

Abstract
Sodium transport via epithelial sodium channels (ENaC) expressed in alveolar epithelial cells (AEC) provides the driving force for removal of fluid from the alveolar space. The membrane-bound channel-activating protease 1 (CAP1/Prss8) activates ENaC in vitro in various expression systems. To study the role of CAP1/Prss8 in alveolar sodium transport and lung fluid balance in vivo, we generated mice lacking CAP1/Prss8 in the alveolar epithelium using conditional Cre-loxP-mediated recombination. Deficiency of CAP1/Prss8 in AEC induced in vitro a 40% decrease in ENaC-mediated sodium currents. Sodium-driven alveolar fluid clearance (AFC) was reduced in CAP1/Prss8-deficient mice, due to a 48% decrease in amiloride-sensitive clearance, and was less sensitive to β2-agonist treatment. Intra-alveolar treatment with neutrophil elastase, a soluble serine protease activating ENaC at the cell surface, fully restored basal AFC and the stimulation by β2-agonists. Finally, acute volume-overload increased alveolar lining fluid volume in CAP1/Prss8-deficient mice. This study reveals that CAP1 plays a crucial role in the regulation of ENaC-mediated alveolar sodium and water transport and in mouse lung fluid balance.
Keywords: β-agonist, elastase, prostasin, sodium channel, alveolar epithelium
INTRODUCTION
Active transepithelial sodium (Na+) transport by alveolar epithelial cells (AEC) is a driving force for reabsorption of fluid from the alveolar space, a function which accounts for the ability of the lung to remove alveolar fluid at the time of birth and represents the main mechanism for alveolar oedema resolution (Basset et al 1987; Hummler et al, 1996; Matthay et al, 1982, 2002). The amiloride-sensitive epithelial Na+ channel (ENaC) (Canessa et al, 1993, 1994) composed of three homologous subunits (α, β and γ) is expressed in type 1 and 2 AEC and plays a major role in Na+ absorption, representing in vivo a limiting factor for lung fluid clearance (Eaton et al, 2009; Hummler et al, 1996; Johnson et al, 2006; Matthay et al, 2002). The regulation of ENaC-mediated lung Na+ absorption is therefore critical for normal lung fluid balance and function. In the past decade, the concept has emerged that aside from well-known hormonal regulations by glucocorticosteroids or endogenous catecholamines, ENaC could also be activated at the surface of lung epithelial cells by a variety of serine proteases through changes in the channel gating (Hughey et al, 2007; Rossier, 2003; Rossier & Stutts, 2009). First identified in the Xenopus kidney A6 cell line, the channel-activating protease 1 (CAP1, also termed protease serine S1 family member 8, Prss8 or prostasin) is a glycosyl-phosphatidylinositol-anchored serine protease which is coexpressed with ENaC in mammalian epithelia transporting Na+, including the distal lung epithelium (Vallet et al, 1997, 2002; Vuagniaux et al, 2000). Subsequently, two additional serine proteases activating ENaC, mCAP2 (homologue of human transmembrane protease serine 4, TMPRSS4) and mCAP3 (MT-SP1/matriptase or epithin), have been cloned from the mpkCCDc14 mouse kidney cell line (Vuagniaux et al, 2002). These CAPs activate ENaC channels expressed at the cell surface in various expression systems by dramatically increasing their open probability (Po) (Vuagniaux et al, 2000, 2002). Interestingly, nonepithelial soluble serine proteases such as trypsin and human neutrophil elastase (hNE) can also increase the Po of near-silent ENaC channels at the cell surface when present in the apical milieu, suggesting that they act via the same mechanism as epithelial CAPs (Caldwell et al, 2004, 2005; Chraibi et al, 1998). There is increasing in vitro evidence that ENaC activation by CAP2, trypsin or hNE is related to proteolytic cleavage of the γ-ENaC subunit (Adebamiro et al, 2007; Diakov et al, 2008; Garcia-Caballero et al, 2008; Harris et al, 2007). Recent studies performed in transfected MDCK cells or in the Xenopus oocyte expression system suggest that ENaC activation by CAP1 could as well be due to the proteolytic cleavage of the extracellular domain of γ-ENaC subunit at a CAP1/prostasin polybasic cleavage site (Bruns et al, 2007; Carattino et al, 2008; Garcia-Caballero et al, 2008). Several investigators including our group previously reported that CAP1, CAP2 and CAP3 were coexpressed with ENaC in rodent alveolar epithelium, and that CAP1 was present in a secreted form in alveolar epithelial lining fluid (Planès et al, 2005; Verghese et al, 2004). Indeed, inhibition of endogenous CAPs by aprotinin decreased basal ENaC-mediated currents in primary cultures of rat and mouse AEC and suppressed the increase in amiloride-sensitive short-circuit current induced by the β2-agonist terbutaline (Planès et al, 2005). These data suggest that endogenous membrane-bound serine proteases could regulate alveolar Na+ and fluid transport in rodent lung. However, although CAPs seem to be good candidates, the precise identification of the serine protease(s) involved and their relative contribution in this process are still lacking. Also, the physiological importance of ENaC regulation by CAPs in distal lung fluid balance in vivo needs to be clearly established. To address these questions, we generated mice lacking mCAP1/Prss8 in the alveolar epithelium using tissue-specific Cre-loxP-mediated recombination, and studied the consequences of CAP1/Prss8 deficiency in distal lung Na+ and fluid transport. Here, we show that CAP1/Prss8 inactivation targeted to the alveolar epithelium decreased baseline ENaC-mediated alveolar Na+ transport in vitro and in vivo, and impaired the response to β2-agonists. Indeed, it increased alveolar lining fluid volume in an experimental model of acute volume-overload. These data provide for the first time direct evidence that CAP1/Prss8 is an important and physiologically relevant activator of ENaC in the distal lung, and that it plays a critical role in lung fluid balance.
RESULTS
Generation of mice lacking CAP1/Prss8 in the alveolar epithelium
To ablate CAP1/Prss8 specifically in the alveolar epithelium, we crossed SPC-rtTA/(tetO)7-CMV-Cre recombinase transgenic mice harbouring two floxed alleles at the Prss8 gene locus, and Cre-loxP-mediated recombination was induced in utero by doxycycline administration to the pregnant female (Perl et al, 2002) (Fig 1A). In the CAP1/Prss8-floxed gene locus, exons 3–5, which contain two residues of the catalytic triad (His85 and Asp134), are flanked by loxP sites (Rubera et al, 2002). By addition, deletion of exons 3–5 results in a frameshift and leads to a premature stop codon in exon 6, thereby generating a truncated protein at the carboxy terminus.
Figure 1. Generation of alveolar epithelium-specific CAP1/Prss8-deficient mice.
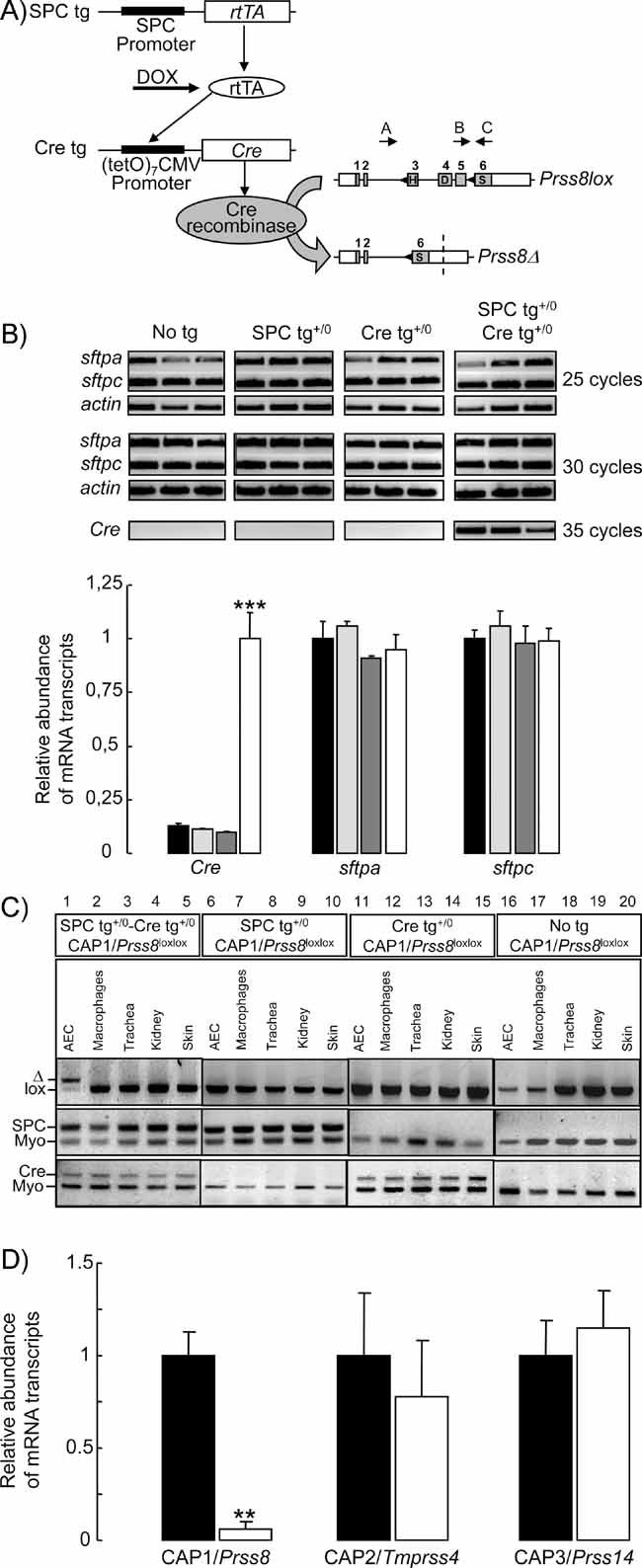
- Triple transgenic mice were generated that harbour the rtTA protein under the control of the SPC promoter in distal lung epithelial cells (SPC tg). Upon doxycycline (DOX) treatment, rtTA activates expression of the (tetO)7-CMV-Cre recombinase transgene (Cre tg). Cre recombinase recognizes the loxP sites (black triangles) in the floxed CAP1/Prss8 gene locus, causing deletion of the exons 3–5 to generate a null allele (Prss8Δ). Coding (grey boxes) and noncoding exon sequences (white boxes) are indicated.
