. Twenty-five microlitres of of 2× Laemmli sample buffer (without Bromophenol Blue) was added and samples were homogenized by sonication. Protein concentration was measured with DC Protein Assay (Bio-Rad), sample volumes were adjusted with 1× Laemmli sample buffer (with Bromophenol Blue) and equal amounts of protein were loaded on SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gels.
Statistics
Statistical data significance was estimated using the Student's t-test (two-tailed and where appropriate with equal or unequal variance). Plotted values represent averages of at least three-independent experiments and error bars represent standard deviation. p-Values below 0.05 were consider significant, where p < 0.05 was indicated with *, p < 0.005 with ** and p < 0.001 with ***.
Acknowledgments
We thank Harm Kampinga, Floris Bosveld and Sarah Pringle for critical reading of the manuscript and Karen Thijssen for technical assistance. The work was supported by a VIDI and VICI (Dutch organization for scientific research) grant to OCMS and a GUIDE (Groningen Graduate school) grant to KS.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
KS, BS, LX, AR, EAAN, SJ, SH and OCMS designed the experiments; KS, BS, AR, LX, LS and JdJ performed the experiments; KS, BS, EAAN, SJ, SH and OCMS wrote the paper.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Afshar K, Gonczy P, DiNardo S, Wasserman SA. Fumble encodes a pantothenate kinase homolog required for proper mitosis and meiosis in Drosophila melanogaster. Genetics. 2001;157:1267–1276. doi: 10.1093/genetics/157.3.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, Gaertig J. MEC-17 is an alpha-tubulin acetyltransferase. Nature. 2010;467:218–222. doi: 10.1038/nature09324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Biedler JL, Roffler-Tarlov S, Schachner M, Freedman LS. Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res. 1978;38:3751–3757. [PubMed] [Google Scholar]
- Bosveld F, Rana A, van der Wouden PE, Lemstra W, Ritsema M, Kampinga HH, Sibon OC. De novo CoA biosynthesis is required to maintain DNA integrity during development of the Drosophila nervous system. Hum Mol Genet. 2008;17:2058–2069. doi: 10.1093/hmg/ddn105. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–243. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Griswold A, Campbell C, Min KT. Individual histone deacetylases in Drosophila modulate transcription of distinct genes. Genomics. 2005;86:606–617. doi: 10.1016/j.ygeno.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Creppe C, Malinouskaya L, Volvert ML, Gillard M, Close P, Malaise O, Laguesse S, Cornez I, Rahmouni S, Ormenese S, et al. Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell. 2009;136:551–564. doi: 10.1016/j.cell.2008.11.043. [DOI] [PubMed] [Google Scholar]
- Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–117. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31:605–617. doi: 10.1016/j.tips.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Foglietti C, Filocamo G, Cundari E, De Rinaldis E, Lahm A, Cortese R, Steinkuhler C. Dissecting the biological functions of Drosophila histone deacetylases by RNA interference and transcriptional profiling. J Biol Chem. 2006;281:17968–17976. doi: 10.1074/jbc.M511945200. [DOI] [PubMed] [Google Scholar]
- Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. 2009;46:73–80. doi: 10.1136/jmg.2008.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katyal S, McKinnon PJ. DNA strand breaks, neurodegeneration and aging in the brain. Mech Ageing Dev. 2008;129:483–491. doi: 10.1016/j.mad.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- Kruer MC, Hiken M, Gregory A, Malandrini A, Clark D, Hogarth P, Grafe M, Hayflick SJ, Woltjer RL. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain. 2011;134:947–958. doi: 10.1093/brain/awr042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdistani SK, Grunstein M. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol. 2003;4:276–284. doi: 10.1038/nrm1075. [DOI] [PubMed] [Google Scholar]
- LeDizet M, Piperno G. Identification of an acetylation site of Chlamydomonas alpha-tubulin. Proc Natl Acad Sci USA. 1987;84:5720–5724. doi: 10.1073/pnas.84.16.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- Leonardi R, Zhang YM, Rock CO, Jackowski S. Coenzyme A: back in action. Prog Lipid Res. 2005;44:125–153. doi: 10.1016/j.plipres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Monti B, Polazzi E, Contestabile A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr Mol Pharmacol. 2009;2:95–109. doi: 10.2174/1874467210902010095. [DOI] [PubMed] [Google Scholar]
- Perdiz D, Mackeh R, Pous C, Baillet A. The ins and outs of tubulin acetylation: More than just a post-translational modification. Cell Signal. 2011;23:763–771. doi: 10.1016/j.cellsig.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Rana A, Seinen E, Siudeja K, Muntendam R, Srinivasan B, van der Want JJ, Hayflick S, Reijngoud DJ, Kayser O, Sibon OC. Pantethine rescues a Drosophila model for pantothenate kinase-associated neurodegeneration. Proc Natl Acad Sci USA. 2010;107:6988–6993. doi: 10.1073/pnas.0912105107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha SN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13:539–550. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Shida T, Cueva JG, Xu Z, Goodman MB, Nachury MV. The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci USA. 2010;107:21517–21522. doi: 10.1073/pnas.1013728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starai VJ, Takahashi H, Boeke JD, Escalante-Semerena JC. A link between transcription and intermediary metabolism: a role for Sir2 in the control of acetyl-Coenzyme A synthetase. Curr Opin Microbiol. 2004;7:115–119. doi: 10.1016/j.mib.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-Coenzyme A synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM. The histone code at DNA breaks: A guide to repair. Nat Rev Mol Cell Biol. 2005;6:757–765. doi: 10.1038/nrm1737. [DOI] [PubMed] [Google Scholar]
- Vempati RK, Jayani RS, Notani D, Sengupta A, Galande S, Haldar D. p300-mediated acetylation of histone H3 lysine 56 functions in DNA damage response in mammals. J Biol Chem. 2010;285:28553–28564. doi: 10.1074/jbc.M110.149393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Li C, Lv S, Zhou B. Pantothenate kinase-associated neurodegeneration: insights from a Drosophila model. Hum Mol Genet. 2009;18:3659–3672. doi: 10.1093/hmg/ddp314. [DOI] [PubMed] [Google Scholar]
- Yuan J, Pu M, Zhang Z, Lou Z. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle. 2009;8:1747–1753. doi: 10.4161/cc.8.11.8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YM, Chohnan S, Virga KG, Stevens RD, Ilkayeva OR, Wenner BR, Bain JR, Newgard CB, Lee RE, Rock CO, et al. Chemical knockout of pantothenate kinase reveals the metabolic and genetic program responsible for hepatic Coenzyme A homeostasis. Chem Biol. 2007;14:291–302. doi: 10.1016/j.chembiol.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden–Spatz syndrome. Nat Genet. 2001;28:345–349. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.