

Abstract
It is widely thought that demyelination contributes to the degeneration of axons and, in combination with acute inflammatory injury, is responsible for progressive axonal loss and persistent clinical disability in inflammatory demyelinating disease. In this study we sought to characterize the relationship between demyelination, inflammation and axonal transport changes using a Plp1-transgenic mouse model of Pelizaeus-Merzbacher disease. In the optic pathway of this non-immune mediated model of demyelination, myelin loss progresses from the optic nerve head towards the brain, over a period of months. Axonal transport is functionally perturbed at sites associated with local inflammation and ‘damaged’ myelin. Surprisingly, where demyelination is complete, naked axons appear well preserved despite a significant reduction of axonal transport. Our results suggest that neuroinflammation and/or oligodendrocyte dysfunction are more deleterious for axonal health than demyelination per se, at least in the short term.
Keywords: axonal transport, multiple sclerosis, myelin, proteolipid protein, visual system
INTRODUCTION
Multiple sclerosis (MS) is an inflammatory demyelinating disorder in which the previously formed myelin sheath breaks down. MS is considered the archetypal myelin disorder and much current research focuses on the associated axonal loss; the major cause of persistent disability (De Stefano et al, 1998; Scolding & Franklin, 1998; Trapp & Nave, 2008). Axonal damage in MS occurs in both the acute-inflammatory and late chronic phases of the disease. The causes are not known, but inflammation and loss of trophic support due to chronic demyelination, respectively, could contribute (Bjartmar et al, 2003; Scolding & Franklin, 1998).
Axonal transport impairment has been implicated in a number of neurodegenerative disorders such as MS (Ferguson et al, 1997) or spastic paraplegia type 2 (Edgar et al, 2004) in which the primary defect is in the oligodendroglial proteolipid protein (PLP1) gene. The aim of the present study was to determine to what extent demyelination impairs axonal transport and whether perturbed transport correlates with the chronic absence of myelin or the acute inflammatory phase of demyelination, or both. We selected as our model the optic pathway of a Plp1-transgenic mouse (line #72) defined by about twofold overexpression of PLP/DM20 (Anderson et al, 1999; Readhead et al, 1994). In the optic nerve, active demyelination with associated inflammation is succeeded by complete demyelination in the absence of inflammation. For comparison, we also examined the dysmyelinating Plp1-mutant rumpshaker (Schneider et al, 1992). In rumpshaker, formation of the myelin sheath is perturbed leading to a reduced amount of myelin, but there is no evidence of demyelination and the inflammatory response is moderate. Both the Plp1-transgenic mouse and rumpshaker are models of Pelizaeus-Merzbacher disease (PMD), an X-linked leukodystrophy most commonly caused by duplication of the PLP1 gene (model: Plp1-transgenic mouse), but also by point mutations (model: rumpshaker).
Here we show that in the Plp1-transgenic mouse, axonal transport impairment occurs at sites where active demyelination is taking place and also at sites where axons remain ensheathed by mutant oligodendrocytes. In contrast, axons in the completely demyelinated region of the nerve appear structurally and functionally preserved. These data suggest that inflammation and/or oligodendrocyte dysfunction in the Plp1-transgenic mouse cause axonal pathology while complete demyelination facilitates axonal survival under stress, at least in the short term.
RESULTS
The optic pathway of Plp1-transgenic mice undergoes temporo-spatially determined demyelination
The optic pathway of the Plp1-transgenic mouse exhibits moderate dysmyelination followed by progressive demyelination that shows both temporal and spatial evolution. By examining the mid point of the pathway, we showed that myelination commenced on schedule and 44 and 60% of axons were myelinated at P20 and P40, respectively; compared to 80 and 96% in the wild type (WT). However, demyelination led to a complete absence of myelin by P120 (Fig 1A). These profiles were mirrored by myelin volume (Mv) measurements (Fig 1B). Myelin sheaths in the transgenic mice were of normal structure and periodicity, but were thinner than in WT (g-ratio of 0.766 ± 0.007 in mutant vs. 0.732 ± 0.003 in controls at P60; p < 0.05). Importantly, there was no significant reduction in absolute axonal number at any age studied. At P120, axon number in the Plp1-transgenic mice was 49,683 (±964 SEM) versus 48,768 (±910 SEM) in WT, and axon diameter frequency distributions were not different from WT (Fig 1C). P120 is the oldest age examined, because the Plp1-transgenic mice rarely live beyond this age.