- Expression of surfactant proteins A (sftpa) and C (sftpc), and of Cre recombinase (Cre) mRNA transcripts by semi-quantitative RT-PCR run at 25, 30 and 35 cycles in distal lung extracts from control (no tg: black bars; SPC tg+/0: light grey bars; Cre tg+/0: dark grey bars) and knockout (SPC tg+/0; Cre tg+/0, white bars) groups. Quantification of signals was performed after 30 cycles for sftpa and sftpc, and after 35 cycles for Cre. Results are expressed as the ratio of sftpa, sftpc or Cre mRNA/β-actin mRNA (n = 3 mice per group). ***: Significantly different from control groups (p < 0.001).
- PCR analysis with primers A–C distinguish between lox (413 bp) and Δ (473 bp) alleles of CAP1/Prss8 gene locus (top). Note the shift of the Prss8lox allele into Prss8Δ allele in AEC harbouring the SPC and Cre transgenes (lane 1). Detection of SPC tg (middle) and Cre tg (bottom), and of myogenin (middle and bottom).
- Quantification of CAP1/Prss8, CAP2 and CAP3 mRNA transcripts by qRT-PCR in AEC isolated from pooled control mice (no transgene or only SPC or Cre transgene: black bars) and knockout mice (SPCtg+/0; Cretg+/0: white bars). Results are expressed as the ratio of CAP1/Prss8, CAP2/Tmprss4 or CAP3/Prss14 mRNA/β-actin mRNA (n = 4 mice per group).**: Significantly different from control group (p < 0.01).
Homologous CAP1/Prss8lox/lox males harbouring the SPC and Cre transgenes were intercrossed with CAP1/Prss8lox/lox females, and females were treated with doxycycline throughout gestation. Analysis of the offspring at weaning (n = 313 mice) showed no significant deviation from the expected Mendelian distribution of genotypes with 25% of mice harbouring the two transgenes, indicating that CAP1/Prss8 inactivation in the alveolar epithelium had no effect on foetal survival. Semi-quantitative RT-PCR experiments performed on distal lung extracts showed that Cre recombinase was specifically expressed in lungs from mice harbouring the two transgenes (SPC tg+/0 /Cre tg+/0, knockout group), but not in mice harbouring none or only one of the transgenes (No tg, SPC tg+/0 and Cre tg+/0: control groups) (Fig 1B). Expression levels of mRNA transcripts encoding surfactant proteins SPA and SPC were similar in the four groups of mice, indicating that expression of the transgenes did not modify gene expression in distal lung cells (Fig 1B). PCR analysis on cells and organs confirmed that Cre-loxP-mediated recombination was only detected under doxycycline treatment in AEC from mice harbouring the two transgenes by the presence of the Prss8Δ-specific PCR-amplified product (Fig 1C, lane 1), but not in alveolar macrophages, trachea, kidney or skin (lanes 2–5). No recombination was observed in AEC or other tissues from mice harbouring none or only one of the transgenes (lanes 6–20). Quantitative qRT-PCR experiments evidenced a 95% decrease in CAP1/Prss8 mRNA transcript expression in AEC isolated from knockout mice, whereas expression of mRNA transcripts encoding CAP2/Tmprss4 or CAP3/Prss14 did not differ between groups (Fig 1D).
Mice from the knockout group showed no abnormal mortality, particularly during postnatal days, and were indistinguishable from control mice in appearance and growth rate (body weight at 6 months: 35.1 ± 1.24 vs. 33.7 ± 2 g in control and knockout males, respectively, NS; 26.8 ± 0.46 vs. 27.1 ± 0.51 g in control and knockout females, respectively, NS: n = 313). Postnatal (8–12 h after birth) and adult (3 months) lung water contents as assessed by lung wet/dry weight ratios were similar in knockout and control groups (Table 1).
Table 1.
Lung wet/dry weight ratios
| Genotype | ||||
|---|---|---|---|---|
| No tg | SPC tg+/0 | Cre tg+/0 | SPC tg+/0; Cre tg+/0 | |
| Postnatal | 6.24 ± 0.12 | 6.37 ± 0.21 | 6.12 ± 0.38 | 6.09 ± 0.32 |
| Adult | 4.05 ± 0.16 | 3.89 ± 0.3 | 4.29 ± 0.13 | 3.96 ± 0.15 |
Postnatal, 8–12 h after birth. Results are means ± SE (n = 4–10 mice per group).
Distal lung histology
Lung macroscopic appearance was similar in control and knockout mice. Histological examination of the lungs by light microscopy revealed no morphological difference between groups (Fig 2A) (n = 10). In particular, bronchioles, alveolar ducts, alveolar epithelium and blood vessels appeared normal in knockout mice. The size of pulmonary alveoli did not differ between groups (Fig 2A). Studies by transmission electron microscopy revealed that ultrastructure of alveolar epithelium was normal and comparable in control and knockout mice, with thin and flattened alveolar type 1 cells and cuboidal alveolar type 2 cells showing microvilli and lamellar bodies (Fig 2B) (n = 6). No aspect of interstitial or alveolar oedema was detected in knockout mouse lung.
Figure 2. Distal lung histology.
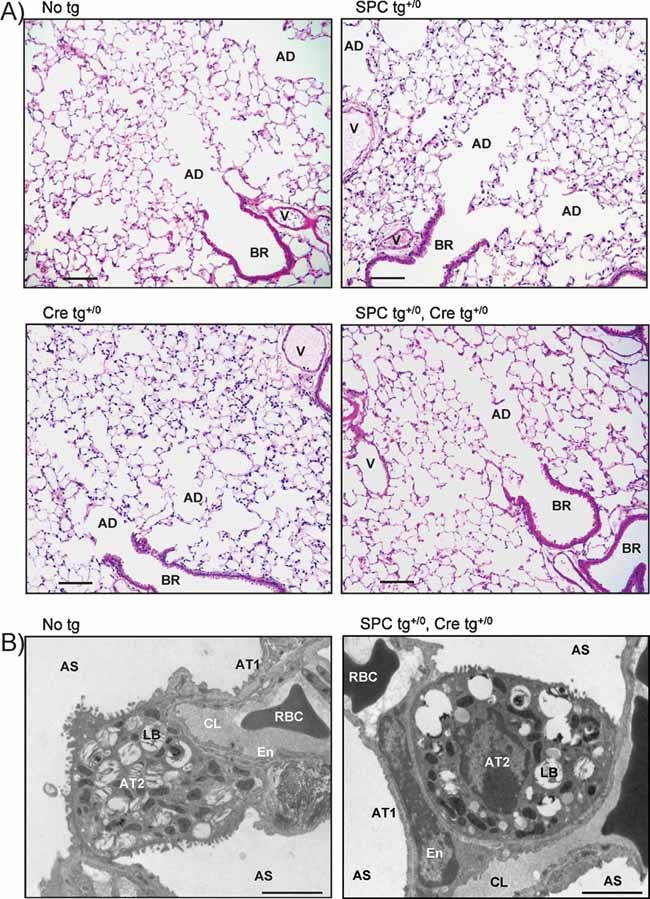
- Light photomicrographs of representative lung sections from control and knockout littermates stained with haematoxylin and eosin. The morphological aspect of blood vessels (V), bronchioles (BR), alveolar ducts (AD) and alveoli was normal in knockout mice. Scale bar, 100 µm.
- Electron micrographs showing that the ultrastructure of alveolar septa and alveolar epithelium was normal in both groups. AS, alveolar space; AT1, alveolar type 1 cells; AT2, alveolar type 2 cells; LB, lamellar body; En, endothelial cell; CL, pulmonary capillary lumen; RBC, red blood cell. Scale bar, 2 µm.
Expression of ENaC subunits in distal lung epithelial cells
Expression levels of ENaC subunit mRNA transcripts as assessed by semi-quantitative RT-PCR in distal lung extracts did not differ between groups (Fig 3A). At the protein level, immunoblotting with anti-α-ENaC in control or knockout distal lung homogenates revealed two distinct bands migrating at 85–90 and 65 kDa, respectively, as previously described (Randrianarison et al, 2008) (Fig 3B). The anti-β-ENaC antibody detected in both groups a single band of 95 kDa, and immunoblotting with anti-γ-ENaC revealed a minor band migrating around 80 kDa and a major band around 70 kDa, as previously reported (Randrianarison et al, 2008). Quantitative analysis showed no significant difference in α-, β- and γ-ENaC protein expression levels between the two groups (Fig 3C).
Figure 3. Expression of ENaC subunits in distal lung epithelial cells.
