

Abstract
The ability to control the alkylation of organic substrates becomes ever more powerful by using metal catalysts. Among the major benefits of metal catalysis is the possibility to perform such processes asymmetrically using only catalytic amounts of the chiral inducing agent which is a ligand to the metal of the catalyst. A unique aspect of asymmetric metal catalyzed processes is the fact that many mechanisms exist for stereoinduction. Furthermore, using the same catalyst system, many types of bonds including but not limited to C-C, C-N, C-O, C-S, C-P, and C-H can be formed asymmetrically. An overview of this process using palladium and molybdenum based metals being developed in my laboratories and how they influence strategy in synthesizing bioactive molecular targets is presented.
INTRODUCTION
Asymmetric catalysis has truly blossomed beginning with the pioneering work on asymmetric hydrogenation and oxidation.1–3 However, the early work did not address an even more fundamental bond forming event in organic chemistry, that is forming the C-C bond asymmetrically. Asymmetric Lewis acid catalyzed reactions constituted one of the earliest directions1 followed by asymmetric phase transfer catalysts4 and organocatalysis5 and more recently asymmetric Bronsted acid catalysis.6
Among asymmetric catalytic processes, metal catalyzed asymmetric allylic alkylations (AAA) are unique in two respects – first, they have multiple mechanisms by which asymmetry can be introduced and, second, they can form many types of bonds among which are C-H, C-O, C-N, C-S, C-P, and, most importantly C-C using the same catalyst system.7 Many different metals may be used for such AAA processes including palladium, platinum, gold, rhodium, ruthenium, iridium, and molybdenum.8 At present, chiral palladium complexes have proven to be the most versatile and have the broadest scope. While the focus of this review is enantioselectivity, the question of regioselectivity is intimately tied. Changing from Pd to other metals is one mechanism to address this type of selectivity. In this vein, asymmetric Mo catalyzed reactions have been pursued as a complement for Pd, notably with carbon based nucleophiles. This account provides an overview of the evolution of the development of asymmetric allylic alkylations (AAA) in my laboratories.
General Features
Scheme 1 outlines the nature of the problem of ligand design for Pd catalyzed AAA. Notably, both the Pd assisted ionization (bond breaking) and nucleophilic attack (bond making) occur outside the coordination sphere of the metal and therefore distal to where the asymmetry resides, the ligands L. The working hypothesis for achieving high enantioselectivity came from enzymes wherein the asymmetric induction arises from the chiral space in which the substrate resides during reaction which, in turn, derives from the conformational chirality of the enzyme. To create such “chiral space” with small molecules, we envisioned the use of a macrocyclic bidentate ligand as depicted in Fig. 1 and 2.9 Fig 2.A and 2B illustrate that the chiral spaces is defined by the helical nature of the three aryl rings to the phosphorous. Recent structural and mechanistic studies reinforces the analogy to an active site of an enzyme in that H-bonding interactions between the N-H of the secondary amides in the backbone with either the leaving group when enantiodiscrimination occurs during ionization or the nucleophile as in Fig. 2C when enantiodiscrimination occurs in the nucleophilic addition are important for the chiral recognition.10
Scheme 1.

Mechanism of Pd Catalyzed Allylic Alkylation
Fig. 1.

Typical ligands for Pd AAA
Fig. 2.

Structural Representatives of Active Catalysts
Whereas Pd based complexes constitute the most extensively studied family of asymmetric catalysts in asymmetric allylic alkylation, other metals such as Ir, Rh, Ru, Mo, W, etc have also been examined. These metals typically involve substrates that require control of regioselectivity as well as enantioselectivity and provides a complement to Pd catalyzed processes. In my laboratories, we have examined Ru,11 Mo,12,13 and W13,14 based systems with the most success derived from Mo catalysts. These complexes also give substitutions with overall retention of configuration but via a double retention mechanism rather than a double inversion mechanism. Since the enantiodiscriminating event occurs within the coordination sphere of the metal, the chiral space required for asymmetric induction requires less steric bulk. Indeed, the typical ligands involve rather simple pyridine coordination as shown in Fig. 3.15 Bidentate coordination with the nitrogen of the pyridine is not required. Indeed, the monopyridine L-7 shows even higher branch selectivity than the bis pyridine L-5. Interestingly, a C2 symmetric diamine is not required.15b
Fig. 3.

Typical Ligands for Mo AAA
Enantiodiscrimination via Ionization
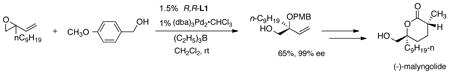
The simplest illustration of enantiodiscrimination via ionization is a kinetic resolution. While, in general, we have avoided effecting kinetic resolutions since it limits the yield to 50%, such a strategy still finds utility. Thus, we demonstrated that kinetic resolutions can be performed with near perfect selectivity as illustrated in eq. 1.16 However, the π-allylpalladium intermediate being symmetric, if both of the enantiomers can be ionized, a kinetic resolution can be avoided. Indeed by appropriate modification of the leaving group, both enantiomers of the starting
 |
(1) |
substrate can be ionized with one enantiomer of the catalyst leading to a DYKAT (dynamic kinetic asymmetric transformation) which is a preferable strategy that we strive for (vide infra).17
Desymmetrizing meso substrates represents another type of enantiodiscimination via ionization. 1,4-Diaryloxy-2-cycloalkenes represent the typical case.18 Using a carbon nucleophile such as
| (2) |
nitromethane provides the monoalkylated product in near perfect ee (eq 2).19 This product can serve as a precursor to D-nucleosides.
