

Abstract
Breast cancer is a molecularly, biologically and clinically heterogeneous group of disorders. Understanding this diversity is essential to improving diagnosis and optimizing treatment. Both genetic and acquired epigenetic abnormalities participate in cancer, but the involvement of the epigenome in breast cancer and its contribution to the complexity of the disease are still poorly understood. By means of DNA methylation profiling of 248 breast tissues, we have highlighted the existence of previously unrecognized breast cancer groups that go beyond the currently known ‘expression subtypes’. Interestingly, we showed that DNA methylation profiling can reflect the cell type composition of the tumour microenvironment, and in particular a T lymphocyte infiltration of the tumours. Further, we highlighted a set of immune genes having high prognostic value in specific tumour categories. The immune component uncovered here by DNA methylation profiles provides a new perspective for the importance of the microenvironment in breast cancer, holding implications for better management of breast cancer patients.
Keywords: breast cancer, DNA methylation, epigenetics, epigenomics, microenvironment
INTRODUCTION
Breast cancer is a global public health issue as it is the most frequently diagnosed malignancy in women in the Western world and the commonest cause of cancer death in European and American women. According to estimates in 2008, there were 1,383,000 new cases of breast cancer diagnosed, 458,000 deaths caused by breast cancer, and more than 4.4 million women living with breast cancer worldwide (Ferlay et al, 2010).
Human breast carcinomas have heterogeneous pathologies and the classification of breast tumours on the basis of histological criteria is confounded by a number of factors (Stingl & Caldas, 2007). Gene expression profiling by microarray analysis has offered a new way to classify human breast tumours. Based on the levels of mRNA expression of specific genes, four subtypes of breast cancers have been identified: basal-like cancers corresponding mostly to ER-negative and HER2-negative cancers, HER2-positive cancers characterized by increased expression of several genes of the HER2 amplicon, and two types of luminal cancers, low-grade luminal A and high-grade luminal B, which are predominantly ER-positive (Perou et al, 2000; Sorlie et al, 2001; Sotiriou et al, 2003; van 't Veer et al, 2002). Correlating this classification system with the traditional method based on tumour histology has revealed that some tumours that are classified according to their morphology correlate with a particular gene expression subset, whereas others do not (Sotiriou & Piccart, 2007). This molecular approach of categorizing breast tumours represents a paradigm shift in how we consider the origins and categorization of breast cancer. Indeed, the heterogeneous nature of breast cancers as well as the presence of distinct molecular entities suggests the existence of multiple ‘cells of origin’ (Lim et al, 2009). However, we are far from having a complete picture of the diversity of breast tumours. Unravelling the complexities of the heterogeneous nature of breast tumours holds important implications for cancer diagnosis, identification of new targets for therapy, and development of new strategies for clinical management.
Altered DNA methylation patterns are hallmarks of human cancers. Normally unmethylated promoters may become densely methylated, and this results in the silencing of critical genes such as tumour suppressor genes (Jones & Baylin, 2007). Other sequences become instead hypomethylated in tumours, leading to the aberrant activation of genes that are normally repressed by DNA methylation (Feinberg, 2007). Hypermethylation events have also been shown to serve as biomarkers in human cancers, for early detection in blood and other bodily fluids, for prognosis or prediction of response to therapy, and to monitor cancer recurrence (Laird, 2003).
Previous studies have documented aberrant methylation events in breast carcinogenesis and it was notably found that specific DNA methylation patterns can be related to some of the known ‘expression breast cancer subtypes’ (Bediaga et al, 2010; Fang et al, 2011; Flanagan et al, 2010; Holm et al, 2010; Sun et al, 2011; Van der Auwera et al, 2010). However such patterns have not been precisely related to novel and specific tumour traits. Here, our goal was to explore the DNA methylation landscapes of phenotypically heterogeneous tumours, to relate this diversity to landscape features, and extract novel biological and clinical meaningful information.
In the present paper, using Illumina's Infinium methylation platform, we have uncovered novel and precise epigenetic portraits in breast cancer, highlighting a key contribution of the DNA methylome to the complexity of the disease. Further, one of the major new finding of the present study is that we showed for the first time that DNA methylation profiles can reflect the cell-type composition of the tumour microenvironment, and in particular a T lymphocyte infiltration of these tumours. Interestingly, we found immune components that are good markers of breast cancer clinical outcome.
RESULTS
Breast tumours display DNA methylation profiles distinct from those of normal breast tissues
We used the Infinium Methylation Platform to perform DNA methylation profiling of two independent sets of frozen breast tissue samples: a ‘main set’ of 123 samples (4 normal and 119 infiltrating ductal carcinomas, IDCs), and a ‘validation set’ of 125 samples (8 normal and 117 IDCs; Fig 1A; Tables SI, SII and SXV of Supporting Information). The high-throughput Infinium technique, based on hybridization of bisulphite-converted gDNA on methylation-specific DNA oligomers, allows quantification of methylation levels at 27,578 CpG sites located within the promoter regions (and preferentially within CpG islands) of 14,475 consensus coding sequences and well-known cancer genes (Bibikova et al, 2009). When applied to the main set of breast tissues, this method revealed 6309 CpGs showing differential methylation between normal samples and IDCs (Fig S2; Table SIII and Supplemental Materials and Methods Section of Supporting Information). Validation of these data is depicted in Fig 1C–E (see also Fig S3 and Table SIV of Supporting Information). In terms of CpG location with respect to CpG islands (CGI), we found the hypermethylated CpGs to be mostly located inside CGI, whereas the hypomethylated CpGs were located principally outside of CGI (Fig 1B, left part). More than a fourth of the CpG island shores presented on the array displayed differential methylation between normal samples and IDCs, suggesting an important role of differential methylation of CpG island shores in cancer, consistent with earlier work (Irizarry et al, 2009). Further, besides the well-described differential methylation of high-CpG-density promoters (HCPs) (Jones & Baylin, 2007), we found even more pronounced methylation changes at intermediate- and low-CpG-density promoters (ICPs and LCPs, respectively) (Fig 1B, right part). Notably, ICPs (also called weak HCPs) seem to be highly susceptible to de novo DNA methylation (Fig 1B, right part), in agreement with previous studies (Weber et al, 2007).
Figure 1. High-throughput DNA methylation profiling in human frozen breast tissues.

- A. Experimental outline for DNA methylation analysis of 248 breast tissues with the Infinium Methylation Assay.