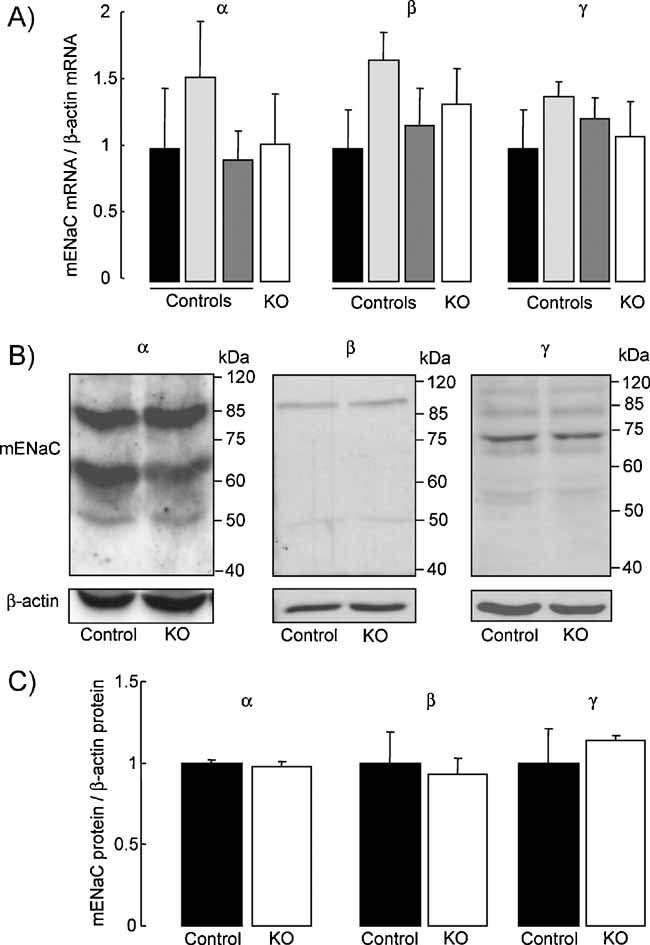
- Quantification of mouse ENaC subunit mRNA transcript expression by semi-quantitative RT-PCR run at 30 cycles in distal lung homogenate extracts from control (no tg: black bars; SPC tg+/0: light grey bars; Cre tg+/0: dark grey bars) and knockout mice (SPC tg+/0; Cre tg+/0, white bars). Results are expressed as the ratio of α-, β- or γ-mENaC mRNA/β-actin mRNA (n = 3 mice per group).
- Representative immunoblots showing the expression of ENaC subunit proteins and β-actin protein in distal lung homogenate extracts from control and knockout mice.
- Quantification of α-, β- and γ-mENaC signals in pooled control (black bars) and knockout (KO, white bars) mouse lung homogenates was obtained using NIH image software. Results are expressed as the ratio of mENaC protein/β-actin protein (n = 4 mice per group). There was no significant difference between the two groups with respect to the mRNA transcript or protein expression levels.
Impairment of transepithelial Na+ transport in primary mouse AEC lacking CAP1/Prss8
We next studied in vitro the effect of CAP1/Prss8 inactivation on transepithelial resistance (Rte) and Na+ transport in well-differentiated primary mouse AEC monolayers (Fig 4). Open-circuit electrophysiological studies showed that both control and knockout AEC monolayers developed high Rte around days 4–5 following isolation, with no difference in Rte values between groups (Fig 4A). By contrast, transepithelial potential difference (PD) was significantly decreased in AEC monolayers from mice lacking CAP1/Prss8 (Fig 4B). Equivalent short-circuit current Ieq calculated from PD and Rte was significantly reduced by 26% in knockout mouse AEC (Fig 4C). The amiloride-insensitive component of Ieq was comparable in both groups, whereas the amiloride-sensitive Ieq, reflecting transepithelial Na+ transport mediated by ENaC, was reduced by 40% in knockout mouse AEC as compared with control (p = 0.02). We also studied whether CAP1/Prss8 inactivation would modify expression of tight junction proteins. Immunocytochemical studies of AEC monolayers at day 5 after isolation revealed that expression and distribution of occludin and ZO-1 were not different in control and knockout mice (Fig 4D). These results indicate that CAP1/Prss8 inactivation in mouse AEC did not affect occludin and ZO-1 expression and the formation of tight junctions, but significantly impaired ENaC-mediated transepithelial Na+ transport.
Figure 4. Electrophysiological properties and expression of the tight junction protein occludin and ZO-1 in control and CAP1/Prss8-deficient mouse AEC.
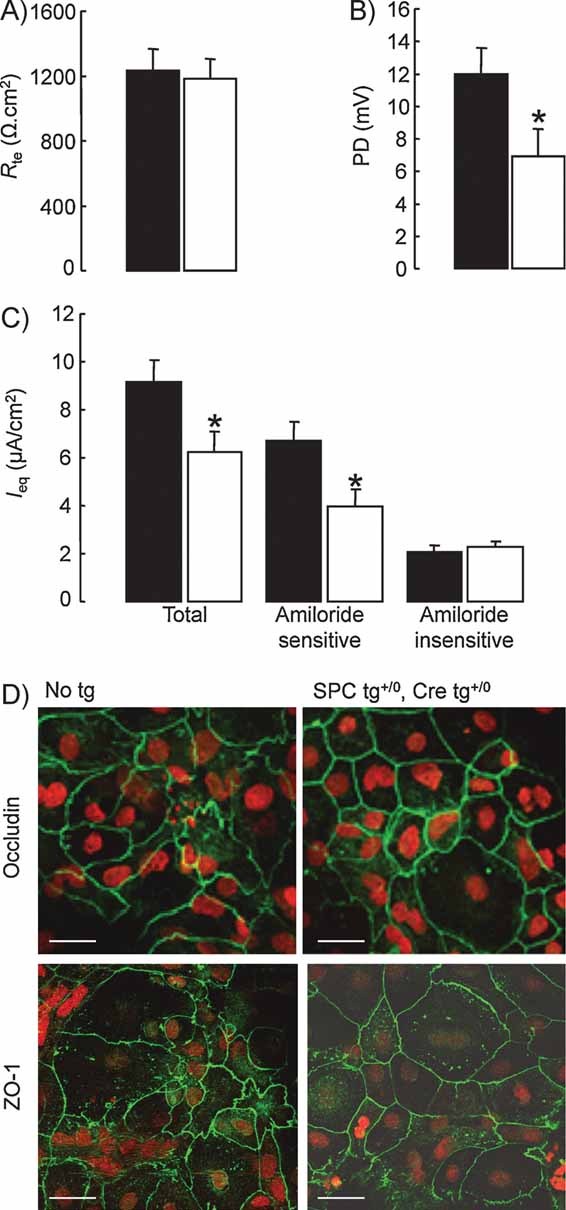
- Transepithelial resistance (Rte) and (B) PD were measured under open-circuit conditions in control (black bars) and knockout (white bars) AEC monolayers grown on transwell filters for 5 days.
- Equivalent short-circuit current (Ieq) was calculated from Rte and PD at baseline (total Ieq), and after apical treatment with amiloride (10 µM) (amiloride-insensitive Ieq) (9–12 filters per group from 3 independent cultures). Amiloride-sensitive Ieq is the difference between Ieq values in the absence and in the presence of amiloride. Transepithelial PD as well as total and amiloride-sensitive Ieq were significantly reduced in knockout AEC monolayers. *: Significantly different from control group (p < 0.05).
- AEC from control (No tg) and knockout mice (SPC tg+/0; Cre tg+/0) were cultured for 5 days on transwell filters before immunofluorescent detection of occludin and ZO-1 was performed. Nuclei are counterstained with Sytox Orange. Occludin staining was localized at the periphery of the cells in both groups. Scale bar, 25 µm.
Impairment of Na+-driven lung fluid clearance in mice lacking CAP1/Prss8
To evaluate whether the decrease in ENaC-mediated transepithelial Na+ transport observed in vitro in AEC lacking CAP1/Prss8 could have a physiological relevance in vivo, we measured sodium-driven alveolar fluid clearance (AFC) in an in situ nonventilated mouse lung model using 125I-albumin as a volume marker (Randrianarison et al, 2007, 2008). As shown in Fig 5A, AFC at baseline was comparable in control mice (harbouring no or only one transgene), but significantly decreased in knockout mice. Therefore, control mice were pooled together to form the control group in following experiments. As shown in Fig 5B, amiloride significantly decreased AFC in control and knockout mice by 61 and 46%, respectively. The amiloride-insensitive component of AFC was similar in both groups. Therefore, the decrease in basal AFC observed in knockout mice was solely due to the impairment of the amiloride-sensitive component of AFC, reflecting Na+-driven lung fluid clearance dependent on ENaC activity. Amiloride-sensitive AFC was reduced by 48% in knockout mice (p < 0.01) (Fig 5C).
Figure 5. Alveolar fluid clearance under basal condition in control and CAP1/Prss8-deficient mice.
