

Abstract
Deficiency of the mitochondrial enzyme 2-methyl-3-hydroxybutyryl-CoA dehydrogenase involved in isoleucine metabolism causes an organic aciduria with atypical neurodegenerative course. The disease-causing gene is HSD17B10 and encodes 17β-hydroxysteroid dehydrogenase type 10 (HSD10), a protein also implicated in the pathogenesis of Alzheimer's disease. Here we show that clinical symptoms in patients are not correlated with residual enzymatic activity of mutated HSD10. Loss-of-function and rescue experiments in Xenopus embryos and cells derived from conditional Hsd17b10−/− mice demonstrate that a property of HSD10 independent of its enzymatic activity is essential for structural and functional integrity of mitochondria. Impairment of this function in neural cells causes apoptotic cell death whilst the enzymatic activity of HSD10 is not required for cell survival. This finding indicates that the symptoms in patients with mutations in the HSD17B10 gene are unrelated to accumulation of toxic metabolites in the isoleucine pathway and, rather, related to defects in general mitochondrial function. Therefore alternative therapeutic approaches to an isoleucine-restricted diet are required.
Keywords: apoptosis, HSD10, ERAB, neurodegeneration, organic aciduria
INTRODUCTION
Organic acidurias are inherited metabolic disorders caused by the deficiency of enzymes involved in the mitochondrial oxidation of coenzyme A (CoA)-activated acyl compounds derived from amino acid breakdown. Clinical symptoms frequently develop at times of increased protein catabolism due to fasting or illness, leading to increased flux through oxidative pathways and accumulation of pathological metabolites. We have previously reported a novel organic aciduria caused by a deficiency of the mitochondrial enzyme 2-methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) involved in isoleucine metabolism (Zschocke et al, 2000). The clinical picture of this condition is very different from other organic acidurias. Most of the affected children do not develop metabolic crises and show few symptoms in the first year of life. From the second year on they follow a neurodegenerative disease course associated with mitochondrial dysfunction, leading to progressive loss of skills, neurological abnormalities including epilepsy, cardiomyopathy, retinal degeneration and death. The causative HSD17B10 gene is located on the X-chromosome and encodes 17β-hydroxysteroid dehydrogenase type 10 (HSD17B10, abbreviated HSD10) (Ofman et al, 2003). Several studies have indicated that this protein (also known as ERAB = endoplasmatic reticulum-associated amyloid-β-binding protein, or ABAD = amyloid-β-binding alcohol dehydrogenase) binds amyloid-β (Aβ) and mediates mitochondrial toxicity in Alzheimer's disease (AD, Lustbader et al, 2004; Yan et al, 1997). HSD10 is thus implicated in two neurodegenerative disorders but the pathogenetic basis has so far remained elusive.
Here we report that the severity of symptoms in patients with MHBD deficiency (MHBDD) is not correlated with residual enzyme activity of the mutated HSD10 proteins, suggesting that the HSD10 protein has other functions in addition to the enzymatic activity. In vertebrate systems, no loss-of-function data are available for HSD10, so far. We present evidence in Xenopus and mouse embryos, as well as in fibroblasts from patients with MHBDD that HSD10 is required for structural and functional integrity of the mitochondria. This function is independent of the enzymatic activity of the protein. Rescue experiments with wild type (WT) and mutant protein in Xenopus embryos in which HSD10 function was knocked-down revealed striking functional differences in properties of mutations found in MHBDD patients. Mutation Q165H, associated with complete loss of enzymatic activity but normal neurological development in humans, prevented apoptosis induced by HSD10 knock-down. In contrast, no rescue of the apoptosis phenotype was observed with human mutations associated with classical disease presentation (R130C, up to 64% residual enzyme activity) or a very severe clinical phenotype (D86G, 30% residual enzyme activity). These results were corroborated by rescue experiments in dendritic cells derived from homozygous HSD10 Tie2-Cre knock-out mice and indicated that the enzymatic activity of HSD10 is not required for cell survival.
Our data indicate that loss of (non-enzymatic) HSD10 function mediated by gene mutation, knock-down or knock-out causes mitochondrial dysfunction and apoptotic cell death. These findings shed new light on the pathogenesis of the clinical features of MHBDD and argue for new approaches to therapy. They also may be of relevance to the understanding of neurodegeneration in other conditions.
RESULTS
Enzymatic activity of mutated HSD10 protein and severity of symptoms in patients do not correlate
In order to gain insight into the pathogenesis of neurodegeneration in MHBD deficiency and to obtain further information on the biological role of HSD10, we investigated additional patients with MHBDD. The condition was retrospectively diagnosed in one child (case 1, Supporting Information) with a very severe neonatal presentation, absent neurological development and death from progressive hypertrophic cardiomyopathy at the age of 7 months. MHBD activity in fibroblasts, as determined in two independent laboratories, was only partially reduced to approximately 30% of normal. This was considerably higher than for other patients with MHBDD. Sequence analysis of the complete coding region of the HSD17B10 gene in this patient revealed hemizygosity for a novel mutation p.D86G (c.257A>G) in exon 3; no other mutation was identified. MHBDD was also diagnosed in a boy (case 2, Supporting Information) who presented with pre- and postnatal failure to thrive but normal cognitive and motor development. Neurological examination in this boy and two affected relatives (brother and cousin) has been entirely normal up to the present age of 8 years. There was no measurable MHBD activity in fibroblasts; molecular studies identified hemizygosity for the novel mutation p.Q165H (c.495A>C) in exon 5 of the HSD17B10 gene.
Observations in these patients indicated that the development and severity of symptoms in MHBDD is unrelated to residual enzyme activity. In order to confirm these findings we carried out in vitro expression analyses of WT HSD10 and the relevant mutations R130C (the common mutation), D86G (associated with a very severe clinical phenotype) and Q165H (associated with normal development). WT and mutants were expressed as stable, soluble proteins (Supporting Information Fig 1). Further insights were gained through the 3D structure determination of human HSD10 (Fig 1A, B), which crystallized in spacegroup P3221 and was determined to high resolution (1.2 Å; data collection and refinement details in Supporting Information Table 1). Kinetic constants were determined with different substrates (Fig 1E, F). Kinetic parameters calculated for the WT enzyme were in line with earlier data reported for this enzyme (Shafqat et al, 2003). Residual dehydrogenase activities of 64 and 28% were measured for mutations R130C and D86G, respectively; however, mutation R130C was unstable at room temperature and steadily lost enzyme activity (Fig 1C). Protein stability was assessed with and without NAD+ or NADH cofactor as a function of temperature by differential scanning fluorimetry (Fig 1D). Cofactor binding of NAD+ or NADH, as reflected by increased thermal stability, was observed for both WT and D86G. In contrast, mutation Q165H located at the active centre of the enzyme showed neither residual activity nor cofactor binding in our experimental settings. Inability of the mutation Q165H to bind the cofactor indicates that regardless of the substrate, no enzymatic reaction requiring NADH or NAD+ (all known reactions) will be sufficiently catalysed by this mutant protein. WT and all mutations alike showed identical mitochondrial distribution (data not shown). The lack of correlation between disease severity and residual enzyme activity in MHBDD patients thus is not due to misdirection of intracellular transport or impaired mitochondrial localization of mutated HSD10. Mitotracker staining revealed a filamentous network-like structure of the mitochondria in WT cells and fibroblasts carrying the Q165H mutation. Cells with the R130C and D86G mutations showed punctate and fragmented mitochondrial organization (Fig 1G–J). Despite the differences in the Mitotracker staining the amount of mitochondrial material is not reduced in patient cells judged by the amount of mitochondrial marker Grp75 protein and TOMM20 mRNA (Supporting Information Fig 2). The amount of HSD10 transcripts was nearly identical in the cells analysed (WT, R130C and Q165H). In agreement with the finding that the R130C mutation is unstable (Fig 1C) less HSD10 protein could be detected in fibroblasts carrying this mutation (Supporting Information Fig 2).