- B. Pie chart depicting the number of CpGs differentially methylated between breast tumour and normal samples of the main set, in terms of: (i) CpG location vs. CGI (as defined in Bock et al, 2007) as well as CpG island shores (as defined in Irizarry et al, 2009); (ii) CpG location vs. promoter classes (as defined in Weber et al, 2007; see also Table SIII of Supporting Information).
- C. Methylation frequencies of representative CpGs examined by bead array and their correlation with previously reported data (see Table SIV of Supporting Information for a more detailes table with references).
- D, E. Validation of the bead array method by conventional Bisulphite Genomic Sequencing (BGS). Panel (D) shows an exemplative analysed locus, CDK3, in 1 normal (N1) and 3 tumour samples (BCs). Red arrows indicate the location of the CpG investigated by the bead array, which seems representative of the surrounding CpGs (see Fig S3 of Supporting Information for further examples). Data representation was done according to (Bock et al, 2005). Panel (E) shows a significant positive correlation (Spearman's rho = 0.82; p < 0.001) between the Infinium Methylation and BGS data.
DNA methylation profiling identifies two major phenotypes of breast cancers that are related to ER status
We next wished to establish DNA methylation profiles that might have biological and clinical relevance. We performed an unsupervised hierarchical cluster analysis of the 119 IDCs of the main set, using a reduced list of CpGs showing differential methylation between normal samples and IDCs (2985 of them; see Supplemental Materials and Methods Section and Table SVII of Supporting Information). There emerged two major clusters (I and II; Fig 2A; Table SVIII of Supporting Information), with a significant correlation between cluster membership and both tumour grade and oestrogen receptor (ER) status (Fig 2B; Fig S4 of Supporting Information). Clusters I and II were enriched in ER-negative and ER-positive tumours, respectively. Importantly, gene expression studies have revealed that clinical biomarkers like ER and HER2 are just the tip of the iceberg, reflecting whole sets of tumour features not obviously related to the marker status (Sotiriou & Pusztai, 2009). This reality can be captured with gene co-expression modules, i.e. comprehensive lists of genes connected to different biological processes and showing highly correlated expression (Desmedt et al, 2008; Wirapati et al, 2008). One of the most discriminating co-expression modules is the ESR1 module (Desmedt et al, 2008). It comprises ER-pathway genes but also genes involved in other biological processes distinguishing ER-positive from ER-negative tumours. We therefore next examined to what extent ESR1 genes might be regulated at the epigenetic level. We divided the previously described ESR1 module (Desmedt et al, 2008) in two sub-modules, an ‘ESR1-positive’ and an ‘ESR1-negative’ module comprising, respectively, the genes whose expression correlates positively or negatively with that of ESR1. As shown in box plots and barcode plots derived from Gene Set Enrichment Analysis, ESR1-positive-module genes showed higher methylation levels in cluster I than in cluster II (Mann–Whitney test: p < 0.001; see Fig 2C and D and Table SIX of Supporting Information). Conversely, ESR1-negative-module genes showed significantly higher methylation levels in cluster II than in cluster I (Mann–Whitney test: p < 0.001; see Fig 2C and D and Table SIX of Supporting Information). Gene expression microarray analysis revealed a significant anti-correlation between the DNA methylation levels of these genes (in red) and their corresponding gene expression levels (in blue; Fig 2C and D).
Figure 2. DNA methylation profiling identifies two main breast tumour categories with different ER status.

- A dendrogram (top) and a heatmap (bottom) of the 119 breast cancer (BC) specimens of the main set obtained by performing a hierarchical cluster analysis show two main clusters, I and II.
- ER status is a main discriminator of the two broad tumour groups (see also Fig S4 of Supporting Information).
- Box plots of ESR1 module scores show that the genes of the ESR1-positive module (left part) showed higher methylation (red) and lower expression (blue) in cluster I than in cluster II. The opposite was observed for the ESR1-negative module (right part). The ESR1 module has been previously described (Desmedt et al, 2008).
- Barcode plots of the ESR1 module (provided by GSEA analysis) showing an anti-correlation of DNA methylation and expression data (see also Table SIX and Supplemental Materials and Methods of Supporting Information). Red and blue bars designate the positions of ESR1 module genes in methylation and expression rankings, respectively. Dotted lines depict the zero.
The above-results are consistent with recent work showing differential methylation between ER-positive and ER-negative tumours (Holm et al, 2010; Ronneberg et al, 2011; Sun et al, 2011). Further, in agreement with Sun and coworkers (Sun et al, 2011), our work shows that whole sets of genes, involved in processes far beyond ER biology and whose expression status distinguishes ER-positive from ER-negative tumours, are epigenetically regulated. This strengthens the idea that ER-positive and ER-negative breast cancers are two distinct diseases.
DNA methylation profiling identifies new subgroups of breast cancers
We next sought to refine the methylation-based taxonomy of our tumour set. As shown in Fig 3A, the unsupervised analysis of recurrent methylation patterns yielded six distinct entities (clusters 1 to 6; see Fig S5 and Supplemental Materials and Methods Section of Supporting Information for the formal procedure for cluster definition). We then wished to relate these methylation clusters with the known breast cancer ‘expression subtypes’. Currently, on the basis of gene expression profiles, four subtypes are distinguished (see also Introduction): basal-like, HER2-positive, luminal A and luminal B breast cancers (Sotiriou & Piccart, 2007). IHC and gene expression profiling (Fig 3A) revealed a significant preponderance of HER2-positive tumours in cluster 2, basal-like tumours in cluster 3, and luminal A tumours in cluster 6. Interestingly, no single ‘expression subtype’ appeared to dominate in methylation clusters 1, 4 and 5: cluster 1 contained HER2, basal-like as well as luminal B tumours; cluster 4 appeared to be a mix of HER2 and luminal B tumours; and cluster 5 contained both luminal A and B tumours (Fig 3A; see also Table SX and Fig S6 of Supporting Information). Hence, this importantly demonstrates the potential of DNA methylation profiling to refine breast cancer classification.
Figure 3. Complexity and heterogeneity of breast cancers as revealed by DNA methylation: a meaningful basis for refining breast tumour taxonomy.

- DNA methylation profiling of the main set identifies six groups of tumours, termed clusters 1–6, displaying differences in terms of ‘expression subtype composition’ and clinical characteristics (see also Fig S6 and Table SX of Supporting Information).