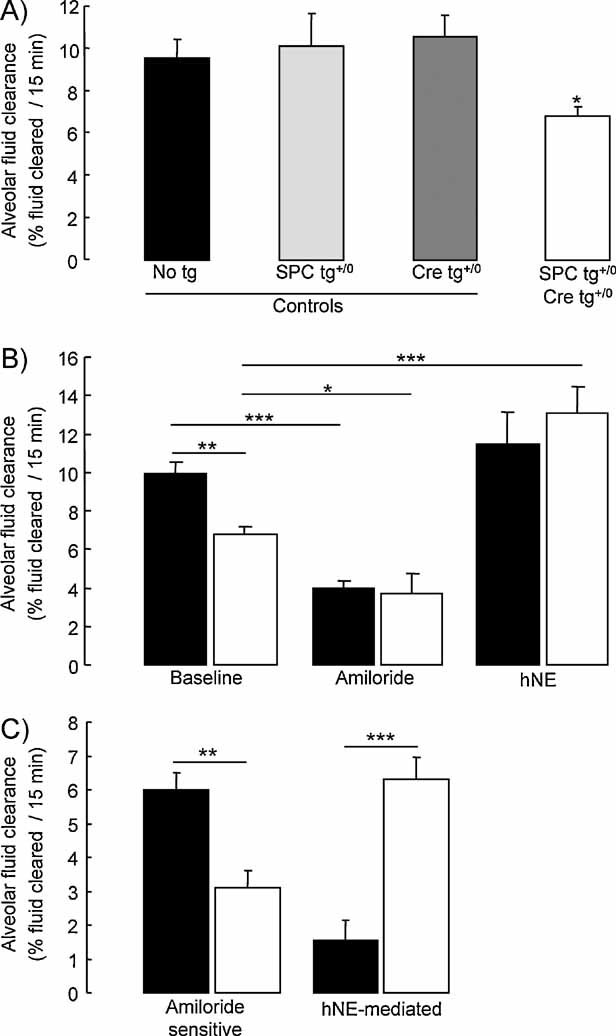
- Sodium-driven AFC was measured at baseline over a 15-min period in control (black, light grey and dark grey bars) and knockout (white bars) CAP1/Prss8lox/lox littermates aged 2–5 months at 37°C using an in situ nonventilated model in which the airspace was instilled with an isoosmolar Ringer's lactate solution containing 125I-albumin as a volume marker. Note that AFC was significantly lower in the knockout group than in the control groups (*: significantly different from control groups, p < 0.05).
- AFC was measured in the absence (baseline) or presence of amiloride (final concentration: 1 mM) or hNE (final concentration: 33 µg/ml) in the alveolar instillate in pooled control littermates (black bars) and knockout (white bars) littermates.
- Calculated values of amiloride-sensitive AFC (difference between AFC values in the absence and in the presence of amiloride) and hNE-mediated AFC (difference between AFC values in the absence and in the presence of hNE). Results are expressed as percentage fluid absorption at 15 min (5–13 mice per group for basal and hNE experiments, 4 mice per group for amiloride experiments. *, **, ***: Significant difference between groups as indicated (*, p < 0.05; **, p < 0.01; and ***, p < 0.001, respectively).
We next tested whether the decrease in AFC in knockout mice could be reversed by intra-alveolar treatment with hNE, a soluble serine protease previously shown to activate ENaC in vitro. Addition of hNE in the alveolar instillate did normalize AFC in the knockout group whereas it was without significant effect in the control group (Fig 5B). hNE-mediated AFC (difference between AFC values in the presence and absence of hNE) was significantly greater in knockout mice than in control mice (p < 0.001) (Fig 5C).
Inhibition of β-adrenergic agonist-induced stimulation of lung fluid clearance in mice lacking CAP1/Prss8
The response of AFC to β-adrenergic stimulation in control and knockout mice was also evaluated (Fig 6). As shown in Fig 6A, AFC measured in the presence of terbutaline in alveolar instillate was comparable in control mice (harbouring no or only one transgene), and significantly higher than in knockout mice. Therefore, control mice were pooled together to form the control group in the following experiments. As shown in Fig 6B, terbutaline treatment increased total AFC by 42% in control mice (p < 0.001) (columns 1 and 3), with no change in the amiloride-insensitive part of the clearance (not shown). Terbutaline induced a small increase in AFC in knockout mice, at the limit of statistical significance (p = 0.06) (columns 2 and 4). Amiloride- sensitive AFC in the presence of terbutaline was reduced by 42% in knockout mice as compared with controls (5.8 ± 0.7 vs. 10 ± 0.5% fluid cleared/15 min in knockout and control mice, respectively. p < 0.001), indicating that the response to β-agonist treatment was impaired in CAP1-deficient mice.
Figure 6. Alveolar fluid clearance under β2-agonist-stimulated condition in control and CAP1/Prss8-deficient mice.
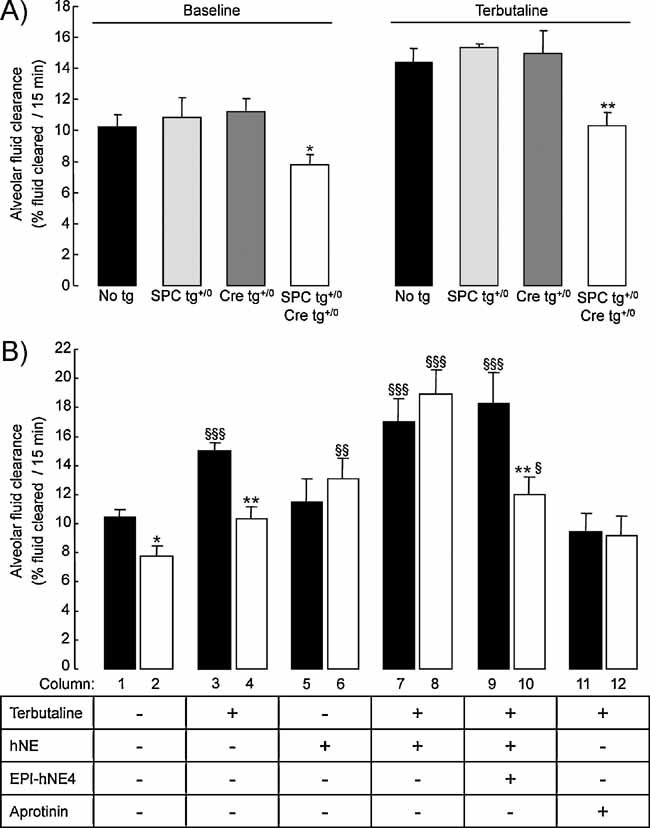
- Sodium-driven AFC was measured at baseline and in the presence of terbutaline (final concentration in alveolar instillate: 10−4 M) over a 15-min period in control (black, light grey and dark grey bars) and knockout (white bars) CAP1/Prss8lox/lox littermates aged 2–5 months at 37°C as described in Methods. AFC values at baseline and in the presence of terbutaline were significantly lower in the knockout group than in control groups (*, **: significantly different from control groups, p < 0.05 and p < 0.01, respectively).
- AFC was measured in the absence or presence of terbutaline, hNE (33 µg/ml), the elastase inhibitor EPI-hNE4 (50 µg/ml), or the serine protease inhibitor aprotinin (100 µg/mL) in the alveolar instillate in pooled control littermates (black bars) and knockout (white bars) littermates. Results are expressed as percentage fluid absorption at 15 min (n = 5–12 mice per group and per condition). *, **: Significantly different from respective control group (p < 0.05, p < 0.01, respectively); §, §§, §§§: significantly different from baseline value (no treatment) in the corresponding group (p < 0.05, p < 0.01 and p < 0.001, respectively).
Next, we examined whether the response of AFC to β-agonists could be modulated by either hNE or serine protease inhibitors. Addition of hNE to the alveolar instillate completely restored the stimulation of AFC induced by terbutaline in knockout mice, as compared with control mice (Fig 6B, columns 7 and 8). The corrective effect of hNE in knockout mice was fully abolished in the presence of EPI-hNE4, a Kunitz-type specific inhibitor of hNE (21) (column 10). Finally, the nonspecific serine protease inhibitor aprotinin completely suppressed the terbutaline-induced stimulation of AFC in control mice, but was without effect in knockout mice (columns 11 and 12). Taken together, these results suggest that CAP1 participates in the stimulation of ENaC and AFC induced by β-agonists, and that in the absence of CAP1, nonepithelial soluble serine proteases such as hNE can substitute for endogenous CAP1.
Hydrostatic volume-overload model of pulmonary oedema
In order to determine whether the decrease in AFC and the absence of effect of β2-agonist treatment could modulate the severity of hydrostatic pulmonary oedema, ventilated control and knockout mice were submitted to acute intravascular volume expansion by saline infusion before pulmonary oedema was quantified by estimation of the volume of alveolar epithelial lining fluid (VELF) (Fig 7). Comparison of body weight before and at the end of the 2 h-saline infusion revealed that the net increase in body weight was 6.94 ± 1.10 and 7.3 ± 0.84 g in control and knockout mice, respectively (NS) (n = 5–7 mice per group). As seen in Fig 7, VELF was near zero in both groups of mice under basal condition (no infusion). Acute volume overload by saline infusion induced an increase in VELF in both groups as compared to baseline, reflecting the accumulation of alveolar lining fluid. The increase in VELF was significantly more important in the knockout group than in the control group (p = 0.01).
Figure 7. Effect of acute volume-overload on lung fluid balance in control and CAP1/Prss8-deficient mice.
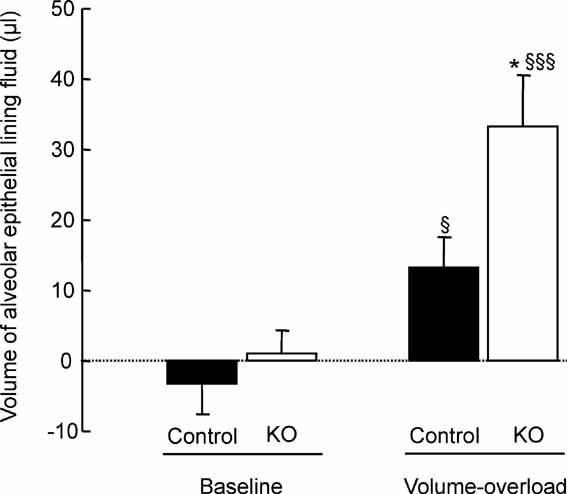
The volume of the alveolar epithelial lining fluid was estimated from the change in 125I-albumin concentration in the alveolar instillate at baseline and at the end of acute saline infusion (40% body weight over 2 h, volume-overload) in control (black bars) and knockout (white bars) littermates aged 2–6 months. Results are expressed in microliter (n = 5–7 mice per group and per condition). *: Significantly different from control group (p < 0.05). §, §§§: Significantly different from baseline value in the corresponding group (p < 0.05 and p < 0.001, respectively).