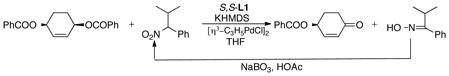
When a sterically crowded nitronate is employed, a strikingly different outcome is observed as shown in eq 3 wherein an O rather that C alkylation ensues.20 The initial O-allylated nitronate undergoes
 |
(3) |
spontaneous fragmentation to generate the ketone and an oxime. Since the nitroalkane is obtained by oxidation of the oxime with sodium perborate, the oxidant is simply readily recycled. This oxidative desymmetrization is a unique and efficient strategy in giving such versatile building blocks. On the other hand, heteroatom nucleophiles serve equally well as illustrated in eq 4.21,22 The dihydrofuran substrate is particularly illustrative of the efficacy of this strategy since the use of enzymatic methods to
 |
(4) |
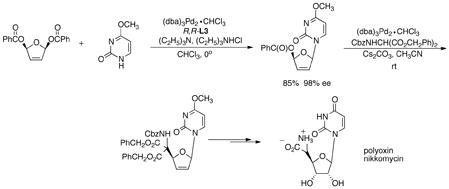
desymmmetrize such diesters is inapplicable here since the hemiacetal product of such processes is chemically unstable and simply falls apart. Of course, the remaining allyl ester serves as a substrate for further Pd catalyzed allylic alkylations as illustrated in eq 4 in an efficient synthesis of the polyoxin-nikkomycin natural products. Since the chirality has already been established, achiral ligands suffice.
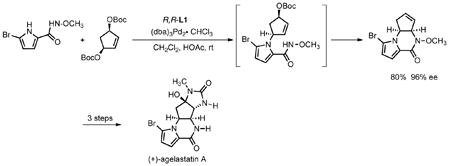
While the initial desymmetrization normally stops at the stage of the monoalkylation since the second alkylation requires a “mismatched” ionization, with certain nucleophiles and leaving groups, a cascade process has been observed. For example, with the bis-N nucleophile of eq 5 and the more reactive carbonate leaving group, the initial monoalkylation product spontaneously cyclizes under the reaction
 |
(5) |
conditions to form the tricycle, an intermediate on the way to agelastatin A.23 With phenylsulfonylnitromethane as nucleophile, a similar cascade was observed (eq 6).24 In this case, it is
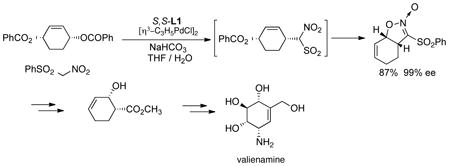 |
(6) |
best to add an achiral catalyst such as tetrakis(triphenylphosphine)palladium to speed up the second stage. The resultant product proved a useful intermediate in the asymmetric synthesis of valienamine via the chiral cis-β-hydroxyester. The higher intrinsic reactivity of the 5-membered ring substrates leads spontaneously to the cyclized product even with the benzoate leaving group.25
Use of malonate type nucleophiles led to a cascade to provide a net asymmetric cyclopropanation or lactone annulation.26 Using a simple malonate (path a, eq 7) with the dibenzoate gave the
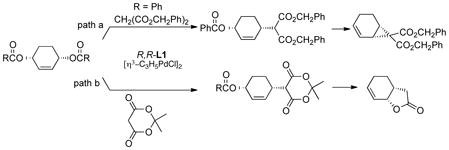 |
(7) |
monoalkylated product in 77% yield and 96% ee. Resubjecting this monoalkylated product to the palladium precatalyst and dppp initially at room temperature and then raising it to reflux gave the cyclopropane in 90% yield and 94% ee. On the other hand, using Meldrum’s acid as the malonate derivative (path b, eq. 7) with the dibenzoate gave the corresponding monoalkylated product in 60% yield and 98% ee. Resubjection of this monoalkylated product to the palladium precatalyst and racemic ligand then effected cyclization not to the cyclopropane but to the lactone in 75% yield. Using the carbonate substrate (R=OCH3) initially in the presence of potassium carbonate and methanol in THF followed by addition of diisopropylethyl amine gave the lactone directly in 70% yield and 99% ee.