- Comparison of the methylation group assigned to each tumour of the main set by the unsupervised cluster analysis and the 86 CpG-classifier established by the nearest centroid classification method (Lusa et al, 2007; Sorlie et al, 2003; see also Fig S8 and Table SXIV of Supporting Information).
- Classification of each tumour of the validation set into one of the six methylation groups by means of the 86 CpG-classifier (see also Fig S9 and Table SXVI of Supporting Information). Note that the 6 groups obtained for the validation set presented the same ‘expression subtype composition’ and clinical characteristics as the groups obtained for the main set (see also Fig S10 and Table SXVII of Supporting Information).
To validate our six methylation clusters, we applied the Infinium methylation assay to an independent validation set of 117 breast tumours and used the efficient nearest centroid classification method (Lusa et al, 2007; Sorlie et al, 2003) to assign, on the basis of DNA methylation profile similarities, each new sample to one of the six clusters (see Supplemental Materials and Methods Section in Supporting Information). Focusing first on the main set, we established an 86 CpG-classifier that consists of a list of 86 key CpGs, this being the minimum number of CpGs required to retrieve the six unsupervised-analysis-based clusters (Fig 3B; Figs S7 and S8, Tables SXI, SXII, SXIII and SXIV, Supplemental Materials and Methods Section of Supporting Information). From this list of 86 CpGs, we calculated 6 centroids (i.e. profiles consisting of the median methylation value for each of the 86 CpGs) for each of the 6 methylation groups. Then, by computing the Spearman correlation of each tumour of the validation set with each calculated centroid, we classified each new sample into one of the 6 methylation clusters (Fig S9 and Table SXVI of Supporting Information). Remarkably, essentially all tumours of the validation set showed a strong correlation with one of the six methylation groups (Fig 3C; Fig S9 and Table SXVI of Supporting Information). Furthermore, IHC performed on the independent validation set showed a very similar ‘expression subtype composition’ for each of the 6 groups as in the case of the main set (Fig 3C; Table SXVII and Fig S10 of Supporting Information). It is noteworthy that the 86 CpG-classifier contained CpGs related to genes well-known to be implicated in breast cancer, such as: the oestrogen-inducible gene (TFF1), cyclin D1 (CCND1), secreted frizzled-related protein 2 (SFRP2), caspase 1 (CASP1), POU class 4 homeobox 1 (POU4F1) and interleukin 1, alpha and beta (IL1A and IL1B) (Table 1; Table SXIII of Supporting Information). Note also that this classifier contained majorly CpGs located in ICPs as well as LCPs (Fig S11 of Supporting Information). Taken together, these results reveal the existence of breast cancer groups that go beyond the currently known ‘expression subtypes’ and suggest that methylation profiling may provide a basis for improving tumour taxonomy.
Table 1.
The 86 key CpGs that characterize the 6 methylation-based clusters
| Illumina ID | Related gene | CpG Islanda | Promoterb |
|---|---|---|---|
| cg27610561 | SLC2A10 | Shore | HCP |
| cg21570818 | FUT5 | False | ICP |
| cg08887581 | C1orf64 | False | ICP |
| cg14023451 | GPLD1 | False | ICP |
| cg05215575 | FLJ25410 | False | ICP |
| cg11037787 | PLA2G2A | False | ICP |
| cg02671171 | RPH3AL | Shore | ICP |
| cg00294382 | IL23A | False | ICP |
| cg02643667 | TFF1 | True | ICP |
| cg21137417 | SPP2 | False | LCP |
| cg05089968 | MGC35308 | Shore | HCP |
| cg19456540 | SIX6 | True | HCP |
| cg14430151 | FLJ35725 | False | LCP |
| cg04457051 | SCOC | False | ICP |
| cg08097882 | POU4F1 | True | HCP |
| cg25942450 | TLX3 | True | HCP |
| cg08658594 | TAS2R13 | False | LCP |
| cg02170525 | CD8A | Shore | HCP |
| cg02880679 | MBTD1 | False | LCP |
| cg13271951 | FAM57B | Shore | ICP |
| cg08285151 | HDAC9 | False | LCP |
| cg05436658 | PRKCB1 | True | HCP |
| cg02148642 | RGPD5 | False | ICP |
| cg26189983 | TNFRSF1B | True | HCP |
| cg10707565 | CUBN | False | LCP |
| cg23801057 | P2RX7 | Shore | ICP |
| cg23092823 | PODN | True | HCP |
| cg03503295 | DNAH5 | False | LCP |
| cg09448880 | PGLYRP3 | False | ICP |
| cg22194129 | CLEC4C | False | ICP |
| cg17108819 | CD8A | True | HCP |
| cg01017147 | DNM3 | True | HCP |
| cg18752854 | TNS1 | False | ICP |
| cg19589427 | TNFSF18 | False | LCP |
| cg21475402 | BCAN | True | HCP |
| cg10300684 | FOXG1B | True | HCP |
| cg17095936 | TBX19 | False | ICP |
| cg01335367 | C12orf34 | True | ICP |
| cg24525573 | C1orf64 | False | ICP |
| cg15604467 | POU4F1 | True | HCP |
| cg05181279 | RIG | False | ICP |
| cg19018097 | FLJ30934 | True | HCP |
| cg06119575 | TAL2 | False | ICP |
| cg14686321 | FLJ31951 | Shore | HCP |
| cg10541755 | EIF5A2 | True | HCP |
| cg10334928 | STON2 | False | LCP |
| cg11354906 | SFRP2 | True | |
| cg06436504 | DOC1 | False | ICP |
| cg17619823 | ADRB3 | True | ICP |
| cg27196745 | PTPRO | True | HCP |
| cg02399455 | SRI | False | ICP |
| cg11802013 | CCND1 | True | |
| cg02595219 | KCNE3 | True | ICP |
| cg00596686 | STS | False | ICP |
| cg27491887 | KCNQ1 | True | |
| cg05158615 | NPY | True | HCP |
| cg20980592 | MEP1A | False | LCP |
| cg13696012 | BPIL1 | Shore | ICP |
| cg00953256 | CCND1 | True | |
| cg07426960 | CCND1 | True | |
| cg01109219 | RASGRP3 | False | LCP |
| cg10968815 | BPIL1 | Shore | ICP |
| cg15046693 | CEBPG | Shore | HCP |
| cg23391785 | DNM3 | True | HCP |
| cg00051623 | CASP1 | False | LCP |
| cg13755070 | FLI1 | True | HCP |
| cg02657438 | STON2 | False | LCP |
| cg13144783 | CCR1 | False | ICP |
| cg18129786 | ZNF445 | Shore | HCP |
| cg02723533 | CCND1 | True | |
| cg10964421 | TNFRSF10D | True | |
| cg24199834 | POU4F2 | True | HCP |
| cg14003512 | PLGLB2 | False | ICP |
| cg23642747 | INA | True | HCP |
| cg01424107 | CDX2 | True | HCP |
| cg02100848 | C3orf32 | False | ICP |
| cg05056120 | EBF | True | ICP |
| cg00839584 | IL1A | False | LCP |
| cg02681442 | FOXG1B | Shore | HCP |
| cg06653796 | LIME1 | True | ICP |
| cg21296230 | GREM1 | True | HCP |
| cg11547724 | HPX | False | ICP |
| cg17240454 | SPDEF | False | ICP |
| cg08047907 | C1orf114 | True | ICP |
| cg17667972 | KRT4 | False | ICP |
| cg07935264 | IL1B | False | LCP |
The CpG Island column indicates whether the CpG is located inside a CGI (true), is a CpG island shore (shore), or is neither in a CGI nor a CpG island shore (false).