DISCUSSION
Alveolar epithelium-specific gene inactivation of CAP1/Prss8
The membrane-bound CAP1 (CAP1/Prss8), a member of the S1 serine protease family, is coexpressed with ENaC in rodent AEC, and previous in vitro data suggested that it could be an important activator of Na+ channels in these cells (Planès et al, 2005; Vallet et al, 1997; Vuagniaux et al, 2000). To study in vivo the role of CAP1/Prss8 in ENaC-mediated alveolar Na+ and water transport and lung fluid balance, we generated triple transgenic mice specifically lacking CAP1/Prss8 in lung alveolar epithelium. We used a previously described Cre-loxP-mediated recombination system under the control of the surfactant protein C (SPC) promoter inducible by doxycycline (Perl et al, 2002). In this system, Cre-loxP-mediated recombination ablates exons 3–5 of CAP1/Prss8 floxed gene locus which contain two essential residues of the catalytic triad, but also results in a frameshift leading to a premature stop codon in exon 6, thereby generating a truncated CAP1 protein (Rubera et al, 2002). When such a truncated construct is coinjected with ENaC subunits in Xenopus laevis oocytes, activation of ENaC-mediated sodium transport by CAP1 is completely abolished (Leyvraz et al, 2005). Administration of doxycycline during gestation has been previously shown to induce Cre-loxP-mediated recombination in utero in progenitors of both alveolar epithelial type 1 and 2 cells, since these progenitor cells specifically express SPC during embryonic lung development (Perl et al, 2002). This is an important point as it is now well established that alveolar type 1 cells, which do not express SPC after birth, express ENaC subunits and are responsible for as much as 60% of overall alveolar Na+ transport, the remaining 40% being due to transport by alveolar type 2 cells (Johnson et al, 2006; Ridge et al, 2003). In our model, disruption of CAP1/Prss8 gene locus was confined to AEC, and led to a 95% decrease in CAP1/Prss8 mRNA transcript expression with no change in surfactant proteins A and C, CAP2 and CAP3 mRNA, and ENaC subunit expression. Inactivation of CAP1/Prss8 in alveolar epithelium had no adverse effect on foetal survival. Knockout mice showed no abnormal mortality at birth, and grew and developed normally. The present study represents the first genetic approach to inactivate CAP1/Prss8 gene inactivation in a Na+-transporting epithelium. Conditional inactivation of CAP1 and CAP3 in mouse skin has been previously performed, and resulted in early post-natal death due to severe dehydration, suggesting an important role of CAPs in epidermal permeability barrier (Leyvraz et al, 2005; List et al, 2002). Analysis of the constitutive knockout of ENaC revealed an impaired barrier function in epidermis as well, although the direct proof of ENaC regulation by serine proteases in this tissue is still missing (Charles et al, 2008).
CAP1/Prss8 activates ENaC in lung AEC
Here, we provide direct functional evidence that CAP1/Prss8 is a physiologically relevant activator of ENaC in distal lung epithelium. Both in vitro electrophysiological studies in primary mouse AEC and in vivo measurements of Na+-driven AFC revealed that CAP1/Prss8 deficiency impaired basal transepithelial alveolar Na+ and water transport. The decrease in basal alveolar Na+ transport was due to a 40–50% decrease in amiloride-sensitive ENaC-mediated Na+ transport, with no change in whole-cell protein expression levels of ENaC subunits. These findings are in line with our previous report that serine protease inhibitor aprotinin decreased ENaC-mediated currents in primary rat and mouse AEC (Planès et al, 2005). Interestingly, the decrease in AFC in mice lacking CAP1 was completely reversed by intra-alveolar treatment with hNE. This indicates that neutrophil elastase, a soluble trypsin-like serine protease, can substitute for CAP1 to activate ENaC in AEC, most likely by cleaving the extracellular domain of γ-ENaC subunit at the cell surface as previously reported in vitro (Adebamiro et al, 2007; Harris et al, 2007). Of note, elastase treatment did not significantly modify AFC in control mice, suggesting that ENaC is normally maximally activated by endogenous serine proteases.
CAP1/Prss8 expression in AEC is required for optimal stimulation of AFC by β-agonists
One major finding of the present study is that CAP1 inactivation blunted the stimulation of AFC induced by the β2-agonist terbutaline. Activation of β2-adrenergic receptors on AEC, either by endogenous catecholamines or by β2-agonist drugs, is one of the main mechanisms for upregulation of alveolar Na+ transport and fluid clearance in vivo in most mammalian species (Matthay et al, 2002). cAMP-agonists have been shown to increase ENaC activity and transepithelial Na+ transport in various cell types including AEC, mostly by promoting the insertion of ENaC subunits at the cell surface (Chen & Jain, 2002; Planès et al, 2002; Snyder, 2000). In our experiments, the stimulation of AFC by terbutaline was fully restored in knockout mice by intra-alveolar treatment with hNE. On the other hand, the response to β2-agonists was fully abolished in control mice by aprotinin, a serine protease inhibitor. This latter finding is consistent with our previous observation that preincubation of cultured rat AEC with aprotinin completely inhibited the increase in ENaC-mediated Na+ current induced by terbutaline (Planès et al, 2005). From these data, it can be hypothesized that in response to β2-agonists, ENaC channels from intracellular pools are addressed to the apical membrane of AEC in an inactive or partially active state, and that they need then to be fully activated by serine proteases. Therefore, activation of ENaC by CAP1 at the surface of AEC appears to be essential for optimal efficacy of β2-agonists in vivo.
CAP1/Prss8 deficiency increases alveolar epithelial lining fluid accumulation in an experimental model of hydrostatic pulmonary oedema
CAP1/Prss8 deficiency in alveolar epithelium may have important pathophysiological implications, by impairing the ability of the lung to remove fluid from the alveolar space (Matthay et al, 2002). In mammals, excessive accumulation of fluid in alveolar spaces frequently accompanies lung injury or left ventricle dysfunction, leading to alveolar oedema and compromised pulmonary gas exchange. It is now well established that sodium-driven AFC is the primary mechanism for resolution of alveolar oedema, on condition that the structural and functional integrity of alveolar epithelium is preserved (Basset et al, 1987; Matthay et al, 1982, 2002; Verghese et al, 1999; Ware & Matthay, 2001). At baseline, CAP1/Prss8 deficient mice had no sign of alveolar epithelial fluid lining accumulation, consistent with previous observation that moderately-decreased ENaC function in AEC is sufficient to maintain normal lung fluid balance under basal condition (Egli et al, 2004; Randrianarison et al, 2008). Ultrastructure of AEC, including the aspect of tight junctions, appeared normal in knockout mouse lung. Indeed, expression of the tight junction proteins occludin and ZO-1 by immunohistochemistry as well as bioelectrical transepithelial resistance values were not affected in CAP1/Prss8-deficient AEC monolayers. However, when challenged with hydrostatic stress due to acute volume- overload, knockout mice showed increased alveolar epithelial lining fluid accumulation compared to control mice. It is noteworthy that in life-threatening situations such as pulmonary oedema, activation of β2-adrenergic receptors on AEC by endogenous catecholamines is particularly important, as it normally results in increased AFC favouring the removal of oedema fluid (Pittet et al, 1994). Interestingly, clinical studies performed in intensive care units previously reported that among patients mechanically ventilated for pulmonary oedema, those with maximal lung fluid clearance had better clinical outcomes than those with impaired lung fluid clearance (Verghese et al, 1999; Ware & Matthay, 2001). Indeed, it was shown that intravenous treatment with β2-agonists decreases the severity of pulmonary oedema in patients with acute lung injury (Perkins et al, 2006). In our experiments, increased accumulation of alveolar fluid after volume-overload in CAP1/Prss8-deficient mice may certainly be due to the fact that basal AFC is impaired in these mice, but perhaps more importantly to the fact that it cannot be upregulated by endogenous catecholamines.
In conclusion, the present study provides direct evidence that the membrane-bound serine protease CAP1/Prss8 is a constitutive activator of ENaC in vivo in mouse lung alveolar epithelium. Also, proper expression of CAP1/Prss8 in alveolar epithelium is required for optimal stimulation by β2-agonists of ENaC-mediated alveolar Na+ and fluid reabsorption. Finally, we show that CAP1/Prss8 deficiency in alveolar epithelium favours the development of experimental hydrostatic pulmonary oedema, most likely by impairing the ability of the lung to remove fluid from the alveolar space. Taken together, our results highlight the crucial role of CAP1/Prss8 in the regulation of ENaC-mediated alveolar Na+ transport and in mouse lung fluid balance.