Figure 1. Demyelination progresses in a rostral to caudal direction with increasing age in optic pathway of Plp1-transgenic mice.
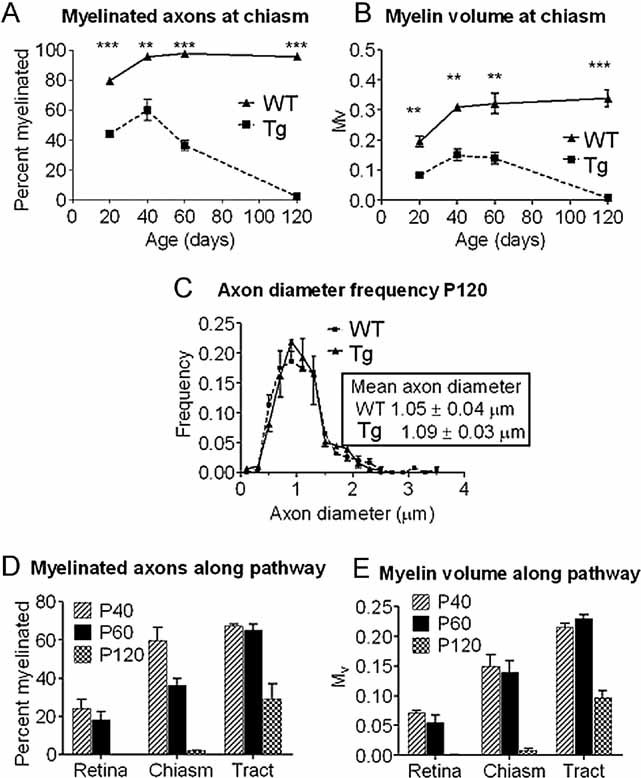
- A, B. The percentage of myelinated axons (A) and the relative volume of myelin (B) at the chiasmal end of the optic nerve in wild type (WT) and Plp1-transgenic (Tg) mice between P20 and P120.
- C. Axon diameter frequency distribution at the chiasmal end of the optic nerve of WT and Plp1-transgenic mice aged P120. Axonal diameter distributions are not significantly different.
- D, E. The percentage of myelinated axons (D) and the relative volume of myelin (E) along the optic pathway of Plp1-transgenic mice at P40, P60 and P120 showing the temporal and spatial progression of demyelination.
To determine the spatial pattern of demyelination we quantified the proportion of myelinated fibres and Mv at the retinal and chiasmal ends of the optic nerve and at the mid optic tract of transgenic mice. The data (Fig 1D, E) showed a distinct pattern of demyelination progressing from the retinal end towards the brain. At P30, the retinal end of the nerve had 40% of axons myelinated (data not shown). At P120 the pathway shows two distinct zones; a proximal (retinal) completely demyelinated region and a distal actively demyelinating portion. Electron microscopy of the proximal nerve revealed numerous naked axons that appeared morphologically normal, with interspersed astrocytic processes (Fig 2A and Supporting Information Fig S1A). Microglia/macrophages were infrequent. In contrast, the distal (chiasmal) end of the nerve, although largely demyelinated, contained numerous active microglia/macrophages (Fig 2B, C). The optic tract was similar, but with a greater proportion of myelinated axons (Fig 2D and Supporting Information Fig S1B). In both the chiasmal end of the nerve and the tract, some axons showed disruption of the cytoskeleton and/or accumulations of membranous organelles (Fig 2B–D).
Figure 2. Distribution of myelin and axonal pathology along the optic pathway of Plp1-transgenic mice aged P120.
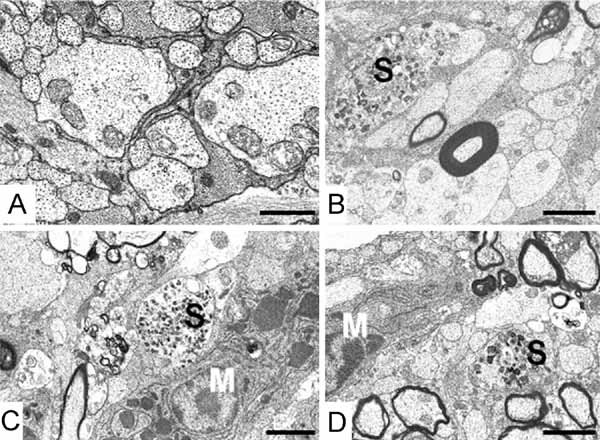
- A. Retinal end of optic nerve: all myelin has been removed leaving naked axons, which appear morphologically normal and are interspersed with astrocyte processes. Bar: 1 µm.