Promoter column referred to the class of the promoter in which the CpG is located (HCP, high-CpG-density promoter; ICP, intermediate-CpG-density promoter; LCP, low-CpG-density promoter). See also Table SXIII of Supporting Information for further details.
Methylation patterns can be linked to specific stem cell/progenitor populations
Breast cancer research is currently undergoing a paradigm shift from tumour classification to an effort to trace distinct groups of breast cancers back to specific stem cell/progenitor populations likely to reflect their cellular origins (Stingl & Caldas, 2007; Visvader, 2009). In that respect, recent work has shed light on normal human mammary epithelial cell hierarchy (Prat & Perou, 2009), and gene expression profiling has shown that mammary stem cells (MaSC), luminal progenitors, and mature luminal cells have distinct gene expression signatures (Lim et al, 2009). To see to what extent the methylation patterns distinguished here might reflect the cellular origins of the studied tumours, we computed overall scores of these three gene expression signatures for each of the six methylation clusters described above; this being an effective way to measure concordance between sample profiles and subpopulation signature profiles. We observed no obvious concordance of any of the clusters with the MaSC signature (Fig 4A). Most strikingly, the score for the luminal progenitor signature (Fig 4B) was highest in cluster 3, rich in basal-like tumours (p = 0.001 vs. clusters 2 and 4; p < 0.001 vs. other clusters). It was low in clusters 1, 4, 5 and also 6, the cluster rich in luminal A-type tumours. Plots obtained for the mature luminal signature (Fig 4C) were almost a mirror image of the previous one: cluster 3 showed the lowest score, and clusters 1, 4, 5 and 6 the highest (p < 0.001 for each of these clusters vs. clusters 2 and 3, except for cluster 4 vs. cluster 2 where p = 0.019). Cluster 2, rich in HER2-overexpressing tumours, showed no clear association with any normal epithelial cell population. Remarkably, the anti-correlation between gene expression and DNA methylation increased from the top to the bottom of the mammary hierarchy (Fig 4, compare panels A, B and C with panels D, E and F, respectively), Spearman's coefficient being −0.435 (p < 0.0001) for the MaSC signature, −0.568 (p < 0.0001) for the luminal progenitor signature, and −0.726 (p < 0.0001) for the mature luminal cell signature. Thus, these observations suggest that the methylation patterns we have identified might be related to the cell type of origin of the tumours concerned (see Fig S12 of Supporting Information).
Figure 4. The identified methylation patterns might be related to the cell type of origin of the tumours concerned.
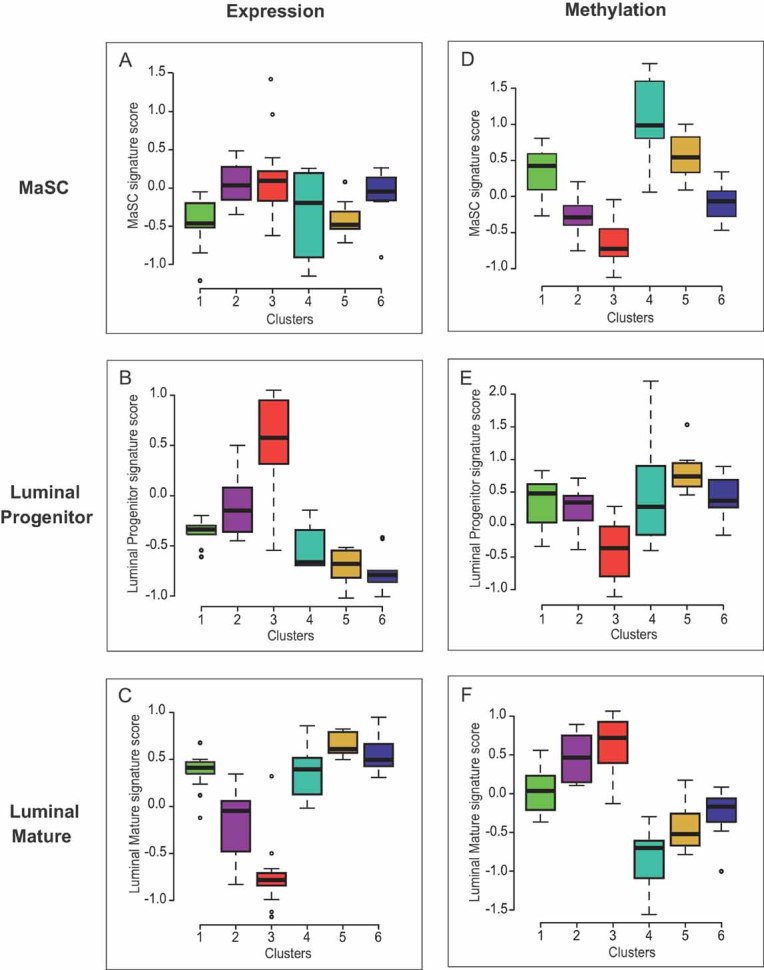
Comparison of gene expression signatures of several normal mammary epithelial subpopulations (Lim et al, 2009) with gene expression and DNA methylation profiles of our six DNA methylation-based groups of patients in the main set.