MATERIALS AND METHODS
Animals
Generation of alveolar epithelium-specific CAP1/Prss8-deficient mice
Mice in which CAP1/Prss8 was selectively disrupted in the alveolar epithelium were generated by doxycycline-inducible Cre- loxP-mediated recombination under the control of SPC. To do so, mice harbouring two floxed CAP1/Prss8 alleles (Prss8lox/lox mice) (Rubera et al, 2002) were interbred with double transgenic SPC-rtTA/(tetO)7-CMV-Cre mice (kindly provided by Dr J. A. Whitsett, University of Cincinatti, OH, USA) (Perl et al, 2002). Homologous CAP1/Prss8 floxed males harbouring the two transgenes (SPC-rtTA: SPC tg, and (tetO)7-CMV-Cre: Cre tg) were intercrossed with CAP1/Prss8 floxed females (Prss8lox/lox), and the females were treated with doxycycline (1 mg/ml in drinking water) throughout gestation. In developing embryos harbouring the two transgenes (SPC tg+/0 /Cre tg+/0), doxycycline activates the rt-TA that is expressed in the developing pulmonary epithelium under the control of SPC promoter, and consequently Cre recombinase (Fig 1A). This results in the excision of exons 3–5 of CAP1/Prss8 gene encoding 2/3 of the catalytic site of mCAP1 (Prss8Δ/Δ/SPC tg+/0/Cre tg+/0, knockout group). Littermates harbouring no transgene (Prss8lox/lox no tg) or only one transgene (Prss8lox/lox/SPC tg+/0 and Prss8lox/lox/Cre tg+/0) were used as controls. Animals were housed in standard cages and light conditions, and fed standard diet with free access to water. Experiments were performed on litter-matched mice aged 2–6 months, with investigators blinded to genotype information for all comparative measurements. The experiments were approved by the institutional reviewing board on animal experimentation (Université de Lausanne, Switzerland and Université Paris 7, France), and in accord with animal welfare guidelines (Ministère Français de la Pêche et de l'Agriculture, agreement # 75-1045 and Swiss Veterinarians).
Genotyping
Genotyping was performed on genomic DNA from biopsies (tail and organs), as described previously (Rubera et al, 2002). PCR analysis was performed using the following primers: Prss8 sense, A (5′-CTG TCA TGT GGA GAG GTT GC-3′); Prss8 sense, B (5′-CAG CAG CTC GAG GTA CCA CT-3′); Prss8 antisense, C (5′-CCA GGA AGC ATA GGT AGA AG-3′) to detect Prss8 wild type- (379 bp), lox- (413 bp) and Δ-specific (473 bp) PCR-amplified products. Forty cycles were run, each consisting of 1 min at 94, 58 and 72°C. The presence of the (tetO)7-CMV-Cre transgene was detected by PCR using Cre-specific primers (sense, 5′-CCT GGA AAA TGC TTC TGT CCG-3′; and antisense, 5′-CAG GGT GTT ATA AGC AAT CCC-3′) to amplify a 350-bp fragment (40 cycles as described above). The presence of the SPC-rtTA transgene was detected by PCR using specific primers (sense, 5′-GAC ACA TAT AAG ACC CTG GTC A-3′; and antisense, 5′-AAA ATC TTG CCA GCT TTC CCC-3′) to amplify a 370-bp fragment (40 cycles as described above) (Perl et al, 2002). Myogenin-specific primers (sense, 5′-TTA CGT CCA TCG TGG ACA GC-3′; and antisense, 5′-TGG GCT GGG TGT TAG TCT TA-3′) were used to control the DNA integrity of each sample.
Histological analysis
Lung histology was performed by light microscopy and transmission electron microscopy as previously described (Randrianarison et al, 2008). Animals were euthanized with intraperitoneal pentobarbital (250 mg/kg). The trachea was cannulated and connected to a syringe before a thoracotomy was performed. For light microscopy, lungs were inflated with paraformaldehyde 4% at a pressure of 20 cm H2O before the trachea was tied and the heart and lungs were removed en bloc and placed in paraformaldehyde 4% overnight. The lungs were embedded with paraffin and sections were cut at 4-µm thickness stained with haematoxylin and eosin. For electron microscopy, the same fixation procedure was used except that paraformaldehyde was replaced by glutaraldehyde 2.5%. Small blocks of lung tissue were postfixed in osmium tetroxyde 1% in PBS, dehydrated in graded series of ethanol, and embedded in Epon 812 (Fluka). Ultrathin sections stained with uranyl acetate and lead citrate were examined in a transmission electron microscope (JEM 1010, JEOL).
Determination of lung wet/dry weight ratio
Lung water content was determined by the lung wet/dry weight ratio in neonates (8–12 h after birth) and in adult mice (aged 3-month). Briefly, littermate mice were anaesthetized as described above and killed by exsanguination. Lungs were removed, weighed, then placed in an incubator at 80°C for 24 h for dessication and weighed again to calculate the wet-to-dry lung weight ratio as described (Hummler et al, 1996).
Isolation and culture of mouse AEC
Mouse AEC were isolated from mice aged 2–3 months by dispase digestion of lung tissue, followed by sequential filtration and differential adherence on culture dishes coated with rat anti-mouse CD 45 and rat anti-mouse CD 16/32 (BD Pharmingen), as previously described (Planès et al, 2005). The yield was 4–5 × 106 cells/mouse, with a percentage of alveolar type II cells ≥78% and a cell viability >95%. Cells were either used for RNA extraction, or resuspended in defined DMEM/Ham's F12 (1:1 v/v) culture medium containing 2% decomplemented mouse serum (Sigma, St Louis, MO) and seeded on type IV collagen (Sigma)-coated Transwell (polycarbonate membrane with a pore size of 0.4 µm, Costar, Cambridge, MA) filters (Ø 0.33 cm2, 1 × 106 cells/cm2, 600/100 µl medium in basolateral/apical compartments). Mouse AEC on filters were cocultured with mouse embryonic fibroblasts (previously treated with mitomycine) grown in the same medium on 24-well plastic dishes in a 5% CO2–95% air atmosphere. Experiments were performed at day 5 following isolation.
RT-PCR analysis
Semi-quantitative RT-PCR analysis
Frozen lungs were homogenized using tissue lyzer (Qiagen, Basel, Switzerland) and RNA was extracted with the Qiagen RNeasy Mini kit following the manufacturer instructions. After dosage using NanoDrop (witec ag ND-1000 Spectrophotometer) and quality control on 1% agarose gel, 1.5 µg of RNA were treated by RQ1 RNase-free DNase and retro-transcribed using M-MLV Reverse Transcriptase RNase H Minus Point Mutant (Promega) following the datasheet protocol. cDNA was diluted 15-fold and PCR amplification was performed with primer pairs specific for Sftpa, Sftpc, Scnn1a, Scnn1b, Scnn1g and Cre summarized in Table 2. A total of 25, 30 or 35 cycles were run, each consisting of 30 s at 95, 56 and 72°C. Amplified PCR products were separated on a 2% agarose gel and visualized by ethidium bromide staining. Densitometry has been carried out using Photoshop.
Table 2.
Primers used for semi-quantitative RT-PCR (Scnn1a, Scnn1b, Scnn1g, Sftpa, Sftpc and Cre) and quantitative real-time PCR (Tmprss4 and Prss14)
| Gene | Type | Sequence |
|---|---|---|
| Scnn1a | FOR | 5′-CTT CAG CTA CCC CGT GAG TC-3′ |
| REV | 5′-CCT GGC GAG TGT AGG AAG AG-3′ | |
| Scnn1b | FOR | 5′-CAT CCA GGC CTG TCT TCA TT-3′ |
| REV | 5′-AGT CAG CCA TGG AGA TGG TC-3′ | |
| Scnn1g | FOR | 5′-GTG CTT GTC CAT CAG CAG AA-3′ |
| REV | 5′-AAG GCA GAT CTG GAG GGA AT-3′ | |
| Sftpa | FOR | 5′-CTG CAA ACA ATG GGA GTC CT-3′ |
| REV | 5′-AGG AGT CTG GCC TTC AAT CA-3′ | |
| Sftpc | FOR | 5′-AGC AGA CAC CAT CGC TAC CT-3′ |
| REV | 5′-TCG GAC TCG GAA CCA GTA TC-3′ | |
| Cre | FOR | 5′-CAG GGT GTT ATA AGC AAT CCC-3′ |
| REV | 5′-CCT GGA AAA TGC TTC TGT CCG-3′ | |
| Tmprss4 | FOR | 5′-AGTAGGCATCGTGAGCTGGG-3′ |
| REV | 5′-GGACGGCAGCGTTACATCTC-3′ | |
| Probe | 5′-FAM-ATGGATGCGGCGGCCCAA-BHQ1-3′ | |
| Prss14 | FOR | 5′-GAAGCTTTGATGTCGCTCCC-3′ |
| REV | 5′-GGAGGGTGAGAAGGTGCCA-3′ | |
| Probe | 5′-FAM-CCACGCTGTGGTGCGGCTG-BHQ1-3′ |
FOR, forward primer; REV, reverse primer; FAM, 6-carboxy fluorescein; BHQ1, black hole quencher 1.
The paper explained
PROBLEM
The amiloride-sensitive ENaC plays a major role in Na+ absorption in AEC and its regulation is critical for normal lung fluid balance. Increasing in vitro evidence points to an important role of proteases such as CAPs in the regulation of ENaC but the precise identification of the serine protease(s) involved and their contribution in vivo is unclear.
RESULTS
This study addresses this question using conditional lung-specific deletion in triple transgenic mice to study the role of the membrane-bound serine protease CAP1/Prss8 in ENaC-mediated fluid absorption across alveolar epithelia in vitro and in vivo. Deletion of CAP1/Prss8 in the lung reduces basal and amiloride-sensitive alveolar fluid absorption, and the protease is critical to β-adrenergic activation of ENaC-mediated sodium and fluid absorption. Importantly, lack of CAP1/Prss8 results in impaired resolution of hydrostatic pulmonary oedema.
IMPACT
The data establish a role for CAP1/Prss8 in alveolar fluid balance and potentially in lung diseases, in which it is perturbed. Excessive accumulation of fluid in alveolar spaces frequently accompanies lung injury or left ventricle dysfunction leading to alveolar oedema. Activation of β2-adrenergic receptors by endogenous catecholamines on AEC is an important response to this life-threatening situation as it favours the removal of oedema fluid. As CAP1/Prss8 is required for an efficient β-adrenergic response, decreased alveolar CAP1/Prss8 expression may significantly delay the resolution of pulmonary oedema in cases of lung injury.