- B, C. Chiasmal end of optic nerve: demyelination is on-going although most axons have lost their sheaths. Microglia (M) and axonal spheroids (S) are present. Bar: 2 µm.
- D. Optic tract: demyelination is present but not as advanced as at the chiasm. Microglia and axonal spheroids are evident. Bar: 3 µm.
Electrical conduction in the dysmyelinated and demyelinated optic nerve
To assess conduction in dysmyelinated and in demyelinated nerves, stimulus-evoked compound action potentials (CAP) were recorded from P40 and P120 WT and Plp1-transgenic mice (Fig 3A, B). In WT mice, the CAP has a stereotyped triple-peaked profile (Brown et al, 2003; Stys et al, 1998). In transgenic mice the duration of the CAP was longer (6 ms in mutants vs. 3 ms in controls) signifying a decrease in conduction velocity, consistent with hypomyelination. However, the CAP could still be resolved into three peaks. The CAP recorded from P120 transgenic mice differed from P40 mice in that the amplitudes of the peaks were smaller. In the absence of axonal loss (see above), reduced amplitudes were most likely caused by conduction blocks along demyelinated axons.
Figure 3. Electrophysiological recordings from WT and Plp1-transgenic optic nerves.
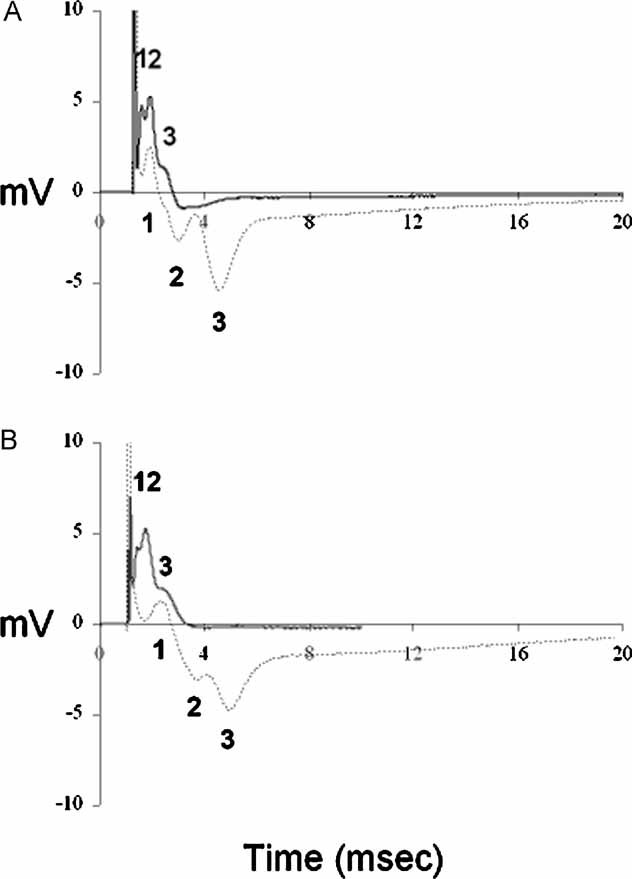
- Representative recordings from P40 WT (——) and Plp1-transgenic (——) mice. In WT mice, the CAP has the stereotyped triple-peaked profile that reflects populations of axons with different conduction velocities. The CAP recorded from Plp1-transgenic mice resolved into three peaks, but the duration of the CAP was longer than that of the control (6 ms vs. 3 ms) signifying a decrease in the conduction velocity.
- Representative recordings from P120 WT (——) and Plp1-transgenic (——) mice. The CAP recorded from P120 Plp1-transgenic mice was similar to that of the P40 transgenic animals, but the peaks were less well defined and their amplitudes were smaller. A total of 10 WT and 10 Plp1-transgenic nerves were examined.