- A-C. Box plots of MaSC (A), luminal progenitor (B) and luminal mature (C) signature scores for each of the six methylation breast cancer groups, based on their gene expression profiles. Cluster 3 displayed the highest luminal progenitor signature score (p = 0.001 vs. clusters 2 and 4; p < 0.001 vs. other clusters; (B)), whereas the luminal mature signature score was higher for clusters 1, 4, 5 and 6 (p < 0.001 for each of these clusters vs. clusters 2 and 3, except for cluster 4 vs. cluster 2 where p = 0.019; (C)). Cluster 2 was not associated with any of the three signatures.
- D-F. Box plots of MaSC (D), luminal progenitor (E), and luminal mature (F) signature scores for each of the six methylation breast cancer groups, based on their DNA methylation profiles. A strong anti-correlation was observed between gene expression and DNA methylation data for the luminal progenitor and mature signatures (compare (E) with (B) and (F) with (C), respectively) (respective Pearson's coefficients: −0.59, p = 1.10−9 and −0.70, p = 6.10−14). It was weaker for the MaSC signature (compare (D) with (A); Pearson's coefficient: −0.47, p = 4.10−6).
DNA methylation profiles reflect the cell-type composition of the tumour microenvironment
Next, to further probe the biological significance of our six clusters, we quantified the number of differentially methylated targets (as compared to normal samples) characterizing each of the above clusters in the main set (see Supplemental Materials and Methods Section of Supporting Information). The number of targets was found to vary greatly between clusters, being lowest for cluster 3 (276 CpGs) and highest for cluster 4 (1,378 CpGs; Fig 5A; Table SXVIII of Supporting Information). We then performed a gene ontology (GO) analysis focusing on the genes in each cluster showing both differential methylation (as compared to normal samples) and a significant anti-correlation between methylation and expression (Tables SXIX, SXX and SXXI of Supporting Information). This revealed differential methylation of several genes involved in immunity, with different clusters showing distinct ‘epigenetic immune profiles’ (Fig 5B). In particular, tumours of clusters 2 (HER2-enriched) and 3 (basal-like-enriched) showed hypomethylation of several immune genes (Fig 5B). Because in this study we considered whole tumour tissues, our samples were constituted principally of epithelial cells, but also of cells from the surrounding stroma, including immune cells. Hence, we hypothesized that the observed hypomethylation of immune genes in clusters 2 and 3 indicated an infiltration of these tumours by immune cells, such as lymphocytes. This hypothesis proved correct. As shown in Fig 5C, we performed histological analysis, as previously described (Denkert et al, 2010), to determine stromal and intratumoural lymphocyte infiltration. Remarkably, the tumours of clusters 2 and 3 were much more infiltrated by lymphocytes than those of the other clusters (Fig 5D). Furthermore, the methylation status of most of the immune genes highlighted by the GO analysis correlated inversely with the level of lymphocyte infiltration (Fig 5E; Table SXXII of Supporting Information). In addition, DNA methylation profiling of normal and breast cancer epithelial cell lines as well as ex vivo T and B lymphocytes and lymphoid cell lines revealed that a high number of the studied immune genes were highly methylated in breast cancer and normal epithelial cell lines but barely methylated in lymphocytes (Fig 5F; Fig S13 of Supporting Information). These data strongly suggest that hypomethylation of immune genes detected in cluster-2 and -3 tumours reflects the cell-type composition of the tumour microenvironment, and in particular a lymphocyte infiltration of these tumours. A closer look at these genes revealed, in cluster 2, hypomethylation of genes involved in T cell biology, e.g. genes encoding T cell markers, like the CD6 antigen and T cell activation markers, like the LCK tyrosine kinase or the PTPN22 tyrosine phosphatase involved in T cell receptor signalling (Salmond et al, 2009; Wu et al, 2006) (Table SXXI of Supporting Information). These data might indicate that cluster-2 tumours, more readily than those of the other clusters, induce an antitumour T-cell response, with mobilization of T lymphocytes in the neoplastic environment.
Figure 5. DNA methylation profiles reflect the cell-type composition of the tumour microenvironment.

- Histograms showing the heterogeneity of breast tumours in terms of the number of CpGs differentially methylated compared to normal samples.
- Differential methylation of genes involved in immunity as revealed by GO analysis, with high hypomethylation content in clusters 2 and 3.
- Histological patterns of breast tumours displaying no lymphocyte infiltration (1) or both stromal and intratumoural infiltration (2). Panel 3 provides a closer look at the intratumoural infiltration presented in panel 2. Black arrows indicate epithelial cells, whereas green and blue arrows indicate stromal and intratumoural lymphocytes, respectively.
- Box plots depicting the higher lymphocyte infiltration in main set tumours belonging to clusters 2 and 3 as compared to tumours belonging to other clusters.
- Box plots illustrating the inverse correlation between LCK and ITGAL methylation and lymphocyte infiltration (Jonckheere–Terpstra test for trends; see also Table SXXII of Supporting Information).
- Methylation status, assessed by DNA methylation profiling, of immune genes highlighted by GO analysis in breast epithelial cell lines as well as in ex vivo lymphocytes and lymphoid cell lines (see also Fig S13 of Supporting Information).
Epigenetically regulated immune components are good markers of breast cancer clinical outcome
We then sought to assess the clinical relevance of the above-mentioned epigenetic changes in breast carcinogenesis. To this end, we performed an univariate survival analysis of all 6,309 CpGs identified in this study (i.e. as being differentially methylated between normal breast samples and tumours). As suspected, our main set appeared too small to allow interpretable results (Table SXXIII of Supporting Information). We therefore turned to the more abundant gene expression data publicly available and selected only untreated patients in order to evaluate the true prognostic value of biomarkers (between 730 and 952 samples, depending on the gene considered; Table SXXIV of Supporting Information). We selected 55 genes (see Supplemental Materials and Methods Section of Supporting Information) showing a strong anti-correlation between their methylation and expression status, and subjected them to a univariate Cox regression analysis. Strikingly, no less than 32 of these genes (58%) emerged as significant prognostic markers (Table SXXV of Supporting Information). Furthermore, 13 of the 32 genes are involved in immunity and 9, particularly, in T lymphocyte biology (CD3D, CD3G, CD6, LCK, LAX1, SIT1, RHOH, UBASH3A and ICOS; Fig 6A). To our knowledge several of them, like for example LAX1, SIT1, or UBASH3A, have never been highlighted before as survival markers in breast cancer. Consistently with the data presented in Fig 5D–F, low methylation of the above genes correlated with high lymphocyte infiltration (except for RHOH and BST2, so these were not subsequently considered; Fig 6B; Table SXXVI of Supporting Information). When looking at the expression levels of these genes, the opposite was found, that is, high gene expression correlated with high lymphocyte infiltration (Fig 6B; Table SXXVII of Supporting Information). This anti-correlation between the methylation and expression status of the immune genes was also found in breast epithelial cell lines as well as in ex vivo lymphocytes and T lymphoid cell lines, as determined by DNA methylation and gene expression profiling (Fig 6C). This is in keeping with the strong anti-correlation observed between methylation and expression status of these genes in the whole tumour samples (Table SXIX of Supporting Information). Furthermore, some of these genes (CD3D, CD3G, ICOS and UBASH3A) appeared highly methylated in ex vivo B lymphocytes and not in T lymphocytes samples (Fig 6C), again indicating that the observed lymphocyte infiltration mostly involves T lymphocytes, as suggested in Fig 6A.