Azide proved to be a most interesting nitrogen nucleophile. Using the benzoate substrate
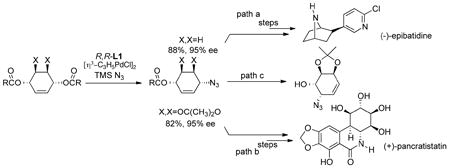 |
(8) |
provided the expected alkyl azide (path a, eq 8) in excellent yield and ee. An advantage of the azide is its excellent nucleophilicity and ease of reduction. Thus, exposing the azido benzoate to trimethylphosphine followed by di-t-butyldicarbonate provides the Boc protected amine directly which led to a facile synthesis of (−)-epibatidine (eq 8, path a).27 Interestingly, the much more heavily substituted substrate where X,X = OC(CH3)2O also reacts quite efficiently to produce the desired alkyl azide which was a key intermediate towards the antitumor agent pancratistatin (eq 8, path b).28 Thus, the acetonide did not hinder the approach of the palladium to the double bond even though the palladium must approach the same face of the double bond proximal to the acetonide. Subjecting the initial azide to base hydrolysis in hot methanol led to isolation of the rearranged azide (path c, eq 8) which could be isolated in 70% overall yield from the starting dicarbonate.29 While such a 3,3-sigmatropic rearrangement can be the result of a thermal process, there is evidence that it also may be promoted by palladium catalysis. In any event, both the 1,4- and 1,2-disubstituted amino alcohols can be accessed.
A second class of substrates for desymetrization of leaving groups is the gem-dicarboxylates(eq 9).30 Thus, the desymmetrical product is produced in high yield and ee.
 |
(9) |
A third type of meso substrate derives from the meso diasteromer of 3,4-dihydroxyhexa-1,5-diene (eq 10a). The cyclic carbonate gives the monoalkylated product using phthalimide as the nucleophile in
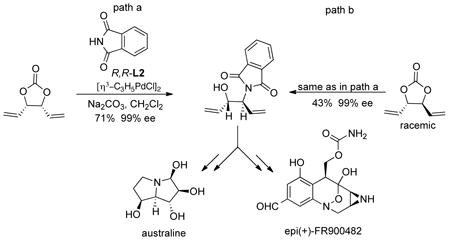 |
(10) |
perfect regio- and enantioselectivity.31 Surprisingly, the racemic d,l cyclic carbonate undergoes a clean kinetic resolution giving the same enantiomer also with perfect selectivity. Thus, using a mixture of both the meso and d,l diastereomers, only a single phthalimide enantiomer results precluding the need to separate the commercially available mixture of diols. This product proved to be a useful chiral ligand for Rh catalyzed asymmetric conjugate additions.32 It also served as a key intermediate for the synthesis of the pyrrolizidine alkaloid australine and an analog of the antitumor agent FR900482 which is epimeric at one center but of comparable biopotency.
Enantiodiscrimination by Deracemization of 1,3-Disubstituted Allyl Substrate
As pointed out in eq 1, the racemic tetraester can undergo a nearly perfect kinetic resolution; however, the yield is limited to 50%. Interestingly, examining the structure of the π-allyl-palladium intermediate
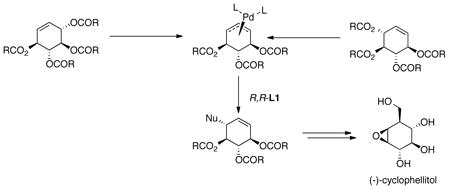 |
(11) |
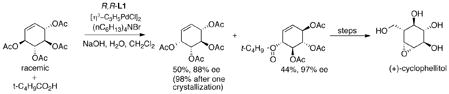
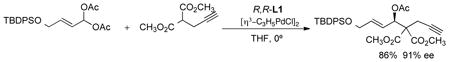
ignoring any asymmetry associated with the ligands reveals that it has a plane of symmetry (eq 11). Thus, the stereochemistry of the product is solely determined by the enantiodiscrimination in the nucleophilic addition. If the rate difference for the two enantiomers of the substrate in the initial ionization can be overcome, both enantiomers of the starting material, by going through the identical intermediate will lead to the same enantiomeric product. By using a trichloroethoxycarbonyl (Troc) leaving group, the rate of the mismatched ionization now becomes fast enough that both enantiomers do react to form mainly a single enantiomer of the product.33 In the case of phenylsulfonylnitromethane as the pronucleophile, the resultant product, an intermediate on the way to (−)-cyclophellitol, is formed in 81% yield and 88% ee.
A non-cyclic allyl substrate also performs quite well. The side chain of amphidimolide A was constructed using a 1,3-dimethylallyl system (eq 12), a particularly challenging substrate, in 90% ee.34
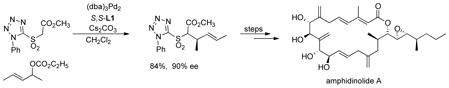 |
(12) |
The same allyl system also works well with an oxygen nucleophile. In the case of a “diosphenol” (a cyclic α-diketone), a good catalyst controlled diastereoselectivity was observed as shown in eq 13 on the way to a total synthesis of terpestacin.35
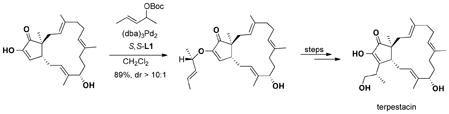 |
(13) |
Phenol nucleophiles have functioned well. A particularly intriguing illustration is shown with ester substituted allyl substrate shown in eq 14.36 Interestingly, the stilbene diamine chiral scaffold proved
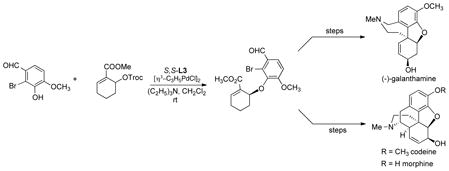 |
(14) |
most effective for the highly chemoselective allylation. The resultant product proved to be a common intermediate towards galanthamine, an inhibitor of Ache for treatment of Alzheimer’s, and morphine, one of the most important analgesics.