The inflammatory response is associated with active demyelination
Since active demyelination is associated with inflammation (Ip et al, 2006), we used the pan-leukocyte marker CD45 to identify inflammatory cells in the Plp-transgenic mouse. Whereas in the more distal portions of the P120 optic nerves the numbers of CD45+ cells per optic nerve section were elevated >10-fold compared with WT; the completely demyelinated retinal portion showed no significant increase (Fig 4A). At P40, prior to overt demyelination the cell numbers were strikingly lower than at P120, although elevated compared with WT, probably reflecting the underlying dysmyelination (Fig 4B). Direct correlation of active demyelination and inflammation was achieved by immunostaining for myelin basic protein (MBP) and CD45 (Fig 4C, E).
Figure 4. Inflammatory cell numbers change in relation to chronic or active demyelination.
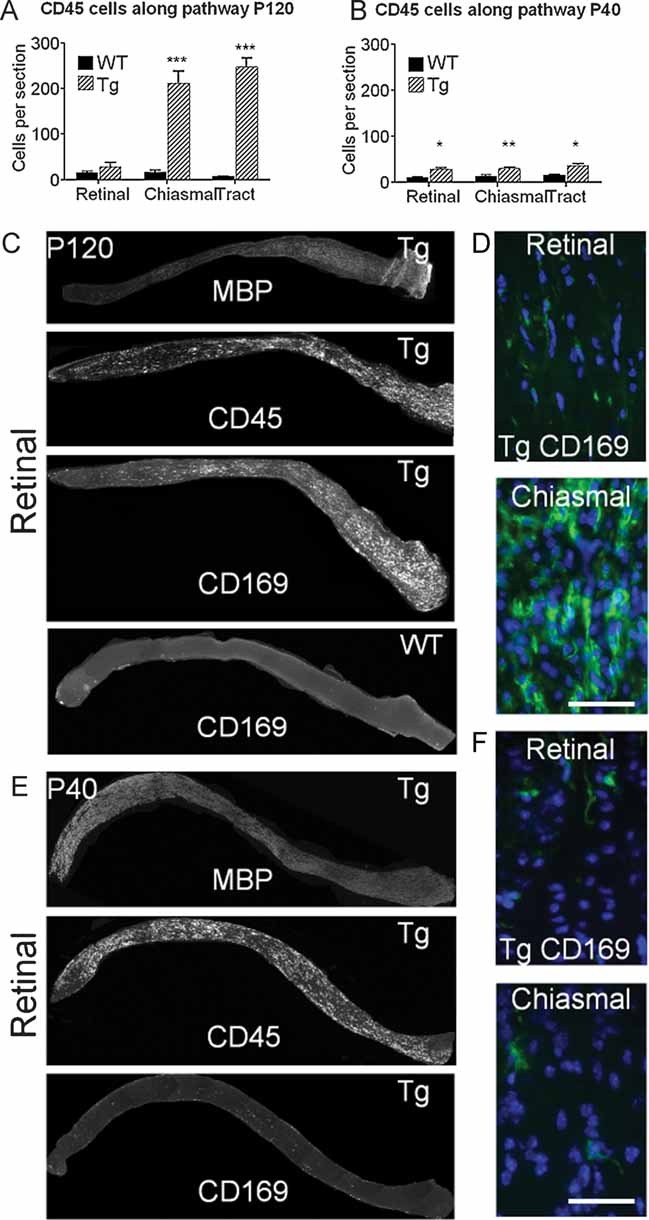
- CD45+ cell numbers along the optic pathway of WT and Plp1-transgenic (Tg) mice aged P120. Whereas the demyelinated retinal region has no increase in these cells there is a massive increase in the actively demyelinating distal regions of the nerve.
- At P40, prior to overt demyelination, the CD45+ cell numbers are similar along the entire pathway. The increased number, relative to WT, reflects the developmental dysmyelination.
- Longitudinal sections of optic nerve from the retinal end (left) to the chiasm from a Plp1-transgenic (Tg) mouse aged P120 immunostained for MBP, CD45 and CD169, showing the gradient in myelin and macrophages between the two ends of the optic nerve. Only CD169 is shown for the WT mouse.
- Enlarged images from the retinal and chiasmal ends of the optic nerve from the Plp1-transgenic mouse illustrate the marked difference in the densities of CD169+ cells between the two sites.
- Longitudinal sections of optic nerve, as for C, from a Plp1-transgenic (#72) aged P40. MBP immunoreactivity shows that the entire length of the nerve (except the unmyelinated lamina cibrossa) is myelinated at this age. Although CD45+ cells are present along the nerve, very few CD169+ cells are evident.