Figure 6. Epigenetically regulated immune components are good markers of breast cancer clinical outcome.

- Pie chart depicting the high proportion of immune genes, and in particular of genes involved in T cell biology, among all the genes that appeared significant prognostic markers (FDR < 0.1) (see Table SXXV of Supporting Information).
- Box plots illustrating the correlation of methylation (in red) and expression (in blue) status of LAX1 and CD3D with stromal lymphocyte infiltration (Jonckheere–Terpstra test for trends; see also Tables SXXVI and SXXVII of Supporting Information).
- Anti-correlation between the methylation and expression status of the 11 prognostic immune markers in breast epithelial cell lines as well as in ex vivo lymphocytes and lymphoid cell lines, as determined by DNA methylation and gene expression profiling.
- High expression of 11 immune genes is associated with a better clinical. Forest plots showing the log2 hazard ratio (squares) with the 95% confidence interval (bars) of the relapse-free survival analysis. A negative hazard ratio reveals that a high expression level of the indicated variable is associated with a good outcome, and conversely.
- Immune markers appear significant in a multivariate analysis with all the classical markers used in clinic, as shown for the LAX1 and CD3D genes used as examples (see also Table SXXVIII of Supporting Information for the complete analysis). n, Number of patients; CI, Confidence Interval.
- Subtype-specific prognostic value of immune markers for breast cancer. Exemplative Kaplan–Meier curves for different levels of expression of the LAX1 and CD3D genes in each known ‘expression subtype’ (see also Table SXXIX of Supporting Information for the detailed continuous univariate survival analysis for each subtype).
We next focused on the association between the above 11 immune genes and clinical outcome. High expression of all of them was associated with a better outcome (Fig 6D), and interestingly, a multivariate analysis revealed that all of them, except CD6, seem to have an independent prognostic value to currently used clinical indicators (Fig 6E; Table SXXVIII of Supporting Information). A detailed survival analysis of the 11 immune genes revealed a subtype-specific prognostic value of these genes. Most of them showed high prognostic value in HER2-overexpressing and luminal B tumours, but none of them had an impact in luminal A tumours; only a few seemed to have prognostic value in basal-like tumours (Fig 6F; Table SXXIX of Supporting Information). Overall, our results suggest that the presence of these markers, associated with a better prognosis, might reflect an antitumour T-cell response, specific for certain tumour categories. In addition, these data highlight the importance of DNA methylation analyses in revealing components of breast cancers, like the immune component described here, that were not that apparent on the basis of classical gene expression analyses (the latter having revealed principally the cell proliferation component as the major prognostic marker for breast cancer (Sotiriou & Pusztai, 2009)).
DISCUSSION
In this report, on the basis of the epigenetic portraits we have drawn here, several novel findings emerge for breast cancer research and management.
Recent work have identified aberrant methylation events in breast tumours, showing that specific DNA methylation patterns can be related to some of the known ‘expression breast cancer subtypes’ (Bediaga et al, 2010; Holm et al, 2010; Sun et al, 2011; Van der Auwera et al, 2010). For example, Holm et al analysed 807 cancer-related genes to investigate whether the known ‘expression subtypes’ also display DNA methylation profiles (Holm et al, 2010). They found that three of the previously described ‘expression subtypes’, basal-like, luminal A and B, harbour different methylation profiles. Of particular interest, another recent study showed that on the basis of their DNA methylation profiles, luminal A tumours can be separated into two distinct entities (Ronneberg et al, 2011), suggesting that despite the strong concordance existing between breast cancer groups determined from gene expression and DNA methylation profiling, DNA methylation analysis provides additional information. Although it will be necessary to perform DNA methylation profiling of many more breast tumours, our work supports this hypothesis and demonstrates the existence of previously unrecognized breast cancer groups that go beyond the currently known ‘expression subtypes’. In other words, we have highlighted that DNA methylation profiling can provide a basis for refining tumour taxonomy.
Portraying the epigenetic facets of mammary tumours might also be relevant for understanding the cellular origins of the various subsets of breast cancers (Stingl & Caldas, 2007; Visvader, 2009). Noteworthy in this respect is the high luminal progenitor signature score observed for cluster 3, rich in basal-like tumours. We propose that based on their DNA methylation profiles, distinct groups of breast cancers can be traced back to specific progenitor populations, probably reflecting their cellular origins and thereby the biological heterogeneity of breast cancers. Thus, our discovery might potentially link cancer epigenetics to the epigenetics of normal and neoplastic stem/precursor cells.
Besides providing a fundamental ground towards an improved taxonomy of breast cancers, DNA methylation profiling uncovered the identification of clinically relevant markers and thus could contribute to cancer screening and prognosis. Beyond its technical advantages over RNA expression profiling (e.g. DNA is more stable, easily stored and could be collected from any quality of biopsy) (Laird, 2003), we show that it can reveal strong survival markers, that single use of gene expression profiling has not highlighted. An example is the heretofore-undervalued epigenetically regulated immune component discovered here, notably T-cell markers which are associated with a better clinical outcome in specific tumour categories. Another example is related to our findings that the two major phenotypes of breast cancers determined by ER status are widely epigenetically controlled, offering the prospect of a novel approach to treating ER-positive tumours. Indeed, being reversible, epigenetic changes are ideal candidate targets for drug development. Several inhibitors of enzymes controlling epigenetic modifications are already being clinically tested, alone or in combination with endocrine therapy, with a view to reversing endocrine resistance due to ER non-expression (Pathiraja et al, 2010). The present results suggest that such agents might be used, instead, to stimulate genes whose lack of expression in ER-positive/HER2-negative tumours (representing 50–60% of all breast cancers) might contribute to the poor response of these tumours to chemotherapy (Liedtke et al, 2009).