Figure 1. Crystal structure, stability and activity of the HSD10 homotetramer and mitochondrial morphology in fibroblasts from MHBDD patients.
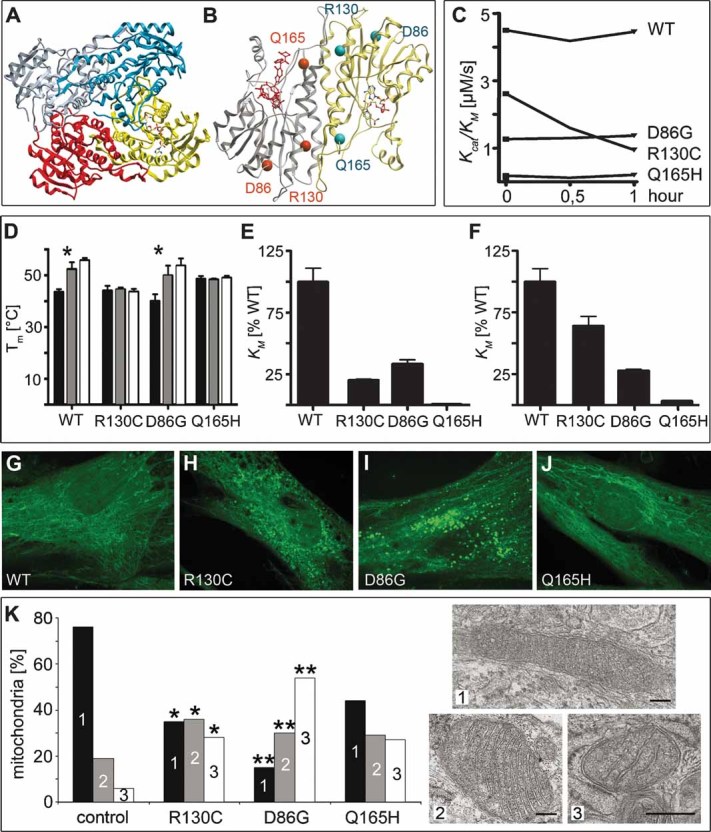
Crystal structure of HSD10. (A) Diagram of the homotetramer. A NADH cofactor is bound to one monomer. (B) Diagram of a HSD10 dimer with the location of the mutations. The active centre is marked by the substrate in red.
- C. Enzyme activity of HSD10 over time. Kcat/KM of HSD10 WT and mutations measured in a 30 min interval; substrate 2-methyl-3-hydroxybutyryl-CoA.
- D. Stability of HSD10 WT and mutations. Mean value of Tm determined by differential scanning fluorimetry (DSF)-experiments and standard deviation are shown. Black column: no cofactor/substrate, grey column: cofactor NAD+, white column: cofactor NADH (* indicates significant cofactor binding).
- E, F. Enzyme activity of HSD10 with different substrates. (E) Kcat/KM of HSD10 mutations with hydroxybutyryl-CoA as substrate (WT enzyme is taken as 100%). (F) Kcat/KM of HSD10 mutations with 2-methyl-3-hydroxybutyryl-CoA as substrate in relativity to the WT enzyme.
- G–J. Mitochondrial staining in patient fibroblasts. 300 nM Mitotracker Green FM were used on cells fixed with 3.7% formaldehyde on coverslips and mitochondria were visualized on a Perkin Elmer spinning disc confocal ERS-FRET on Nikon TE2000 inverted microscope. (G) Control, (H) R130C, (I) D86G, (J) Q165H.
- K. Fibroblasts were sectioned for electron microscopy. Pictures of 10–43 random systematically chosen visual fields were taken in a magnification of 11.5 × 103, scale bars: 100 nm. Mitochondria were classified into three groups (1—dense, dark; 2—loosely packed; 3—depleted cristae) with group 3 not only being morphologically distinct but also characterized by smaller mitochondria. Total numbers per sample and an overview of the cells are given in Supporting Information Fig 3. ** Indicate significance at p < 0.0001, * gives significance at p = 0.0366–0.0857 adjusted for multiple comparisons within the experiment compared to control fibroblasts. Normal human dermal fibroblasts served as controls.
Mutations D86G and R130C cause severe disruption of mitochondrial morphology
Since HSD10 WT and mutations are localized to mitochondria and patients with MHBDD show signs of mitochondrial dysfunction, we analysed mitochondrial morphology by EM in fibroblasts derived from patients compared with control fibroblasts (Fig 1K and Supporting Information Fig 3). The majority of mitochondria in control cells showed WT morphology (dense, dark). Fibroblasts carrying the Q165H mutation maintained WT morphology of 45% of the mitochondria, 30% displayed an intermediate phenotype with loosely packed and/or swollen cristae and 27% showed depletion of cristae and appeared ‘empty’ (for details, see Material and Methods). In contrast, in cells carrying the D86G and R130C mutation 65–85% of the mitochondria displayed an aberrant phenotype (group 2 and 3). The EM analysis of mitochondria of HSD10 mutant human cells thus showed that HSD10 is required for mitochondrial integrity. This function is not correlated with residual enzyme activity.
Mitochondrial disintegration after conditional Hsd17b10 knock-out in mice
Since knock-out of Hsd17b10 in mice results in early embryonic lethality at gastrula stages (B. Arnold, unpublished data), we established two conditional Hsd17b10 knock-out mouse lines. In one line, Hsd17b10 was eliminated in endothelial cells and cells of the immune system by mating mice carrying a floxed allele of Hsd17b10 with a Tie2 promoter driven Cre recombinase strain. These mice are viable and fertile, but have defects in spleen and vasculature. The animals die rapidly around week 25. Supporting Information Fig 4 shows the recombination efficiency in spleen cells and dendritic cells of Tie2-Cre × Hsd17b10 mice as an example.
To generate the second conditional knock-out line, mice carrying a floxed allele of Hsd17b10 were mated with a DBH-Cre line which eliminates the Hsd17b10 gene in noradrenergic neurons. These mice are viable and fertile but die around week 26.