A similar allylating agent was employed in an intramolecular version using a nitrogen nucleophile (eq 15).37 The problem with this substrate was its propensity to undergo a background reaction. With our
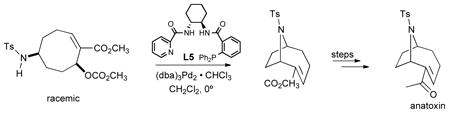 |
(15) |
typical ligands, L-1 to L-4, the sluggishness of the catalyzed processes required elevated temperatures (~100°) for reaction to occur and thus led to ee’s between 0 and 23%. However, by reducing the steric demand and increasing the donor property of the ligand by modifying the ligand as in L-5 (eq 15) allowed reaction to proceed at 0° which gave satisfactory results and led to a facile synthesis of anatoxin-a, a compound known as “very fast death factor” because of its potency in inducing respiratory paralysis.
The use of a lactone as the allylating agent in eq 16 raised a most unusual issue. Using phthalimide as
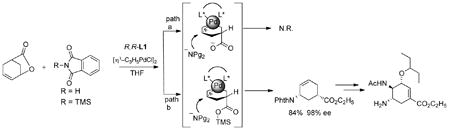 |
(16) |
the pro-nucleophile led to no reaction, either with or without base. The intermediate for path a requires a negatively charged nucleophile to come within close proximity to the carboxylate anion. The resultant charge-charge repulsion makes such a scenario unlikely. On the other hand, use of N-trimethylsilylphthalimide as the substrate in path b allows silyl transfer to neutralize the carboxylate anion and eliminate such charge-charge repulsion. Indeed, the reaction now proceeds quite well to give the desired alkylated phthalimide in 98% ee, a key intermediate for a very short synthesis of the antiviral agent tamiflu.38
Phthalimide also served as an excellent nucleophile for acyclic 1,3-disubstituted cases (typically >95% ee). A particularly intriguing example involved the use of epoxide of eq 17, available in two steps from cyclopentadiene and singlet oxygen, which led to a synthesis of (+)-polyoxamic acid.39
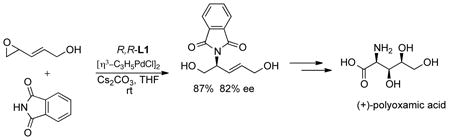 |
(17) |
Enantioselectivity with 1-Substituted or 1,1-Disubstituted Allyl Systems
Based upon the general notion that Pd catalyzed allylic alkylations normally lead to nucleophilic attack at the unsubstituted terminus of terminally monosubstituted allyl systems, focus for such substrates was concentrated on the use of Mo catalysis. As shown in eq 18, good yields of branched
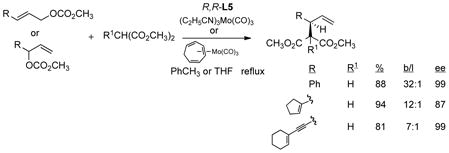 |
(18) |
products of high ee are obtained wherein the R substituent is an aryl, vinyl, or alkynyl group.15,40 While the Mo precatalysts were typically the (tris-propionitrile)- or cycloheptatriene-molybdenum tricarbonyl (eq 18), on large scale (2 kg) the more conveniently available Mo(CO)6 proved satisfactory (eq 19) in a
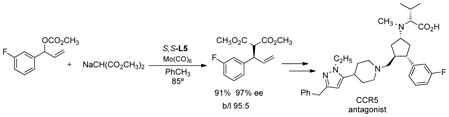 |
(19) |
synthesis of the CCR5 antagonist.41,42 Interestingly, a particularly sterically hindered 2,4,6-trisubstituted aryl ring used for a synthesis of (−)-tetrahydrocannabinol did not diminish the regioselectivity.43 The
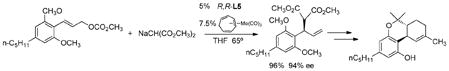 |
(20) |
chemoselectivity of the reaction is nicely highlighted in its success in a synthesis of the protease inhibitor tipranavir (eq 21).44 Running this reaction in a microwave at 180° with either L-5 or L-6 gave
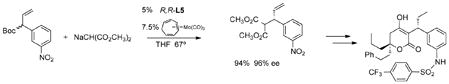 |
(21) |
the same yields and ee within a half hour compared to the 24h for the reaction at 67°. Remarkably, the ee was invariant with temperature up to 180°!