In summary, we have established an 86 CpG-classifier that may provide a meaningful basis for refining breast tumour taxonomy. Further, the identified methylation patterns might be related to the cell type of origin of the tumours concerned. One of the major novel findings of the current work is that DNA methylation profiles can reflect the cell-type composition of the tumour microenvironment, and in particular a T lymphocyte infiltration of these tumours. What's more, immune genes are shown to be good markers of breast cancer clinical outcome. The immune component we have uncovered opens new avenues to better understand the emerging intricate relationship existing between the tumour cells and the surrounding stroma, holding implications for breast cancer prevention, diagnosis and treatment.
MATERIALS AND METHODS
Breast tissue samples
The main set is constituted of 119 archival frozen breast cancer samples from patients diagnosed at the Jules Bordet Institute between 1995 and 2003. These samples were selected according to the following criteria: (i) sufficient presence of invasive cells as defined by pathologist (see Supporting Information); (ii) >2 µg yield of high quality DNA available; (iii) balanced distribution of the four main ‘breast cancer expression subtypes’ determined by IHC; and (iv) balanced distribution of patients with and without relapses within each subtype. Four samples of normal breast tissues with sufficient high-quality DNA were selected as well for this main series.
The validation set is constituted of 117 frozen breast cancer samples from patients diagnosed at the Jules Bordet Institute between 2004 and 2009. Of note, the validation set was slightly enriched in basal-like tumours as compared to the main set. Eight normal breast tissue samples were selected as well.
For complete patient data, see Tables SI, SII and SXV of Supporting Information. The Ethics committee of the Jules Bordet Institute approved the present research project.
DNA methylation profiling
Genomic DNA from the clinical frozen samples was extracted from twenty 10-µm sections using the Qiagen-DNeasy Blood &Tissue Kit according to the supplier's instructions (Qiagen, Hilden, Germany). This included a proteinase K digestion at 55°C overnight. For breast epithelial cell lines and lymphocyte samples, genomic DNA was extracted with the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) including the recommended proteinase K and RNase A digestions. DNA was quantitated with the NanoDrop® ND-1000 UV–Vis Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
Site-specific CpG methylation was analysed using Infinium® HumanMethylation27 bead-array-based technique. This array was developed to assay 27,578 CpG sites selected from more than 14,000 genes. Genomic DNA (1 µg) was treated with sodium bisulphite using the Zymo EZ DNA Methylation Kit™ (Zymo Research, Orange, USA) according to the manufacturer's procedure, with the alternative incubation conditions recommended when using the Illumina Infinium® Methylation Assay. The methylation assay was performed from 4 µl converted gDNA at 50 ng/µl according to the Infinium® Methylation Assay Manual protocol. The quality of bead array data was checked with the GenomeStudio™ Methylation Module software. All samples passed this quality control. Methylation raw data are available online (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=bvonpyugyawqqto&acc=GSE20713).
The paper explained
PROBLEM
Breast cancer is a very heterogeneous disease at histological as well as molecular levels. Despite considerable efforts by legions of clinicians and biologists to classify breast cancers into different groups, patients with the same ‘profile’ of breast cancer can respond differently to chemotherapies or targeted therapies and have completely different clinical outcomes. In order to improve diagnosis and optimize treatment, it is of particular importance to understand the heterogeneity of breast cancers. As it is increasingly clear that epigenetic abnormalities—and in particular aberrant DNA methylation—are involved in carcinogenesis, devoting more energy to explore this epigenetic component should bring new insights to better explain breast cancer diversity. Towards this end, we explored the DNA methylation landscapes of phenotypically heterogeneous breast tumours to extract novel biological and clinical meaningful information.
RESULTS
From DNA methylation profiling of 248 primary breast tumours, several key findings emerged. On the basis of their DNA methylation profiles, breast tumours can be divided into several groups that go beyond the currently known breast tumour ‘expression’ subtypes, suggesting a possibility to refine the classification. Interestingly, our study showed that DNA methylation profiles of breast tumours can reflect the cell type composition of tumour microenvironment, at least a T lymphocyte infiltration. Further, we found that several immune genes are as good prognostic markers of breast cancer clinical outcome in specific tumour categories.
IMPACT
Revealing a novel level of breast tumour heterogeneity, our study offers a perspective to refine the current breast cancer classification. In addition, we showed that DNA methylation profiling of whole breast tumours is able to assess both the epithelial and stromal components of tumours. Therefore, such approach could be useful to increase our understanding of the contribution of the tumour microenvironment in breast cancer. Overall, the epigenetic portraits we describe should help to improve breast cancer patient management.
Gene expression profiling
For tumours of the main set as well as cell lines and ex vivo samples, RNA was isolated by the Trizol method (Invitrogen) or the Tripure method (Roche) according to manufacturers' instructions and purified on RNeasy mini-columns (Qiagen). The quality of the RNA obtained from each tumour sample was assessed on the basis of the RNA profile generated by the Bioanalyzer (Agilent Inc.). Total RNA (100 ng) was first reverse-transcribed into double-stranded cDNA. This cDNA was transcribed in vitro. After purification of the aRNA, 12.5 µg were fragmented and labelled prior to hybridization to the Affymetrix HG133 Plus 2.0 GeneChip. Among the clinical samples of the main set, 30 initially profiled for DNA methylation were not profiled for gene expression because of low tumour-cell content (<70% tumour cells, n = 11), no tumour left at all in the samples (n = 4), low-quality RNA (n = 13), or low RNA quantity (n = 2). In addition, the CD4+ lymphocyte clone R12C9 was not profiled for gene expression because of low RNA quantity. The quality of the microarray data was checked using the ‘yaqcaffy’ package of the R statistical software (http://www.r-project.org/). On the basis of the results, two samples were excluded from further analysis. Gene expression raw data are available online (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=bvonpyugyawqqto&acc=GSE20713).
Author contributions
SD and CD designed experiments, performed research and interpreted data; Infinium Methylation Assays were done by SD and EC; Bisulphite genomic sequencing and pyrosequencing were done by SD, EC, MV, RD and PP; JL assisted in analysing bisulphite genomic sequencing data; SD, EC and JL performed experiments on breast epithelial cell lines; GH, NvB and PC isolated ex vivo lymphocytes and provided lymphoid cells; SD, CD, SKS, BHK, MD and StM conducted bioinformatic and statistical analyses. For clinical samples: selection was done by CD, FL and DL; preparation was done by FL, DL, JT, SH, FR, SaM and KS; characterization was done by DL and GR; annotation was performed by CD and OM; CS and FF designed experiments, interpreted data and directed the study; SD, CS and FF wrote the manuscript.