In the central nervous system (CNS) the locus coeruleus contains noradrenergic neurons and we set out to analyse the morphology of the mitochondria in this region by EM in conditional knock-out mice. In the loci coerulei of DBH-Cre HSD10 deficient mice, almost 30% of the mitochondria showed depletion of cristae and appeared ‘empty’, more than 50% of the mitochondria were loosely packed and had swollen cristae, while normal morphology (dense, dark) was only found in 20% (Fig 2, Supporting Information Fig 5). In the cerebellum of the same mice, which lacks noradrenergic neurons, 50% of the mitochondria had WT morphology, 40% displayed the intermediate phenotype and only 10% appeared ‘empty’ and without cristae. A similar picture was observed in the peripheral nervous system (PNS). Mitochondria were morphologically severely altered in superior cervical ganglia isolated from DBH-Cre conditional knock-out mice (Supporting Information Figs 6 and 7). The EM analysis of two independent primary cell types (neurons of the CNS and PNS) from Hsd17b10 DBH-Cre conditional knock-out mice thus confirmed in a genetic system that HSD10 is required for mitochondrial structural integrity in the CNS and the PNS and that the observed changes in mitochondria are cell type independent.
Figure 2. Mitochondrial morphology in brains of mice with a conditional knock-out in noradrenergic neurons (DBH-Cre).
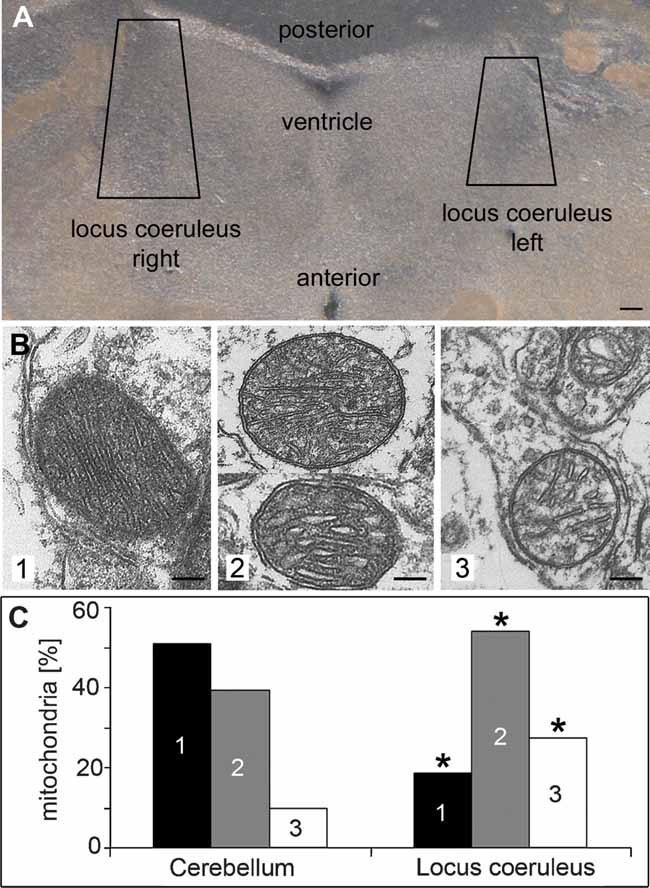
- A. Brains from knock-out mice were Vibratome sectioned (50 µm), the loci coerulei, which could be identified by pigmentation, were dissected and prepared for electron microscopy. Scale bar: 100 µm.
-
B, C. Mitochondria were classified into three groups (1—dense, dark; 2—loosely packed/swollen cristae; 3—depleted cristae). Total numbers per sample and an overview of the cells are given in Supporting Information Fig 5. Sections of the cerebellum, which lacks noradrenergic neurons, served as a control. * Indicates significance (p < 0.0001) of differences to the cerebellum.Pictures of 33 random systematically chosen visual fields were taken in a magnification of 11.5 × 103, scale bars: 100 nm.
HSD10 knock-down in Xenopus impairs mitochondrial integrity and induces apoptosis
In order to study HSD10 loss of function in early vertebrate embryos we turned to Xenopus as an experimental model. We cloned the Xenopus homologue of HSD10 (xHSD10) and performed RT-PCR analysis, as well as whole mount in situ hybridization during the first 3 days of embryonic development. HSD10 mRNA is provided maternally, and zygotic HSD10 transcription was found from neurula stages onward (Supporting Information Fig 8). In Xenopus embryos, translation of mRNAs can be specifically blocked by Morpholino (Mo) antisense oligonucleotides. Two Mo antisense oligonucleotides were designed to inhibit HSD10 mRNA translation. Mo5′UTR targeted the 5′UTR and MoATG the coding region of xHSD10. Both oligos specifically suppressed HSD10 translation in Xenopus embryos (Fig 3A).
Figure 3. Mitochondrial function and morphology in Xenopus animal caps.
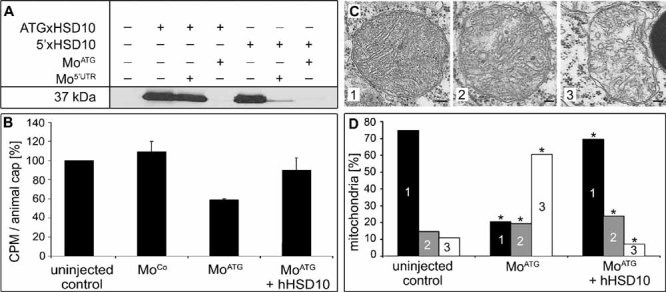
- A. Functionality and specificity of antisense Mo oligonucleotides is shown. Antisense Mo oligonucleotides and Myc-tagged HSD10 cDNA were injected in Xenopus embryos. Protein extract from these embryos was subjected to Western blot using 9E10 anti-Myc antibody. Antisense Mo oligonucleotide Mo5′UTR can block the translation of 5′xHSD10 cDNA including the 5′UTR, but not the translation of ATGxHSD10 cDNA starting with the start codon. MoATG can block translation of both constructs.
- B. Effect of HSD10 knock-down on mitochondrial function. Animal caps were dissected from injected Xenopus embryos and the turnover of 1-C14 pyruvate was measured. Counts per minute (CPM) is shown in relation to uninjected control (100%) and the standard error is given.
- C, D. Animal caps were dissected from injected Xenopus embryos and sectioned for electron microscopy. Pictures of 24 random systematically chosen visual fields were taken in a magnification of 6.6 × 103, scale bars: 100 nm (C). Mitochondria were classified into three groups (1—dense, dark; 2—loosely packed; 3—depleted cristae) and the distribution is shown in (D). * Indicates significance at p < 0.0001 compared to uninjected control animal caps. Total numbers per sample and an overview of the cells are given in Supporting Information Fig 9.