The inability to use heteroatom nucleophiles with Mo catalysts led us to reconsider the question of regioselectivity with Pd catalysts. In contrast to the general wisdom, obtention of branched products could be accomplished. The most direct way to achieve such a regioselectivity is to tether the nucleophile. Indeed cyclization of the phenol of eq 22 to form the chromanes gave attack at the more
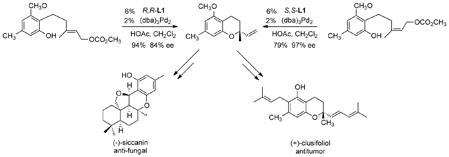 |
(22) |
substituted tertiary terminus.45,46 Interestingly, the Z olefin substrate gave higher ee than the corresponding E-olefin isomer. With such sterically small nucleophiles, use of bulky large bite angle ligands can direct the nucleophile to the more substituted terminus even in an intermolecular process as shown in the alkylation of eq 23 directed to a synthesis of calanolide B.47,48
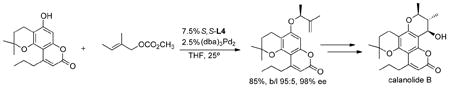 |
(23) |
Vinyl epoxides and aziridines have proven to be particularly useful in providing attack at the more substituted terminus. Thus, both nitrogen and oxygen nucleophiles can provide high selectivity in either monosubstituted (eq 2449 and 2550) or 1,1-dusubstituted allyl systems (eq 26).51 In the case of simple
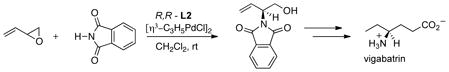 |
(24) |
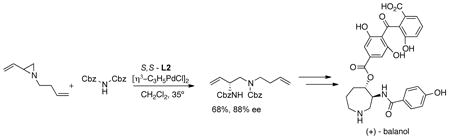 |
(25) |
 |
(26) |
alcohols as the nucleophilic partner, triethylboron is employed as a co-catalyst to improve reactivity and regioselectivity.
Deracemization of 1,3-Disubstituted Systems
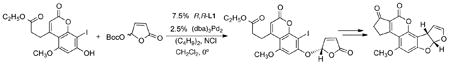
The ability to perform dynamic kinetic asymmetric transformations (DYKAT) requires a mechanism for interconversion of the facial complexation of the metal to the π-allyl unit. Butenolides provide such a mechanism via a π–σ–π interconversion through σ–binding to the carbonyl oxygen. This enantiodiscriminating mechanism is illustrated in the asymmetric inducing event in the synthesis of (−)- alfatoxin B (eq 27).52 The addition of a soluble chloride ion source as a co-catalyst facilitates the
 |
(27) |
conversion of the π– to σ–allyl complexes and thus the rate of racemization of the intermediate allyl complex.
Enantioselectivity of Prochiral Nucleophiles
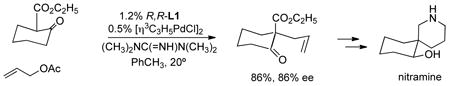
Inducing stereochemistry at the pro-chiral nucleophiles is an even more daunting challenge considering the mechanism of this process which places the incoming nucleophile very distant to the location of the chiral ligands and therefore the chiral space for the Pd catalyzed process. Remarkably, this mechanism for enantiodiscrimination can work quite well. In the first instance, β-ketoesters were
 |
(28) |
employed as prochiral nucleophiles with surprisingly high ee which led to a synthesis of the alkaloid nitramine (eq 28). 53
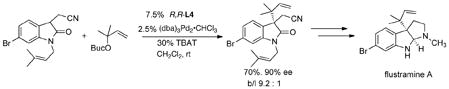
With substituted allyl systems, the reaction can become ligand controlled for both enantio- and regioselectivity. A particularly impressive example is the creation of two adjacent quaternary centers in a synthesis of the interesting flustramine alkaloids (eq 29).54
 |
(29) |
The asymmetric ligands impart increased scope of the type of nucleophiles that can be employed in addition to inducing asymmetry as exemplified by the use of ketone enolates. Thus, the 2-methylcyclopentanone derivative shown in eq 30 undergoes asymmetric allylation in excellent ee.55–57
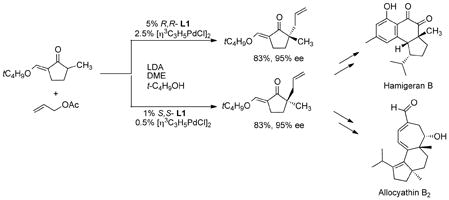 |
(30) |
One of the advantages of asymmetric chemical catalysis is the availability of both enantiomers by simply changing the enantiomer of the chiral ligand. In the example of eq 30, use of the R,R ligand provides access to hamigeran B; whereas, the synthesis of allocyathin B2 requires the enantiomeric allylic cyclopentanone which is equally easily available.