Quantitative qRT-PCR analysis
Total RNA was prepared from freshly isolated mouse AEC using the RNeasy extraction kit (Qiagen, Hilden, Germany). The RNAs (1 µg/sample) were treated with DNase (Turbo DNase treatment, Ambion, Austin, TX, USA,) and reverse-transcribed at 37°C for 1 h using the Superscript II RNase H-reverse-transcriptase (Invitrogen, Basel, Switzerland) and oligo-dT(20) primers (Invitrogen). The products were then diluted five times before proceeding with the real-time PCR reaction. Real-time PCRs have been performed by TaqMan® PCR with the Applied Biosystems 7500. The primers/probes mix 20× (Mm00504792_m1 for mCAP1, and 4352341E for β-actin) have been purchased with the Universal TaqMan mix 2× and used according to the manufacturer instructions (Applied Biosystems, Foster City, CA, USA). The other primers have been designed with the primer express software (Applied Biosystems) (Table 2). Quantification of fluorescence was performed, and the method used is the ΔΔCT normalized on β-actin. Each measurement was performed in triplicate.
Western blot analysis and immunocytochemistry
Distal lung protein extraction and Western blot analysis
Lungs were removed from thorax and homogenized for 3 min in ice-cold lysis RIPA buffer (pH 8) containing 20 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate and protease inhibitors. The lysate was centrifuged (15,000 rpm, 10 min) at 4°C and supernatants were aliquoted and frozen before use. Samples of protein extracts (100–200 µg/sample, the amount necessary to detect minor bands of ENaC proteins) were separated by SDS-PAGE on 8% acrylamide gels, electrically transferred to nitrocellulose paper, and subsequently probed for ENaC subunits and β-actin by using previously characterized rabbit polyclonal anti rat α-, β- and γ-ENaC antibodies (dilution 1:2000) (Duc et al, 1994), and mouse monoclonal anti-β-actin (dilution 1:1000) (Sigma). The anti-rabbit IgG secondary antibody (Amersham Pharmacia Biotech, UK) was used at dilution 1:5000 and the anti-mouse IgG (Sigma) at the dilution 1: 10,000. The signal was developed with the ECL + system (Amersham Pharmacia Biotech). Quantification of ENaC subunits and β-actin levels was obtained using NIH image software.
Immunocytochemistry
Immunohistochemical studies were performed on AEC monolayers cultured for 5 days on transwell filters after permeabilization with 0.1% Triton X-100, using rabbit polyclonal antibodies raised against murine occludin (1:50, Zymed Laboratories) or ZO-1 (1:100, Zymed Laboratories), and Alexa 488 conjugated goat anti-rabbit IgG as secondary antibodies (Molecular Probes). Nuclei were counterstained with Sytox Orange (Molecular Probes). Specimens were examined using a confocal microscope (CLSM-510-META, Zeiss, Mannheim, Germany).
Functional experiments
Bioelectric measurements under open-circuit conditions
Transepithelial resistance (Rte) and voltage (PD) were measured in mouse AEC grown on semi-permeable transwell filters for 5 days under open-circuit conditions using dual silver/silver chloride electrodes connected to the Millicell Electrical resistance Clamp apparatus, as previously described (Planès et al, 2005). Equivalent short-circuit current (Ieq) was calculated with Ohm's law from Rte and PD. Measurements of Rte and PD were performed at 37° before and after apical addition of amiloride (Sigma) (final concentration: 10 µM) for 10 min to block ENaC activity.
Measurement of AFC
Sodium-driven AFC was measured in vivo at 37°C over a 15-min period using an in situ nonventilated model in which the airspace was instilled with an isoosmolar Ringer's lactate solution containing 125I-albumin, as previously described (Planès et al, 2005; Randrianarison et al, 2007, 2008). AFC (percentage fluid absorption at 15 min) was calculated from the increase in alveolar fluid albumin as follows: AFC (%) = (Cf − Ci)/Cf × 100, where Ci and Cf represent the initial and final concentrations of 125I-albumin in the aspirate at 1 and 15 min respectively, as assessed by radioactivity measurements. In some experiments, amiloride was added to the instillate and AFC was measured. Amiloride was used at a final concentration of 1 mM in the alveolar instillate to block ENaC-dependent AFC because an important fraction of amiloride binds to the bovine serum albumin present in the alveolar instillate, so that the effective concentration of free amiloride is much lower than 1 mM (approximately 10−4 M) (Garat et al, 1997). In another set of experiments, the effects of the β-adrenergic agonist terbutaline (Sigma) (10−4 M) and of the serine proteases trypsin (Sigma) (100 µg/ml) or hNE (Serva Electrophoresis) (33 µg/ml) were studied as described above in the absence or presence of aprotinin (Sigma), a nonspecific serine protease inhibitor (100 µg/ml) or of EPI-hNE4 (kindly provided by Debiopharm S.A., Lausanne, Switzerland) (33 µg/ml), a specific hNE inhibitor (Harris et al, 2007).
Hydrostatic volume-overload studies
A standard model of acute hydrostatic oedema was used, as previously described (Randrianarison et al, 2007). Littermate mice matched for body weight were anaesthetized with intraperitoneal ketamine (80 mg/kg) and xylasine (12 mg/kg), and ventilated with a constant volume ventilator (Harvard Apparatus) with a tidal volume of 8 ml/kg, a positive end-expiratory pressure of 3 cm H2O and 100% oxygen. After a 20-min baseline period, a saline infusion via a catheter inserted into the jugular vein was given by an infusion pump over 2 h (total volume = 40% of body weight, with 40% of the total volume given over the first 20 min, the remaining 60% volume given over 100 min). At the end of infusion, the animal was weighed again in order to assess the change in body weight induced by saline infusion, before being killed by exsanguination and further processed for quantification of alveolar oedema fluid.
The volume of fluid in the airspace compartment was estimated from the change in albumin concentration in the alveolar instillate, as previously described (Randrianarison et al, 2007). Briefly, volume of alveolar epithelial lining fluid was calculated by instilling 0.3 ml of fluid (V0) containing a known concentration of 125I-albumin (C0) into the airspace. After a 1 min-mixing in the lungs inflated at 7 cm H2O continuous positive airway pressure, the instillate was aspirated and the concentration of 125I-albumin (C1) was measured. The volume of alveolar epithelial lining fluid (VELF) was calculated as follows: VELF = V0(C0/C1) − V0.
Statistical analysis
Results are presented as means ± SE. For functional data, one-way variance analyses were performed and, when allowed by the F value, results were compared by the modified least significant difference (Statview software, Abacus Concepts, Berkeley, CA). For RT-PCR experiments and Western blot experiments (performed on raw densitometric data), differences between groups were evaluated with unpaired t-test. p < 0.05 was considered significant.
Acknowledgments
We would like to thank Dr Jeffrey A. Whitsett (Cincinatti Children's hospital Medical Center, Cincinatti, OH) for kindly providing the SPC-rtTA/(tetO)7-CMV-Cre mice. We also thank Dr Alain Vandewalle and Dr Evelyne Ferrary for helpful suggestions, as well as Dr Frédéric Jaisser, Marcelle Benz, Nicole Fowler-Jaeger, Anne-Marie Mérillat and Olivier Thibaudeau for helpful technical assistance. This work was supported by INSERM, by Chancellerie des Universités de Paris (Fondation du Legs Poix), by Société de Pneumologie de Langue Française (SPLF) and by the Swiss National Foundation (grant FNRS # 3100AO-102125/1 to E. Hummler and grant FNRS # 3100-061966 to B. C. Rossier).
The authors declare that they have no conflict of interest.
Author contributions
C. P. and N. R. performed cell isolation, functional studies and Western-blot analyses. R. P. C. and S. F. performed the molecular biology experiments. F. C. and P. S. performed the immunohistochemistry and lung histology study. G. V. provided reagents. C. C. and B. R. contributed to the experimental design and discussions. C. P. and E. H. conceived and designed the study, supervised the experiments and wrote the manuscript.