Next we tested whether HSD10 knock-down impaired mitochondrial function. Antisense MoATG was microinjected into the prospective ectoderm and the injected region (animal cap) was explanted and cultured. Explanted animal caps differentiate into a homogeneous tissue, the so-called atypical epidermis. After 2 days in culture, when control embryos had reached tailbud stage, pyruvate turnover was measured as a parameter of mitochondrial function. MoATG-injected samples displayed a 40% reduction in pyruvate turnover compared to the uninjected or control Mo (MoCo) injected tissue. Impaired mitochondrial function was specific because co-injection of a human HSD10 expression construct which could not be blocked by the HSD10 MoATG rescued pyruvate turnover almost to the level of uninjected controls (Fig 3B).
EM analysis revealed that the morphology of mitochondria in animal cap explants was changed after HSD10 knock-down (Fig 3C, D, Supporting Information Fig 9). Mitochondria showed severe reduction of cristae and a generally irregular shape. This phenotype was rarely observed in uninjected samples or in tissue in which the MoATG was injected together with a human HSD10 expression construct. This set of experiments demonstrated by LOF analysis that HSD10 is required for functional and structural integrity of mitochondria.
Next we asked whether Mo-mediated HSD10 knock-down would interfere with the establishment of the early body plan in Xenopus embryos. HSD10 MoATG was injected into dorsal animal (d/a) blastomeres at the four-cell stage and the embryos were grown for 2 days until they had reached tailbud stages. The progeny of the d/a blastomeres contributes mainly to neural tissue and indeed we found reduced brain tissue and eyes in MoATG-injected embryos (Fig 4A). This phenotype was specific because a human HSD10 expression construct which did not harbour the Mo binding site completely rescued the eye and brain defects. Despite the adverse effects of xHSD10 knock-down on neural tissue, the anterior/posterior (a/p) patterning of the brain was unaffected in these embryos. The spatial expression patterns of the forebrain marker BF1 and the fore-/midbrain marker otx2 were unchanged, but expression levels were reduced in MoATG-injected embryos (Fig 4B, C). Transferase dUTP nick end labelling (TUNEL) staining of embryos which received the xHSD10 MoATG only into the right side of the body showed enhanced apoptosis on the injected side (Fig 4D, Supporting Information Fig 10). Selective targeting of the Mo is possible in Xenopus embryos because the dorsal/ventral and the left/right axes can be seen already at the four-cell stage due to pigmentation differences of the animal blastomeres. Induction of apoptosis after siRNA-mediated knock-down of HSD10 was confirmed in human SHSY5Y cells (Supporting Information Fig 11).
Figure 4. Loss-of-function analysis of xHSD10 using Mo antisense oligonucleotides.
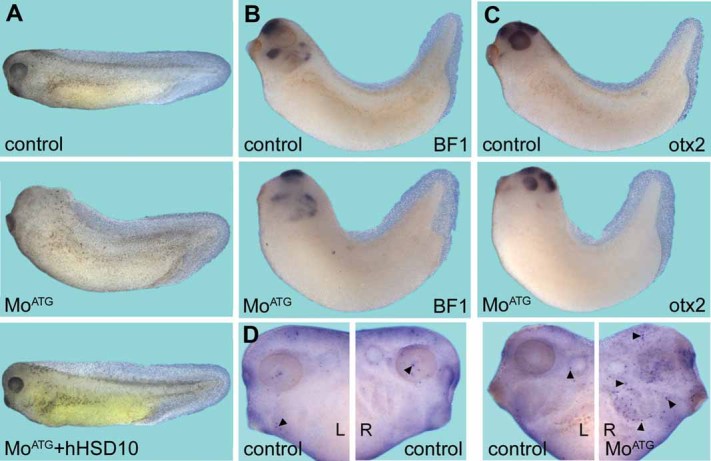
- A. The phenotype of embryos injected with xHSD10 MoATG and its rescue with hHSD10 are shown. The injection of MoATG resulted in small or no eyes and reduced anterior structures in 99% (n = 60) of the injected embryos compared with control embryos. Differences in pigmentation are due to slightly differing developmental stages of the MoATG injected and the rescued embryo. Rescue of this phenotype was shown in two independent experiments (n = 40) where 92% of the embryos were WT and only 8% showed the knock-down phenotype.
- B, C. The reduction of forebrain and mid-/hindbrain tissue is shown by in situ hybridization of embryos injected with MoATG in comparison to uninjected controls using BF1 and otx2 as a markers. Fourteen out of 15 embryos (otx2) and 13 out of 15 embryos (BF1), respectively, showed reduction of forebrain and mid-/hindbrain tissue but brain patterning is not disturbed.
- D. The increase of apoptosis after knock-down of xHSD10 is shown by TUNEL staining (arrowheads). Embryos were injected with HSD10 MoATG on the right side (R). The left side (L) served as an internal control.
HSD10 MoATG-induced apoptosis could be rescued by human HSD10 WT which also rescued the defects in neural tissue (Fig 5). The mutated HSD10 proteins found in human patients, however, showed remarkable differences in their ability to rescue the apoptotic phenotype. The Q165H mutation, which has less than 3% residual enzymatic activity but was identified in three neurologically normal boys, partially rescued HSD10 MoATG-induced apoptosis. In contrast, injection of the mutations R130C and in particular D86G failed to rescue, and instead further enhanced apoptosis. D86G has considerable residual enzyme activity despite being associated with a very severe clinical phenotype. The inability of the R130C and D86G mutants to rescue apoptosis was not due to protein mislocalization. In human HeLa and Xenopus A6 cells WT and all mutated proteins colocalized with mitochondria (data not shown and Supporting Information Fig 12). Next we performed rescue experiments using dendritic cells derived from Tie2-Cre HSD10 mice. Loss of HSD10 induced apoptosis was detected and quantified by TUNEL assay. In agreement with our Xenopus data, WT HSD10 and the Q165H mutant, but not the R130C and the D86G mutants, partially rescued apoptosis (Fig 5, Supporting Information Fig 4).
Figure 5. Quantitative analysis of the effect of HSD10 loss-of-function on the apoptosis rate and rescue.
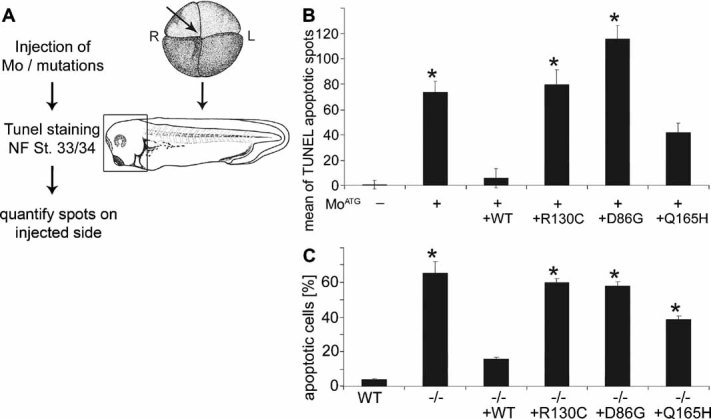
- Experimental scheme.
- Quantification of TUNEL staining on the right (injected) side of tailbud stage embryos (NF St. 33/34) normalized against the left (control) side. The standard error of four individual experiments (n = 45–52 embryos) is shown. * Denotes significance of mean difference from uninjected control at p < 0.0001.