A particularly effective procedure for asymmetric ketone alkylations is the use of a decarboxylative asymmetric allylic alkylation (DAAA).58 Simply subjecting the enol allyl carbonate to Pd(0) effects
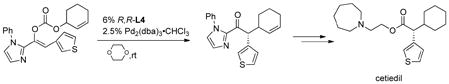 |
(31) |
loss of CO2 and formation of the allylated product in near quantitative yields and in excellent enantioselectivities under totally neutral conditions (eq 31).59 In the illustrated example, both the allyl and nucleophile are prochiral and only one diastereomer results, obviously controlled by the catalyst.
The fact that Mo catalyzed reactions are inner sphere processes make them even more likely catalysts for induction of stereochemistry at prochiral nucleophiles. Thus, simple asymmetric allylation of the oxindole in eq 32 proceeds in high yield and ee.60 Performing this asymmetric allylation with
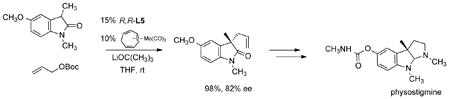 |
(32) |
Pd catalysts gave the product in a maximum of 70% ee. Use of a substituted allyl generates the product wherein two stereocenters are created in the Mo AAA. As shown in eq. 33, excellent control at both asymmetric centers is observed even in sterically congested systems.61
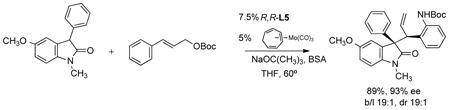 |
(33) |
CONCLUSION
The Pd and Mo AAA reactions constitute powerful tools for enantioselective synthesis. This power derives from the ability to form almost any kind of bond (C-C, C-O, C-N, C-F, C-P, etc) and to invoke a braod range of enantiodiscriminating mechanisms. The reactions achieve excellent selectivities under practical conditions of temperature, typically rt for Pd and 60–80° for Mo. The ligands are typically commercially available in one step from inexpensive commercially available materials. The processes also scale-up well, having been run on multi-kilo scales without problems. The reactions show excellent chemoselectivity. As illustrated by the examples herein, a broad diversity of structures of particular relevance with respect to biological properties are readily accessed wherein the AAA reaction played a key role in facilitating enhanced efficiency in the strategic approach.
Acknowledgments
I thank my numerous coworkers identified in the individual references who are the ones who have performed all of the work described. Their talent and enthusiasm made it all possible. Thanks also goes to the funding agencies, notably the National Science Foundation (CHE-0948222 and CHE-0846427) and the National Institutes of Health (GM33049) for their generous support of our programs. We thank Professor Guy Lloyd-Jones for structure C of Fig. 2.
References
- 1.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Synthesis. Springer-Berlin; 1999. [Google Scholar]; Ojimi I, editor. Catalytic Asymmetric Synthesis. 3. Wiley; New York: 2010. [Google Scholar]
- 2.Knowles WS. Angew Chem Int Ed. 2002;41:1998–2007. [Google Scholar]; Noyori R. Angew Chem Int Ed. 2002;41:2008–2022. [PubMed] [Google Scholar]
- 3.Sharpless KB. Angew Chem Int Ed. 2002;41:2024–2032. [PubMed] [Google Scholar]
- 4.Ooi T, Maruoka K. Angew Chem Int Ed. 2007;46:4222–4266. doi: 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]
- 5.Dondoni A, Massi A. Angew Chem Int Ed. 2008;47:4638–4660. doi: 10.1002/anie.200704684. [DOI] [PubMed] [Google Scholar]
- 6.Terada M. Synthesis. 2010:1929–1982. [Google Scholar]
- 7.Busch FR, Berchtold GA. J Am Chem Soc. 1983;105:3346–3347. [Google Scholar]; Trost BM, Van Vranken DL. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- 8.Trost BM, Lee C. Asymmetric Allylic Alkylation Reactions. In: Ojima I, editor. Catalytic Asymmetric Synthesis II. [Google Scholar]
- 9.Trost BM. Acc Chem Res. 1996;29:355–364. [Google Scholar]; Trost BM, Machacek MR, Aponick A. Acc Chem Res. 2006;39:747–760. doi: 10.1021/ar040063c. [DOI] [PubMed] [Google Scholar]
- 10.Butts CP, Filali E, Lloyd-Jones GC, Norrby P-O, Sale DA, Schramm Y. J Am Chem Soc. 2009;131:9945–9957. doi: 10.1021/ja8099757. [DOI] [PubMed] [Google Scholar]
- 11.Trost BM, Fraisse PL, Ball ZT. Angew Chem Int Ed. 2002;41:1059–1061. doi: 10.1002/1521-3773(20020315)41:6<1059::aid-anie1059>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 12.Trost BM, Lautens M. Organometallics. 1983;2:1687–1689. [Google Scholar]
- 13.Trost BM. J Organomet Chem. 1986;300:263–280. [Google Scholar]
- 14.Trost BM, Hung MH. J Am Chem Soc. 1983;105:7757–7759. [Google Scholar]; Trost BM, Hung MH. J Am Chem Soc. 1984;106:6837–6839. [Google Scholar]
- 15.a) Trost BM, Hachiya I. J Am Chem Soc. 1998;120:1104–1105. [Google Scholar]; b) Trost BM, Dogra K, Hachiya I, Emura T, Hughes DL, Krska S, Reamer RA, Palucki M, Yasuda N, Reider PJ. Angew Chem Int Ed. 2002;41:1929–1932. [PubMed] [Google Scholar]
- 16.Trost BM, Hembre EJ. Tetrahedron Lett. 1999;40:219–222. [Google Scholar]
- 17.Trost BM, Patterson DE, Hembre EJ. J Am Chem Soc. 1999;121:10834–10835. [Google Scholar]
- 18.Trost BM, Van Vranken DL. Angew Chem Int Ed. 1992;31:228–230. [Google Scholar]; Trost BM, Van Vranken DL, Bingel C. J Am Chem Soc. 1992;114:9327–9343. [Google Scholar]
- 19.Trost BM, Surivet J-P. Angew Chem Int Ed. 2000;39:3122–3124. doi: 10.1002/1521-3773(20000901)39:17<3122::aid-anie3122>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 20.Trost BM, Richardson J, Yong K. J Am Chem Soc. 2006;128:2540–2541. doi: 10.1021/ja057163d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trost BM, Shi Z. J Am Chem Soc. 1996;118:3039–3040. [Google Scholar]
- 22.Also see Trost BM, Madsen R, Guile SD, Brown B. J Am Chem Soc. 2000;122:5947–5956.