Acknowledgments
We thank Michel Georges, Nadine Cambisano, Naima Ahariz and Latifa Karim for technical assistance and for their helpful advices. SD, EC, JL and JT were supported by the Belgian F.N.R.S. CS and FF are ‘Chercheur Qualifié’ and ‘Maître de Recherche’ from the F.N.R.S., respectively. CD and SKS were supported by the Brussels Region. This work was funded by grants from the F.N.R.S. and Télévie, the ‘Plan National Cancer’, the Brussels Region ‘BruBreast’ and the ‘Interuniversity Attraction Poles’ (IAP P6/28), by the E.U. grant CANCERDIP FP7-200620 and by a European Molecular Biology Organization Young Investigator Programme (EMBO YIP).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Bediaga NG, Acha-Sagredo A, Guerra I, Viguri A, Albaina C, Ruiz Diaz I, Rezola R, Alberdi MJ, Dopazo J, Montaner MM, et al. DNA methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res. 2010;12:R77. doi: 10.1186/bcr2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics. 2009;1:177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulphite sequencing. Bioinformatics. 2005;21:4067–4068. doi: 10.1093/bioinformatics/bti652. [DOI] [PubMed] [Google Scholar]
- Bock C, Walter J, Paulsen M, Lengauer T. CpG island mapping by epigenome prediction. PLoS Comput Biol. 2007;3:e110. doi: 10.1371/journal.pcbi.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, et al. Tumour-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol. 2010;28:105–113. doi: 10.1200/JCO.2009.23.7370. [DOI] [PubMed] [Google Scholar]
- Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, Delorenzi M, Piccart M, Sotiriou C. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008;14:5158–5165. doi: 10.1158/1078-0432.CCR-07-4756. [DOI] [PubMed] [Google Scholar]
- Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011;3:75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- Flanagan JM, Cocciardi S, Waddell N, Johnstone CN, Marsh A, Henderson S, Simpson P, da Silva L, Khanna K, Lakhani S, et al. DNA methylome of familial breast cancer identifies distinct profiles defined by mutation status. Am J Hum Genet. 2010;86:420–433. doi: 10.1016/j.ajhg.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm K, Hegardt C, Staaf J, Vallon-Christersson J, Jonsson G, Olsson H, Borg A, Ringner M. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010;12:R36. doi: 10.1186/bcr2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- Liedtke C, Hatzis C, Symmans WF, Desmedt C, Haibe-Kains B, Valero V, Kuerer H, Hortobagyi GN, Piccart-Gebhart M, Sotiriou C, et al. Genomic grade index is associated with response to chemotherapy in patients with breast cancer. J Clin Oncol. 2009;27:3185–3191. doi: 10.1200/JCO.2008.18.5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, et al. Aberrant luminal progenitors as the candidate target population for basal tumour development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- Lusa L, McShane LM, Reid JF, De Cecco L, Ambrogi F, Biganzoli E, Gariboldi M, Pierotti MA. Challenges in projecting clustering results across gene expression-profiling datasets. J Natl Cancer Inst. 2007;99:1715–1723. doi: 10.1093/jnci/djm216. [DOI] [PubMed] [Google Scholar]
- Pathiraja TN, Stearns V, Oesterreich S. Epigenetic regulation in estrogen receptor positive breast cancer-role in treatment response. J Mammary Gland Biol Neoplasia. 2010;15:35–47. doi: 10.1007/s10911-010-9166-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Prat A, Perou CM. Mammary development meets cancer genomics. Nat Med. 2009;15:842–844. doi: 10.1038/nm0809-842. [DOI] [PubMed] [Google Scholar]
- Ronneberg JA, Fleischer T, Solvang HK, Nordgard SH, Edvardsen H, Potapenko I, Nebdal D, Daviaud C, Gut I, Bukholm I, et al. Methylation profiling with a panel of cancer related genes: association with estrogen receptor, TP53 mutation status and expression subtypes in sporadic breast cancer. Mol Oncol. 2011;5:61–76. doi: 10.1016/j.molonc.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmond RJ, Filby A, Qureshi I, Caserta S, Zamoyska R. T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunol Rev. 2009;228:9–22. doi: 10.1111/j.1600-065X.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumour subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al. Repeated observation of breast tumour subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou C, Piccart MJ. Taking gene-expression profiling to the clinic: when will molecular signatures become relevant to patient care. Nat Rev Cancer. 2007;7:545–553. doi: 10.1038/nrc2173. [DOI] [PubMed] [Google Scholar]
- Sotiriou C, Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med. 2009;360:790–800. doi: 10.1056/NEJMra0801289. [DOI] [PubMed] [Google Scholar]
- Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A, Martiat P, Fox SB, Harris AL, Liu ET. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA. 2003;100:10393–10398. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingl J, Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat Rev Cancer. 2007;7:791–799. doi: 10.1038/nrc2212. [DOI] [PubMed] [Google Scholar]
- Sun Z, Asmann YW, Kalari KR, Bot B, Eckel-Passow JE, Baker TR, Carr JM, Khrebtukova I, Luo S, Zhang L, et al. Integrated analysis of gene expression, CpG island methylation, and gene copy number in breast cancer cells by deep sequencing. PLoS One. 2011;6:e17490. doi: 10.1371/journal.pone.0017490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera I, Yu W, Suo L, Van Neste L, van Dam P, Van Marck EA, Pauwels P, Vermeulen PB, Dirix LY, Van Laere SJ. Array-based DNA methylation profiling for breast cancer subtype discrimination. PLoS One. 2010;5:e12616. doi: 10.1371/journal.pone.0012616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Visvader JE. Keeping abreast of the mammary epithelial hierarchy and breast tumourigenesis. Genes Dev. 2009;23:2563–2577. doi: 10.1101/gad.1849509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, Desmedt C, Ignatiadis M, Sengstag T, Schutz F, et al. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008;10:R65. doi: 10.1186/bcr2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, Tang J, Jeffery D, Mortara K, Sampang J, Williams SR, et al. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem. 2006;281:11002–11010. doi: 10.1074/jbc.M600498200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.