- Dendritic cells from WT mice and mice with a conditional knock-out in endothelial cells and haematopoietic stem cells (Tie2) were transfected with plasmids (pT-Rex-DEST30) bearing human HSD10 WT or mutations for 48 h before TUNEL assay was performed. The percentage of TUNEL positive cells of three independent experiments and standard error are shown. Statistical significance (*) of difference from WT cells was determined at p < 0.0001. The increase in apoptosis rate caused by HSD10 knock-down can be rescued by human HSD10 WT and the Q165H mutation but not by R130C and D86G cDNA.
These experiments demonstrate that loss of HSD10 results in induction of apoptosis independent of the cell type and organism. The rescue experiments furthermore show that apoptosis after HSD10 loss of function is not dependent on the enzymatic activity of HSD10, again arguing for a non-enzymatic function of this protein which is required in mitochondria.
DISCUSSION
MHBD deficiency causes an atypical organic aciduria
Mitochondrial dysfunction is a commonly observed feature of primary disorders of mitochondrial intermediary metabolism such as the classical organic acidurias. Neurological abnormalities in these conditions are usually attributed to accumulation of toxic substrates proximal to a specific enzymatic block, causing secondary interference with energy metabolism. Primary apoptotic nerve cell death is not a feature of organic acidurias. Here we report that neurodegeneration in MHBDD, a recently identified organic aciduria, is not due to loss of enzyme activity and, presumably, the accumulation of toxic metabolites, but rather involves a fundamentally different pathomechanism. Although the respective enzyme, HSD10 has an important role in mitochondrial metabolism, we show that the adverse effects of its deficiency are primarily caused by a non-enzymatic effect triggering mitochondrial disintegration, apoptosis and cell death.
Several lines of evidence presented here support this view. Measurements of enzymatic activity in patient fibroblasts revealed that the R130C and D86G mutations associated with classical and neonatal neurodegenerative forms of MHBDD, had up to 30% residual activity compared to the WT protein. In contrast, the Q165H mutation that was identified in three children with normal neurological development was associated with less than 3% residual enzymatic activity. Thus the severity of the clinical phenotype is not at all correlated with the enzymatic activity of the mutated HSD10 proteins. We subsequently set out to corroborate this observation with experimental evidence. Mitochondrial integrity and cell viability could be restored after HSD10 knock-down in Xenopus and in conditional knock-out mice by Q165H mutant HSD10 protein, but not by R130C or D86G. Complete loss of the HSD10 protein in mouse embryos causes lethality at very early stages of embryogenesis (day 5 p.f.). It is unlikely that the embryonic lethality is due to the lack of enzyme activity, because it was shown for defects in comparable metabolic enzymes that toxic metabolites are efficiently eliminated by the placenta.
Additional clinical observations support our conclusions. Attempts to improve the condition of MHBDD patients by a restrictive diet that avoids accumulation of potentially toxic precursor metabolites failed (unpublished data). Slow progressive neurodegeneration without metabolic crises is atypical for organic acidurias caused by defects in mitochondrial intermediary metabolism, and deficiency of the enzyme 2-methylacetoacetyl-CoA thiolase, positioned immediately after MHBD in the isoleucine pathway, causes accumulation of the same metabolites in sometimes even higher concentrations but is associated with a completely different disease picture (episodic ketoacidosis, no neurodegeneration). In contrast to most other metabolic disorders, no null mutations that completely eliminate the protein have been identified in MHBDD. Indeed, one particular mutation, R130C, is found in more than half of cases and has usually occurred de novo in the respective patients. There is no evidence that the corresponding nucleotide change c.388C>T represents a hypermutable position, raising the possibility that most other mutations in the HSD17B10 gene are not observed because they are incompatible with life. This again is highly unusual for organic acidurias that usually manifest after birth only, since prenatally toxic metabolites are removed via the placenta. Finally, a splicing variant of the HSD17B10 gene that causes moderately reduced production of normal HSD10 without significant loss of enzyme activity and without production of an abnormal protein was found to cause an unusual non-progressive syndrome of mild mental retardation, choreoathetosis and abnormal behaviour (Lenski et al, 2007).
HSD10 has a non-enzymatic, protective function in mitochondria
Reduction or loss of HSD10 in Xenopus, mouse and humans has adverse effects on mitochondrial integrity and leads to enhanced apoptotic cell death and reduced viability. Thus, HSD10 may be a component serving to protect mitochondria at times of increased metabolic stress. This interpretation is compatible with the observation that infections or other stress situations in patients with MHBDD may trigger clinical deterioration. In response to metabolic stress, HSD10 protein translocates from the mitochondrial matrix to the inner mitochondrial membrane and this might be important for maintaining mitochondrial integrity as this is an essential site for assembly of the mitochondrial membrane permeability transition pore (Tieu et al, 2004, and unpublished own data). A protective effect of overexpressed HSD10 under conditions of oxidative stress was found in the setting of acute brain damage (Yan et al, 2000) and in a mouse model of Parkinson disease (Tieu et al, 2004). It appears plausible that interaction of HSD10 with an integral protein of the mitochondrial membrane could mediate its protective effect and is disturbed by altered HSD10 protein structure due to specific mutations.
Cellular energy failure caused by mitochondrial dysfunction is thought to play an important role in the development of AD, and several studies indicated that this effect is caused by a direct interaction of Aβ with HSD10 (Lustbader et al, 2004; Yan et al, 1997). HSD10 protein or transcript was found to be increased in affected neurons in human AD as well as in AD mouse models and it has been suggested that overexpression of HSD10 amplifies Aβ-mediated mitochondrial toxicity (Chen & Yan, 2007). However, a recent study showed that a mitochondrial bioenergetic deficit precedes HSD10 overexpression and Alzheimer's pathology in an AD mouse model (Yao et al, 2009). The results of our present study, the first to examine the effect of HSD10 loss-of-function, indicate that adverse effects observed as a consequence of the binding of Aβ to HSD10 are more likely due to Aβ-mediated loss of a non-enzymatic HSD10 function, and that increased amounts of (non-functional) HSD10 protein may be produced as a secondary or compensatory mechanism. It thus remains possible that the mitochondrial interaction of Aβ with HSD10, triggering early dysfunction of energy homeostasis, is one of the first steps in the pathogenesis of AD.
Therapeutic implications
Our conclusion that toxic metabolites are not responsible for the clinical symptoms observed in MHBDD patients implies that treatment targeting reduction of precursor metabolites through dietary measures is not indicated. This is in line with the clinical observation that dietary reduction of the isoleucine load has no beneficial effect on the course of this disease. In contrast, treatment of patients with MHBDD should aim at reducing mitochondrial stress and maintaining mitochondrial homeostasis through proactive management of infections and fever and possibly the administration of vitamins and cofactors. Our results may also contribute to a better understanding of neurodegeneration in the context of AD. Strategies to reduce Aβ toxicity through inhibition of its interaction with HSD10 could make HSD10 an interesting protein for Alzheimer's therapy.