- 23.Trost BM, Dong G. J Am Chem Soc. 2006;128:6054–6055. doi: 10.1021/ja061105q. [DOI] [PMC free article] [PubMed] [Google Scholar]; Trost BM, Dong G. Chem Eur J. 2009;15:6910–6919. doi: 10.1002/chem.200900794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trost BM, Chupak LS, Lübbers T. J Am Chem Soc. 1998;120:1732–1740. [Google Scholar]
- 25.Trost BM, Li L, Guile SD. J Am Chem Soc. 1992;114:8745–8747. [Google Scholar]
- 26.Trost BM, Tanimori S, Dunn PT. J Am Chem Soc. 1997;114:2735–2736. [Google Scholar]
- 27.Trost BM, Cook GC. Tetrahedron Lett. 1996;37:7485–7488. [Google Scholar]
- 28.Trost BM, Pulley SR. J Am Chem Soc. 1995;117:10143–10144. [Google Scholar]
- 29.Trost BM, Pulley SR. Tetrahedron Lett. 1995;36:8737–8740. [Google Scholar]
- 30.Trost BM, Lee CB. J Am Chem Soc. 2001;123:3670–3686. doi: 10.1021/ja003774o. [DOI] [PubMed] [Google Scholar]; Trost BM, Lee CB. J Am Chem Soc. 2001;123:3687–3696. doi: 10.1021/ja003775g. [DOI] [PubMed] [Google Scholar]
- 31.Trost BM, Aponick A. J Am Chem Soc. 2006;128:3931–3933. doi: 10.1021/ja0578348. [DOI] [PMC free article] [PubMed] [Google Scholar]; Trost BM, Aponick A, Stanzl BN. Chem Eur J. 2007;13:9547–9560. doi: 10.1002/chem.200700832. [DOI] [PubMed] [Google Scholar]
- 32.Trost BM, Burns AC, Tautz T. Org Lett. 2011;13:4566–4569. doi: 10.1021/ol201754c. [DOI] [PubMed] [Google Scholar]
- 33.Trost BM, Patterson DE, Hembre EJ. Chem Eur J. 2001;7:3768–3775. doi: 10.1002/1521-3765(20010903)7:17<3768::aid-chem3768>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 34.Trost BM, Wrobleski ST, Chisholm JD, Harrington PE, Jung M. J Am Chem Soc. 2005;127:13589–13597. doi: 10.1021/ja0533646. [DOI] [PubMed] [Google Scholar]
- 35.Trost BM, Dong G, Vance JA. J Am Chem Soc. 2007;129:4540–4541. doi: 10.1021/ja070571s. [DOI] [PubMed] [Google Scholar]; Trost BM, Dong G, Vance JA. Chem Eur J. 2010;16:6265–6277. doi: 10.1002/chem.200903356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trost BM, Tang W. J Am Chem Soc. 2002;124:14542–14543. doi: 10.1021/ja0283394. [DOI] [PubMed] [Google Scholar]; Trost BM, Tang W, Toste FD. J Am Chem Soc. 2005;127:14785–14803. doi: 10.1021/ja054449+. [DOI] [PubMed] [Google Scholar]
- 37.Trost BM, Oslob JD. J Am Chem Soc. 1999;121:3057–3064. [Google Scholar]
- 38.Trost BM, Zhang T. Angew Chem Int Ed. 2008;47:3759–3761. doi: 10.1002/anie.200800282. [DOI] [PubMed] [Google Scholar]; Trost BM, Zhang T. Che Eur J. 2011;17:3630–3643. doi: 10.1002/chem.201003454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trost BM, Krueger AC, Bunt RC, Zambrano J. J Am Chem Soc. 1996;118:6520–6521. [Google Scholar]
- 40.Trost BM, Hildbrand S, Dogra K. J Am Chem Soc. 1999;121:10416–10417. [Google Scholar]
- 41.Palucki M, Um JM, Conlon DA, Yasuda N, Hughes DL, Mao B, Wang J, Reider PJ. Adv Synth Catal. 2001;343:46–50. [Google Scholar]
- 42.Yasuda N. CCRS Receptor Antagonist. In: Yasuda N, editor. The Art of Process Chemistry. Chapter 2. Wiley-VCH; Weinheim: 2011. pp. 45–76. [Google Scholar]