References
- Adebamiro A, Cheng Y, Rao US, Danahay H, Bridges RJ. A segment of γENaC mediates elastase activation of Na+ transport. J Gen Physiol. 2007;130:611–629. doi: 10.1085/jgp.200709781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basset G, Crone C, Saumon G. Significance of active ion transport in transalveolar water absorption: a study on isolated rat lung. J Physiol (Lond) 1987;384:311–324. doi: 10.1113/jphysiol.1987.sp016456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski J, Hughey RP, Kleyman TR. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ-subunit. J Biol Chem. 2007;282:6153–6160. doi: 10.1074/jbc.M610636200. [DOI] [PubMed] [Google Scholar]
- Caldwell RA, Boucher RC, Stutts MJ. Serine protease activation of near-silent epithelial Na+ channels. Am J Physiol Cell Physiol. 2004;286:C190–C194. doi: 10.1152/ajpcell.00342.2003. [DOI] [PubMed] [Google Scholar]
- Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol. 2005;288:L813–L819. doi: 10.1152/ajplung.00435.2004. [DOI] [PubMed] [Google Scholar]
- Canessa C, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature (Lond) 1993;361:467–470. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]
- Canessa C, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature (Lond) 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- Carattino MD, Hughey RP, Kleyman TR. Proteolytic processing of the epithelial sodium channel γ subunit has a dominant role in channel activation. J Biol Chem. 2008;283:25290–25295. doi: 10.1074/jbc.M803931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles RP, Guitard M, Leyvraz C, Breiden B, Haftek M, Haftek-Terreau Z, Stehle JC, Sandhoff K, Hummler E. Postnatal requirement of the epithelial sodium channel for maintenance of epidermal barrier function. J Biol Chem. 2008;283:2622–2630. doi: 10.1074/jbc.M708829200. [DOI] [PubMed] [Google Scholar]
- Chen XJ, Jain L. Alveolar epithelial ion and fluid transport. β-adrenergic regulation of amiloride-sensitive lung sodium channels. Am J Physiol Lung Cell Mol Physiol. 2002;282:L609–L620. doi: 10.1152/ajplung.00356.2001. [DOI] [PubMed] [Google Scholar]
- Chraibi A, Vallet V, Firsov D, Kharoubi Hess S, Horisberger JD. Protease modulation of the activity of the epithelial sodium channel expressed in Xenopus oocytes. J Gen Physiol. 1998;111:127–138. doi: 10.1085/jgp.111.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diakov A, Bera K, Mokrushina M, Krueger B, Korbmacher C. Cleavage of the γ-subunit of the epithelial sodium channel (ENaC) plays an important role in the proteolytic activation of near-silent channels. J Physiol (Lond) 2008;586:4587–4608. doi: 10.1113/jphysiol.2008.154435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duc C, Farman N, Canessa CM, Bonvalet JP, Rossier BC. Cell-specific expression of epithelial sodium channel α, β and γ subunits in aldosterone-responsive epithelia from the rat: localization by in situ hybridization and immunocytochemistry. J Cell Biol. 1994;127:1907–1921. doi: 10.1083/jcb.127.6.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol. 2009;71:403–423. doi: 10.1146/annurev.physiol.010908.163250. [DOI] [PubMed] [Google Scholar]
- Egli M, Duplain H, Lepori M, Cook S, Nicod P, Hummler E, Sartori C, Scherrer U. Defective respiratory amiloride-sensitive sodium transport predisposes to pulmonary oedema and delays its resolution in mice. J Physiol. 2004;560:857–865. doi: 10.1113/jphysiol.2004.066704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garat C, Meignan M, Matthay MA, Luo DF, Jayr C. Alveolar epithelial fluid clearance mechanisms are intact after moderate hyperoxic lung injury in rats. Chest. 1997;111:1381–1388. doi: 10.1378/chest.111.5.1381. [DOI] [PubMed] [Google Scholar]
- Garcia-Caballero A, Dang Y, He H, Stutts MJ. ENaC proteolytic regulation by channel-activating protease 2. J Gen Physiol. 2008;132:521–535. doi: 10.1085/jgp.200810030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M, Firsov D, Vuagniaux G, Stutts MJ, Rossier BC. A novel neutrophil elastase inhibitor prevents elastase activation and surface cleavage of the epithelial sodium channel expressed in Xenopus laevis oocytes. J Biol Chem. 2007;282:58–64. doi: 10.1074/jbc.M605125200. [DOI] [PubMed] [Google Scholar]
- Hughey RP, Carattino MD, Kleyman TR. Role of proteolysis in the activation of epithelial sodium channels. Curr Opin Nephrol Hypertens. 2007;16:444–450. doi: 10.1097/MNH.0b013e32821f6072. [DOI] [PubMed] [Google Scholar]
- Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher RC, Rossier BC. Early death due to defective neonatal lung liquid clearance in αENaC-deficient mice. Nat Genet. 1996;12:325–328. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- Johnson MD, Bao HF, Helms MN, Chen XJ, Tigue Z, Jain L, Dobbs LG, Eaton DC. Functional ion channels in pulmonary alveolar type I cells support a role for type I cells in lung ion transport. Proc Natl Acad Sci USA. 2006;103:4964–4969. doi: 10.1073/pnas.0600855103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyvraz C, Charles RP, Rubera I, Guitard M, Rotman S, Breiden B, Sandhoff K, Hummler E. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J Cell Biol. 2005;170:487–496. doi: 10.1083/jcb.200501038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- List KR, Haudenschild CC, Szabo R, Chen W, Wahl SM, Swaim W, Engelholm LH, Behrendt N, Bugge TH. Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene. 2002;21:3765–3779. doi: 10.1038/sj.onc.1205502. [DOI] [PubMed] [Google Scholar]
- Matthay M, Landolt CC, Staub NC. Differential liquid and protein clearance from the alveoli of anesthetized sheep. J Appl Physiol. 1982;53:96–104. doi: 10.1152/jappl.1982.53.1.96. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- Perkins GD, McAuley DF, Thickett DR, Gao F. The beta-agonist lung injury trial (BALTI): a randomized placebo-controlled clinical trial. Am J Respir Crit Care Med. 2006;173:281–287. doi: 10.1164/rccm.200508-1302OC. [DOI] [PubMed] [Google Scholar]
- Perl AKT, Wert SE, Nagy A, Lobe CG, Whitsett JA. Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci USA. 2002;99:10482–10487. doi: 10.1073/pnas.152238499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planès C, Blot Chabaud M, Matthay MA, Couette S, Uchida T, Clerici C. Hypoxia and β2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem. 2002;277:47318–47324. doi: 10.1074/jbc.M209158200. [DOI] [PubMed] [Google Scholar]
- Planès C, Leyvraz C, Uchida T, Angelova MA, Vuagniaux G, Hummler E, Matthay MA, Clerici C, Rossier BC. In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1099–L1109. doi: 10.1152/ajplung.00332.2004. [DOI] [PubMed] [Google Scholar]
- Pittet JF, Wiener-Kronish JP, McElroy MC, Folkesson HG, Matthay MA. Stimulation of lung epithelial liquid clearance by endogenous release of catecholamines in septic shock in anesthetized rats. J Clin Invest. 1994;94:663–671. doi: 10.1172/JCI117383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randrianarison N, Escoubet B, Ferreira C, Fontayne A, Fowler-Jaeger N, Clerici C, Hummler E, Rossier BC, Planès C. β-Liddle mutation of the epithelial sodium channel increases alveolar fluid clearance and reduces the severity of hydrostatic pulmonary oedema in mice. J Physiol (Lond) 2007;582:777–788. doi: 10.1113/jphysiol.2007.131078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randrianarison N, Clerici C, Ferreira C, Fontayne A, Pradervand S, Fowler-Jaeger N, Hummler E, Rossier BC, Planès C. Low expression of the β-ENaC subunit impairs lung fluid clearance in the mouse. Am J Physiol Lung Cell Mol Physiol. 2008;294:L409–L416. doi: 10.1152/ajplung.00307.2007. [DOI] [PubMed] [Google Scholar]
- Rossier BC. The epithelial sodium channel. Activation by membrane-bound serine proteases. Proc Am Thorac Soc. 2003;1:4–9. doi: 10.1513/pats.2306007. [DOI] [PubMed] [Google Scholar]
- Rossier BC, Stutts MJ. Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol. 2009;71:361–379. doi: 10.1146/annurev.physiol.010908.163108. [DOI] [PubMed] [Google Scholar]
- Ridge KM, Olivera WG, Saldias F, Azzam Z, Hirowitz S, Rutschman DH, Dumasius V, Factor P, Sznajder JI. Alveolar type 1 cells express the α2 Na, K-ATPase, which contributes to lung liquid clearance. Circ Res. 2003;92:453–460. doi: 10.1161/01.RES.0000059414.10360.F2. [DOI] [PubMed] [Google Scholar]
- Rubera I, Meier E, Vuagniaux G, Mérillat AM, Beermann F, Rossier BC, Hummler E. A conditional allele at the mouse channel activating protease 1 (Prss8) gene locus. Genesis. 2002;32:173–176. doi: 10.1002/gene.10040. [DOI] [PubMed] [Google Scholar]
- Snyder PM. Liddle's syndrome mutations disrupt cAMP-mediated translocation of the epithelial Na+ channel to the cell surface. J Clin Invest. 2000;105:45–53. doi: 10.1172/JCI7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997;389:607–610. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- Vallet V, Pfister C, Loffing J, Rossier BC. Cell-surface expression of the channel-activating protease xCAP-1 is required for activation of ENaC in the Xenopus oocyte. J Am Soc Nephrol. 2002;13:588–594. doi: 10.1681/ASN.V133588. [DOI] [PubMed] [Google Scholar]
- Verghese GM, Ware LB, Matthay BA, Matthay MA. Alveolar epithelial fluid transport and the resolution of clinically severe hydrostatic pulmonay oedema. J Appl Physiol. 1999;87:1301–1312. doi: 10.1152/jappl.1999.87.4.1301. [DOI] [PubMed] [Google Scholar]
- Verghese GM, Tong ZY, Bhagwandin V, Caughey GH. Mouse prostasin gene structure, promoter analysis, and restricted expression in lung and kidney. Am J Respir Cell Mol Biol. 2004;30:519–529. doi: 10.1165/rcmb.2003-0251OC. [DOI] [PubMed] [Google Scholar]
- Vuagniaux G, Vallet V, Fowler-Jaeger N, Pfister C, Bens M, Farman N, Courtois-Coutry N, Vandewalle A, Rossier BC, Hummler E. Activation of the amiloride-sensitive epithelial sodium channel by the serine protease mCAP1 expressed in a mouse cortical collecting duct cell line. J Am Soc Nephrol. 2000;11:828–834. doi: 10.1681/ASN.V115828. [DOI] [PubMed] [Google Scholar]
- Vuagniaux G, Vallet V, Fowler Jaeger N, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucose-regulated kinase (Sgk1) in Xenopus oocytes. J Gen Physiol. 2002;120:191–201. doi: 10.1085/jgp.20028598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]