MATERIALS AND METHODS
Enzymatic methods
Enzyme purification
Bl-21 DE3 cells (Invitrogen) were used for protein expression after transformation with p11 expression vectors containing cDNA of HSD10 WT or mutations. Bacteria were cultivated at 37 °C and 160 rpm until OD600 ≥ 1. Protein expression was induced with 0.5 mM IPTG (Sigma) and continued overnight at 30 °C. Cells were harvested for 15 min at 6500 rpm (4 °C) and resuspended in binding buffer (500 mM NaCl, 5% glycerol, 50 mM HEPES pH 7.5, 5 mM imidazole, 1 mM PMSF, 0.5 mM TCEP, protease inhibitor cocktail tablet, EDTA free). Lysis of cells was achieved with an EmulsiFlex-C5 High Pressure Homogenizer (Avestin). Cell debris was removed by centrifugation (45 min, 16 500 rpm, 4 °C) and the supernatant was subjected to immobilized metal ion affinity chromatography. His-tagged proteins were eluted from Ni2+-sepharose (GE Healthcare) with elution buffer containing 250 mM imidazole. TEV protease (Invitrogen) was used to remove His tags. HSD10 enzymes were further purified by gelfiltration using HiLoad™ 16/60 Superdex 75 prep grade (GE Healthcare) and by ion exchange chromatography using ‘Resource S’ and ‘Mono Q5/50GL’ (GE Healthcare). Enzyme purification was evaluated by SDS-PAGE. Protein mass was determined by mass spectroscopy in an Agilent LC/MSD TOF.
Protein crystallization
Crystallization of HSD10 WT protein was performed by mixing 14.25 mg/ml protein and crystallization solution (25.5% PEG3350, 0.17 M (NH4)2SO4, 15% glycerol) in a 1:1 ratio at 4 °C in sitting drops. Experiments were performed in the presence of 5 mM NADH (Sigma). Crystals were mounted directly from the crystallization solution and flash-cooled in liquid nitrogen. Data were collected on the synchrotron beamline SLS-X10 (SLS, Switzerland). The 3D structure of HSD10 was determined to a resolution of 1.2 Å by molecular replacement with the medium resolution structure of HSD10 as search model (PDB id 1u7t). The structure was deposited with the ProteinDataBank under the accession number 2023. Data collection and refinement statistics are found in Supporting Information Table 1.
Enzyme kinetics
Different amounts of purified enzyme and substrate in the presence of the cofactor NAD+ (Sigma) (400 µM in 100 mM Tris/HCl pH 9, 25 °C) were incubated in a SpectraMax M5/M5e Microplate Reader and the change in nucleotide cofactor absorbance (A340 nm) was measured. Graph Pad Prism program was used to determine KM and Vmax.
Differential scanning fluorimetry (DSF)
Purified proteins (10 µM) in HEPES buffer (10 mM, pH 7.5) were incubated with 150 mM NaCl, 85 µM substrate and 0.5 µl SYPRO Orange (Invitrogen) in the presence or absence of cofactors NAD+ and NADH (200 µM). Fluorescent emission was measured in an Mx3005p RT-PCR machine (Stratagene) and incubation temperature increased from 25 to 75 °C (1 °C/min).
Mitochondrial function
Xenopus animal caps were homogenized in 500 µl homogenization buffer (250 mM sucrose, 50 mM KCl, 5 mM MgCl2, 20 mM Tris/HCl, pH 7.4) using a Potter-Elvehejm system and 7 downstrokes. The suspension was incubated with 50 µl 10 mM malate and 50 µl 1-C14 pyruvate (5 µCi/ml, GE Healthcare) for 1 h at 37 °C in a scintillation vial that contains a tube with 200 µl hyamine. This tube was transferred in a new scintillation vial containing 7 ml UltimaGold (Perkin Elmer) and measured in a scintillation counter.
Electron microscopy
Primary fixation was done with 2.5% glutaraldehyde in PBS. For easier handling tissue culture cells were scraped off and embedded in 2% agarose. All samples were postfixed with 1% osmium tetroxide (Serva Electrophoresis) in 100 mM phosphate buffer pH 7.2 for 1 h on ice, washed extensively with water, block-stained with 1% aqueous uranyl acetate (Serva Electrophoresis) for 1 h at 4 °C, dehydrated in a graded series of ethanol at ambient temperature, infiltrated with mixtures of ethanol/Epon and finally embedded in Epon. Ultrathin sections were stained with uranyl acetate and lead citrate (Serva Electrophoresis) (Venable & Coggeshall, 1965) and viewed in a Philips CM10 electron microscope at 60 kV using a 30 µm objective aperture. Mitochondria were quantitatively classified into groups according to their morphology by examiner who was blinded to the nature (WT/mutant, etc.) of the samples. Group 1 is distinguished by a regular arrangement and shape of the cristae stacks. Group 3 characteristics are a reduced cristae compartment resulting in less electron dense material in the mitochondria and an irregular arrangement and shape of the cristae stacks. Group 2 is represented by an intermediate morphological phenotype (see Figs 1K, 2B and 3C).
All samples were sectioned at random angles resulting in cross and longitudinal sections of mitochondria. For the morphological analyses the plain of sections was not relevant.
Statistical evaluation
For each mitochondrium the density was used as an ordinal variable (dense, medium or depleted). The variable was evaluated as a categorical variable within a logistic model framework. The hypothesis of equal density distribution rates for different treatments was tested controlling for a suitable covariante (animal cap, brain sections or patient) in the respective experiment. All analyses have been performed with SAS 9.1 on PC (SAS Institute, Inc., Cary, NC). Differences were counted as significant if the p-values were lower than 5% adjusted for multiple testing within one experiment.
Generation of a specific conditional HSD10 knock-out in noradrenergic neurons and immune cells
As mice deficient for HSD10 display embryonic lethality, two conditional knock-out mouse lines were generated. The construct used to generate floxed HSD10 alleles contained the coding sequence for HSD10 with exon 1 being flanked by loxP sites. This construct was introduced into embryonic stem cells (E14.1 of the 129P2/Ola Hsd stem) via electroporation. 20 µg DNA were mixed with 107 cells in PBS and electroporated at 240 V and 960 µF. Stable clones were selected with G418 and cultured in DMEM containing LIF (myeloid leukemia inhibitory factor) on a layer of Feeder cells. Blastocysts were gained from pregnant C57Bl/6N mice at day 3.5 p.c. Stable transfected ES cells (20–25) were injected into the blastocoel and the blastocysts were transferred into pseudo-pregnant NMRI females at day 2.5 p.c. Resulting chimeric male animals were backcrossed with C57Bl/6 N females to homozygosity.
To generate an Hsd17b10 DBH-Cre conditional knock-out mouse, a transgenic mouse line carrying the Cre recombinase under the control of the DBH promoter was mated with a mouse line carrying a floxed allele of Hsd17b10. In the DBH-Cre line Hsd17b10 is eliminated specifically in noradrenergic neurons of the peripheral and CNS. The generation of DBH-Cre mice has been described previously (Stanke et al, 2006).