- Enlarged images from the retinal and chiasmal ends of the optic nerve from the Plp1-transgenic (#72) mouse illustrating the scarcity of CD169+ cells prior to overt demyelination. Bar = 50 µm.
Using the T-cell marker CD3 we identified approximately 10–20 cells per 10 µm thick longitudinal section (data not shown). B-cells were rarely observed. We then focused on the microglial/macrophage population using CD169 (sialoadhesin), since CD169 expressing cells contribute to pathogenesis in another Plp-transgenic line (line #66; Ip et al, 2006; Readhead et al, 1994). Very few CD169+ cells were present in the demyelinated retinal end of the P120 optic nerve, in contrast to the abundant population at the chiasmal end (Fig 4C, D). CD169+ cells were absent in WT (Fig 4C) and infrequent in P40 optic nerve, prior to overt demyelination (Fig 4E, F). In fact, the pattern of expression of CD169 closely mirrored the progression of active demyelination at all ages examined (Supporting Information Fig S2). Note that in Fig 4E and Supporting Information Fig S2, the variation in the intensity of MBP staining probably reflects improved antibody penetration into the demyelinating tissue.
Impaired retrograde axonal transport correlates with active inflammation
The occurrence of axonal spheroids (Fig 2B–D) suggests impaired axonal transport, as reported previously in the Plp1 gene knockout mouse (Edgar et al, 2004). To examine retrograde transport, we injected fluorescein isothiocyanate (FITC)-labelled cholera toxin B subunit (CtB) into the right rostral colliculus of P40 or P120 mice and measured fluorescence signal in the contralateral retinal ganglion cells (RGC) after 18 h. At P40 there was no significant difference in either CtB-labelled area or mean signal intensity between transgenic and WT mice (Fig 5A). At P120, mean signal intensity in RGCs was significantly lower in transgenic mice than in WT, whereas the CtB-labelled area (an indication of the number of labelled RGC) was similar (Fig 5A). Since axonal counts are not reduced in the mutants, this suggests that the average amount of material transported along the length of an individual axon within 18 h is reduced in the P120 mutant nerves. To visualize perturbations of retrograde transport directly, we examined longitudinal sections of the left optic nerve of Plp1-transgenic animals. At P40, occasional focal accumulations of CtB in axons occurred more often in the retinal portion of the nerve, where demyelination is commencing, than in the chiasmal portion. However, the difference between the two regions was not significant (Fig 5B). Interestingly, at P120, numbers of focal accumulations of tracer were significantly greater at the inflamed chiasmal end of the nerve compared to the completely demyelinated retinal end (Fig 5B, C).
Figure 5. Impaired retrograde axonal transport is associated with active inflammatory demyelination.
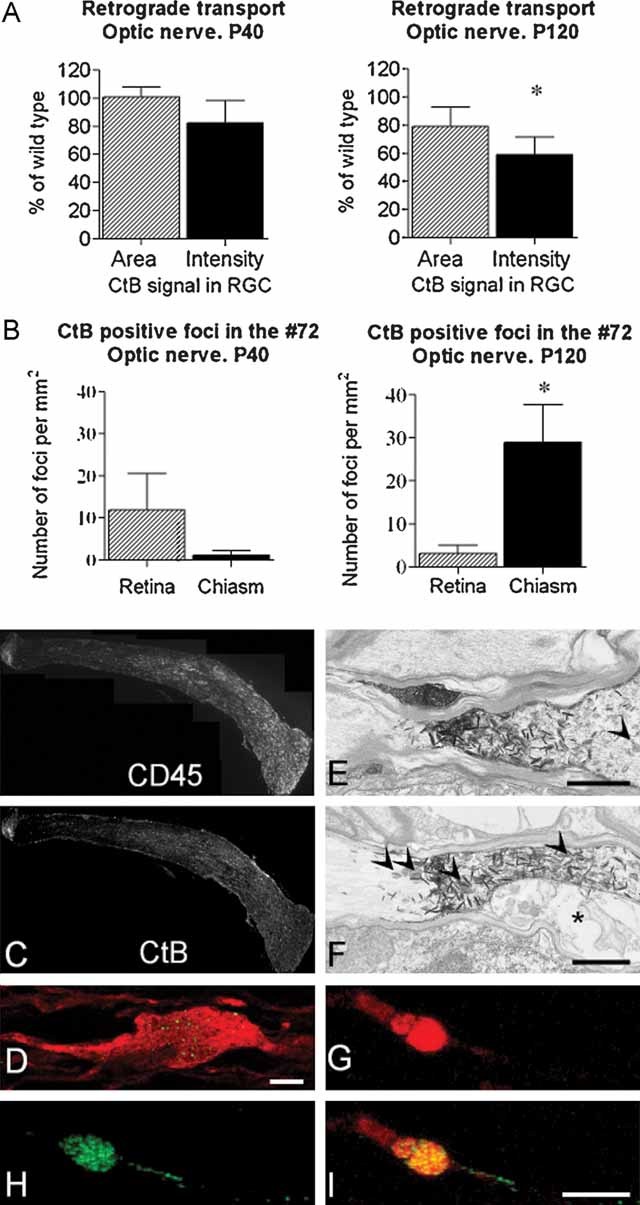
- A. Following injection of CtB into the rostral colliculus, the mean signal intensity in the retinal ganglion cells (RGC) and the area of retina occupied by labelled cells was determined in WT and transgenic littermates aged P40 and P120. At P40, there is no significant difference between transgenic and WT. However, the signal intensity is significantly lower in the P120 transgenic group, when there is active, inflammatory demyelination. As the axon counts in the optic nerves of the two genotypes are also similar the data suggest that the reduced signal is due to impaired transport rather than axonal loss.