- 43.Trost BM, Dogra K. Org Lett. 2007;9:861–864. doi: 10.1021/ol063022k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trost BM, Andersen NG. J Am Chem Soc. 2002;124:14320–14321. doi: 10.1021/ja028497v. [DOI] [PubMed] [Google Scholar]
- 45.Trost BM, Shen HC, Dong L, Surivet JP, Sylvain C. J Am Chem Soc. 2004;126:11966–11983. doi: 10.1021/ja048078t. [DOI] [PubMed] [Google Scholar]
- 46.Trost BM, Shen HC, Surivet JP. J Am Chem Soc. 2004;126:12565–12579. doi: 10.1021/ja048084p. [DOI] [PubMed] [Google Scholar]
- 47.Trost BM, Toste FD. J Am Chem Soc. 1998;120:9074–9075. [Google Scholar]
- 48.Trost BM, Toste FD. J Am Chem Soc. 1999;121:4545–4554. [Google Scholar]
- 49.Trost BM, Bunt, Lemoine RC, Calkins TL. J Am Chem Soc. 2000;122:5968–5976. [Google Scholar]
- 50.Trost BM, Fandrick DR, Brodmann T, Stiles DT. Angew Chem Int Ed. 2007;46:6123. doi: 10.1002/anie.200700835. [DOI] [PubMed] [Google Scholar]
- 51.Trost BM, Tang W, Schulte JL. Org Lett. 2000;2:4013–4015. doi: 10.1021/ol006599p. [DOI] [PubMed] [Google Scholar]
- 52.Trost BM, Toste FD. J Am Chem Soc. 2003;125:3090–3100. doi: 10.1021/ja020988s. [DOI] [PubMed] [Google Scholar]
- 53.Trost BM, Radinov R, Grenzer EM. J Am Chem Soc. 1997;119:7879–7880. [Google Scholar]
- 54.Trost BM, Malhotra S, Chan WH. J Am Chem Soc. 2011;133:7328–7331. doi: 10.1021/ja2020873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trost BM, Schroeder GM. J Am Chem Soc. 1999;121:6759–6760. [Google Scholar]; Trost BM, Schroeder GM, Kristensen J. Angew Chem Int Ed. 2002;41:3492–3495. doi: 10.1002/1521-3773(20020916)41:18<3492::AID-ANIE3492>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; Trost BM, Schroeder GM. Chem Eur J. 2005;11:174–184. doi: 10.1002/chem.200400666. [DOI] [PubMed] [Google Scholar]
- 56.Trost BM, Pissot-Soldermann C, Chen I, Schroeder GM. J Am Chem Soc. 2004;126:4480–4481. doi: 10.1021/ja0497025. [DOI] [PubMed] [Google Scholar]; Trost BM, Pissot-Soldermann C, Chen I. Chem Eur J. 2005;11:951–959. doi: 10.1002/chem.200400558. [DOI] [PubMed] [Google Scholar]
- 57.Trost BM, Dong L, Schroeder GM. J Am Chem Soc. 2005;127:10259–10268. doi: 10.1021/ja051547m. [DOI] [PMC free article] [PubMed] [Google Scholar]; Trost BM, Dong L, Schroeder GM. J Am Chem Soc. 2005;127:2844–2845. doi: 10.1021/ja0435586. [DOI] [PubMed] [Google Scholar]
- 58.Trost BM, Xu J. J Am Chem Soc. 2005;127:2846–2847. doi: 10.1021/ja043472c. [DOI] [PubMed] [Google Scholar]; Trost BM, Xu J, Schmidt T. J Am Chem Soc. 2009;131:18343–18357. doi: 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trost BM, Lehr K, Michaelis DJ, Xu J, Buckl AK. J Am Chem Soc. 2010;132:8915–8917. doi: 10.1021/ja103771w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trost BM, Zhang Y. J Am Chem Soc. 2006;128:4590–4591. doi: 10.1021/ja060560j. [DOI] [PubMed] [Google Scholar]; Trost BM, Zhang Y. Chem Eur J. 2011;17:2916–2922. doi: 10.1002/chem.201002569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trost BM, Zhang Y. J Am Chem Soc. 2007;129:14548–14549. doi: 10.1021/ja0755717. [DOI] [PMC free article] [PubMed] [Google Scholar]; Trost BM, Zhang Y. Chem Eur J. 2010;16:296–303. doi: 10.1002/chem.200902770. [DOI] [PMC free article] [PubMed] [Google Scholar]