The second conditional knock-out (Hsd17b10 Tie2-Cre) in endothelial cells and hematopoietic stem cells was achieved using a transgenic mouse line carrying the Cre recombinase under the control of the Tie2 promoter. Generation of these mice has been described elsewhere (Constien et al, 2001).
Tissue preparation
Animals were killed and immediately dissected to collect brains. Tissues were fixed in 4% formaldehyde, 1% glutaraldehyde in PBS and cut in Vibratome sections (50 µm). Appropriate regions of the brain were identified by pigmentation and dissected from these Vibratome sections prior to electron microscopy.
Isolation and cultivation of dendritic cells
Dendritic cells from WT and HSD10 knock-out mice were isolated from bone marrow and expanded for 10 days in GM-CSF supplemented F1/16 medium. The ratio of CD11 positive cells used for the experiments was measured by FACS analysis and varied between 70 and 90%.
Transfection
Dendritic cells were transfected with 0.5 µg/106 cells plasmid-DNA (pT-Rex-DEST30) coding for HSD10 WT or mutations, respectively by electroporation (Microporator MP-100, Peqlab) using single pulses of 990 V for 40 ms. Transfection efficiency was confirmed by PCR for the neomycin-gene on pT-Rex-DEST30.
The paper explained
PROBLEM
HSD10 is an essential enzyme in the isoleucine breakdown pathway and has also been reported as an important mediator of mitochondrial toxicity in Alzheimer's disease. A deficiency of HSD10 caused by mutations in the HSD17B10 gene can be recognized by specific metabolic changes. However, children with HSD10 deficiency show a neurodegenerative disease picture that does not resemble other disorders of isoleucine metabolism or similar organic acidurias but is more reminiscent of primary mitochondrial disorders. The exact pathomechanism is unknown.
RESULTS
By investigating additional patients with a genetic deficiency of HSD10 we show that there is no correlation between enzyme activity and clinical presentation. Loss-of-function and rescue experiments in Xenopus embryos and cells derived from conditional Hsd17b10−/− mice demonstrate that HSD10 is essential for structural and functional integrity of mitochondria independently of its enzymatic activity. Impairment of this function in neural cells causes apoptotic cell death whilst the enzymatic activity of HSD10 is not required for cell survival.
IMPACT
The exact molecular mechanisms leading to mitochondrial disintegration and neuronal apoptosis in HSD10 deficiency are still unknown but our data show that the clinical effects cannot be attributed to the accumulation of toxic metabolites in the isoleucine pathway or other metabolic effects. Rather, HSD10 has a protective effect on mitochondrial integrity. Delineation of this protective mechanism should provide new therapeutic perspectives for HSD10 dysfunction. Children with a genetic deficiency of HSD10 are unlikely to benefit from an isoleucine-restricted diet, previously suggested as a therapeutic option. Treatment should aim at reducing mitochondrial stress and maintaining mitochondrial homeostasis through proactive management of infections and fever and possibly the administration of vitamins and cofactors.
TUNEL assay
TUNEL assay was performed 48 h after transfection using the In Situ Cell Death Detection TMR kit (Roche). The staining was performed according to the manufacturer's instructions. The cells were counterstained with DAPI nuclear stain (Vector Laboratories) and evaluated with an Olympus AX70 microscope. TUNEL-positive cells were counted in five different visual fields using the visualizing software Cell F (Olympus) and expressed as the percentage of positive cells. Rates in apoptotic cells for different transfections were compared to WT cells by a Chi-square test. Differences were counted as significant if the p-values were lower than 5% adjusted for multiple testing within one experiment.
Embryological methods
Eggs were obtained by injecting human chorionic gonadotropin (Sigma) into female Xenopus laevis and fertilized artificially. Embryos were dejellied using 2% l-cysteine (pH 7.8–8.0, Biomol) and cultured in 0.1× MBSH. Embryo stages were determined according to Nieuwkoop and Faber (1967). Animal caps were dissected at stage 8 or 9 and cultured in 0.5× MBSH until stage NF 33/34.
Gene knock-down
For the specific knock-down of HSD10 in Xenopus embryos the following two Mo antisense oligonucleotides (Gene Tools) were used: Mo5′UTR 5′tgaccaaccaattgcagtcattcat3′ and MoATG 5′ccttcaggttcctcacatgcgccat3′ binding the 5′UTR and the region of the start codon of HSD10 mRNA, respectively. Control Morpholino MoCo 5′cctcttacctcagttacaatttata3′ was used as an inert control.
Gene overexpression
The following DNA constructs were used in rescue experiments: pT-Rex-DEST30 containing cDNA of hHSD10 and the mutations R130C, D86G and Q165H. Constructs were obtained by cloning using Gateway Technology (Invitrogen). Other constructs were pCS2+_myc containing xHSD10 and xHSD10 including the 5′UTR.
Microinjection
Embryos were cultured in 1× MBSH (88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 0.82 mM MgSO4 × 7H2O, 0.41 mM CaCl2 × 2H2O, 0.33 mM Ca(NO3) × 4H2O, 10 mM HEPES, pH 7.5) for microinjection and injected with 5 nl of diluted Mo or DNA into each blastomere.
Protein extract
Xenopus embryos were lysed in RIPA buffer (20 mM Tris/HCl pH 7.5, 0.1% NP-40, 10% glycerol, 10% protease inhibitor tablets), cell debris was pelleted for 5 min at 2300 × g, and the protein fraction was mixed well with 1 volume Freon (Fluka). The protein suspension was obtained by centrifugation for 5 min at 2300 × g. Protein samples were analysed by SDS-PAGE and Western Blot using 9E10 anti-Myc antibody (Merck) to detect overexpressed xHSD10_myc protein.
In situ hybridization
Embryos of different stages were fixed in MEMFA (100 mM MOPS, 2 mM EGTA, 1 mM MgSO4, 3.7% formaldehyde), and stored in methanol at −20 °C. Whole-mount in situ hybridization was performed according to Harland, 1991. DIG-labelled antisense RNA was synthesized using Digoxygenin RNA labelling Kit (Roche) with pCS2+/otx2 and pBluescriptSK+/xBF1 digested with BamHI and XbaI as a template.
TUNEL staining
Embryos were fixed in MEMFA for terminal deoxynucleotidyl TUNEL analysis and stored in methanol at −20°C. The whole-mount staining protocol was previously described (Hensey & Gautier, 1997). Statistical evaluation of the significance of the rescue experiment was achieved by comparing the differences in the amount of apoptosis spots on the injected side of the embryo with the uninjected side within an one way analysis of variance and Scheffe test was used for post hoc pairwise group comparisons. Differences were counted as significant if the p-values were lower than 5% adjusted for multiple testing within one experiment.
Cell culture methods
Fibroblasts from MHBDD patients and control fibroblasts were cultivated in DMEM ReadyMix (Paa Laboratories) at 37°C and 5% CO2. Mitochondrial staining in patient fibroblasts was achieved within 20 min with 300 nM Mitotracker Green FM (Invitrogen) on cells fixed with 3.7% formaldehyde on coverslips. Mitochondria were visualized after mounting in Mowiol (Merck) on a Perkin Elmer spinning disc confocal ERS-FRET on Nikon TE2000 inverted microscope.
The study was approved by the Ethical Committee of the Medical Faculty of Heidelberg University.
Acknowledgments
Dr. Willy Lehnert, University Children's Hospital Freiburg, Germany, carried out the initial metabolic characterization in the patient of case 1. The clinical characterization of patients with MHBD deficiency was kindly supported by the Reimann-Dubbers Foundation, Heidelberg. The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust. The project was supported by the Oxford NIHR Musculoskeletal Biomedical Research Unit. We thank Afsaneh Majdazari for her help with the preparation of murine ganglia.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
All authors were involved in ongoing discussions about the study, read and/or corrected drafts of the manuscript and agreed with the final manuscript.
Katharina Rauschenberger carried out the extensive experimental work in Xenopus embryos as well as other experiments, liaised with other contributors, and co-wrote the manuscript.
Katja Schöler, Kathryn L. Kavanagh, Naeem Shafqat and Udo Oppermann carried out and/or supervised functional analyses of overexpressed human mutations and HSD10 crystallization.
Jörn Oliver Sass and Ronald J. A. Wanders carried out metabolite and enzyme analyses for diagnosis or confirmation of diagnosis in patients with HSD10 deficiency.
Sven Sauer, Jürgen G. Okun and Stefan Kölker analysed mitochondrial function in homogenized Xenopus animal caps and human fibroblasts.
Zdenka Djuric, David Stern, Peter Nawroth and Angelika Bierhaus carried out or supervised siRNA studies in murine cells and/or were involved in the generation of Hsd17b10 knock-out mice.
Cordula Rumig, Günter Hämmerling and Bernd Arnold generated and bred the Hsd17b10 conditional knock-out mice.
Nicole I. Wolf, Julian H. P. Shield and Udo Wendel diagnosed and/or clinically characterized the patients with HSD10 deficiency.
Heinz Schwarz supervised and carried out electron microscopy.
Christine Fischer carried out statistical analyses.
Beate Grziwa, Heiko Runz and Astrid Nümann carried out or assisted in different experiments.
Georg F. Hoffmann and Claus R. Bartram contributed to the design of the study and the interpretation of the data.
Herbert Steinbeisser co-initiated and supervised the experimental work in Xenopus embryos, was involved in most experimental aspects of the study and co-wrote the manuscript.
Johannes Zschocke initiated and coordinated the study, was involved in all clinical and experimental aspects of the study, co-wrote the manuscript and serves as guarantor.
For more information
OMIM, Online Mendelian Inheritance in Man:
HSD17B10
http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=300256
NCBI, Entrez Gene:
HSD17B10
http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene&cmd=Retrieve&dopt=full_report&list_uids=3028
Ensembl genome browser:
HSD17B10:
http://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000072506
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Chen JX, Yan SD. Amyloid-β-induced mitochondrial dysfunction. J Alzheimers Dis. 2007;12:177–184. doi: 10.3233/jad-2007-12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constien R, Forde A, Liliensiek B, Gröne HJ, Nawroth P, Hämmerling G, Arnold B. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001;30:36–44. doi: 10.1002/gene.1030. [DOI] [PubMed] [Google Scholar]
- Harland RM. In situ hybridization: an improved whole-mount method for Xenopus embryos. Methods Cell Biol. 1991;36:685–695. doi: 10.1016/s0091-679x(08)60307-6. [DOI] [PubMed] [Google Scholar]
- Hensey C, Gautier J. A developmental timer that regulates apoptosis at the onset of gastrulation. Mech Dev. 1997;69:183–195. doi: 10.1016/s0925-4773(97)00191-3. [DOI] [PubMed] [Google Scholar]
- Lenski C, Kooy RF, Reyniers E, Loessner D, Wanders RJ, Winnepenninckx B, Hellebrand H, Engert S, Schwartz CE, Meindl A, et al. The reduced expression of the HADH2 protein causes X-linked mental retardation, choreoathetosis, and abnormal behavior. Am J Hum Genet. 2007;80:372–377. doi: 10.1086/511527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal Table of Xenopus laevis. 2nd edition. Amsterdam: North Holland Publishing, Co; 1967. [Google Scholar]
- Ofman R, Ruiter JP, Feenstra M, Duran M, Poll-The BT, Zschocke J, Ensenauer R, Lehnert W, Sass JO, Sperl W, et al. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency is caused by mutations in the HADH2 gene. Am J Hum Genet. 2003;72:1300–1307. doi: 10.1086/375116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafqat N, Marschall HU, Filling C, Nordling E, Wu XQ, Björk L, Thyberg J, Mårtensson E, Salim S, Jörnvall H, et al. Expanded substrate screenings of human and Drosophila type 10 17beta-hydroxysteroid dehydrogenases (HSDs) reveal multiple specificities in bile acid and steroid hormone metabolism: characterization of multifunctional 3alpha/7alpha/7beta/17beta/20beta/21-HSD. Biochem J. 2003;376:49–60. doi: 10.1042/BJ20030877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Duong CV, Pape M, Geissen M, Burbach G, Deller T, Gascan H, Otto C, Parlato R, Schutz G, et al. Target-dependent specification of the neurotransmitter phenotype: cholinergic differentiation of sympathetic neurons is mediated in vivo by gp130 signaling. Development. 2006;133:141–150. doi: 10.1242/dev.02189. [DOI] [PubMed] [Google Scholar]
- Tieu K, Perier C, Vila M, Caspersen C, Zhang HP, Teismann P, Jackson-Lewis V, Stern DM, Yan SD, Przedborski S. L-3-hydroxyacyl-CoA dehydrogenase II protects in a model of Parkinson's disease. Ann Neurol. 2004;56:51–60. doi: 10.1002/ana.20133. [DOI] [PubMed] [Google Scholar]
- Venable JH, Coggeshall R. A simplified lead citrate stain for use in electron microscopy. J Cell Biol. 1965;25:407–440. doi: 10.1083/jcb.25.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, et al. An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer's disease. Nature. 1997;389:689–695. doi: 10.1038/39522. [DOI] [PubMed] [Google Scholar]
- Yan SD, Roher A, Chaney M, Zlokovic B, Schmidt AM, Stern D. Cellular cofactors potentiating induction of stress and cytotoxicity by amyloid beta-peptide. Biochim Biophys Acta. 2000;1502:145–157. doi: 10.1016/s0925-4439(00)00041-7. [DOI] [PubMed] [Google Scholar]
- Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschocke J, Ruiter JP, Brand J, Lindner M, Hoffmann GF, Wanders RJ, Mayatepek E. Progressive infantile neurodegeneration caused by 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency: a novel inborn error of branched-chain fatty acid and isoleucine metabolism. Pediatr Res. 2000;48:852–855. doi: 10.1203/00006450-200012000-00025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.