- B. Quantification of CtB foci in retinal or chiasmal portions of the P40 or P120 nerve. At P40, only a few foci are observed and they tend to be more prevalent in the retinal half of the nerve. However, the difference between retinal and chiasmal halves is not significant. At P120, sites of transport stasis are located predominantly in the distal portion of the nerve.
- C. Longitudinal sections of nerve (retinal end left) from a Plp1-transgenic mouse aged P120 immunostained for CD45 and showing CtB positive foci. The sections show the gradient in macrophages and CtB between the two ends of the optic nerve. Lipofucsin is present in these nerves, particularly in the chronically demyelinated region, and is indistinguishable from CtB in these images.
- D. Single z step through an SMI32-labelled (red) axon in which FITC–CtB (green) appears punctuate, suggesting that it may associate with endosomes and multivesicular bodies. Bar: D = 10 µm.
- E, F. Electronmicrographs of horseradish peroxidase (HRP)-conjugated CtB, labelled with TMB, in axons in the optic tract of a P120 mouse. TMB appears as electron dense ‘spikes’. The axons remain patent. Axons in these images remain myelinated, however in (F), the inner tongue (asterisk) of the oligodendrocyte is swollen. Arrowheads indicate axonal organelles. Bar: E,F = 1 µm.
- G, H, I. Eighteen hours after TRITC–CtB (red) and FITC–CtB (green) were simultaneously injected into the eye and the rostral colliculus, respectively, confocal microscopy showed that many axonal swellings contained both tracers. This suggests that the swellings are associated with defects in both retrograde and anterograde axonal transport, and also indicates that the associated axon remains intact. Bar: I = 5 µm.
Within axons, CtB accumulations often showed a punctate appearance (Fig 5D), suggesting an association with endosomes or multivesicular bodies (MVBs). Electron microscopy following 3,3/5,5/-tetramethylbenzidine (TMB) labelling of peroxidase-conjugated CtB showed that accumulations occurred at sites in which axons appeared intact, and in some cases, myelinated (Fig 5E, F).
Simultaneous injection of FITC-labelled CtB into the right colliculus and tetramethyl rhodamine iso-thiocyanite (TRITC)-labelled CtB into the left eye, led to the co-localization of the tracers in some axonal swellings (Fig 5G–I and Supporting Information Fig S3), demonstrating that these foci result from the impairment of both anterograde and retrograde transport and indicating that the associated axons are still intact.
Inflammation correlates with transport defects
The microglial/macrophage population in the transgenic mouse optic nerve revealed a biphasic temporal course. There was a low level of inherent microglia activation associated with dysmyelination, followed by a massive activation in areas of overt demyelination. To compare transport defects in solely dysmyelinated optic nerves, we turned to the Plp1 mutant rumpshaker mouse (Schneider et al, 1992) that exhibits no demyelination. The level of hypomyelination in P40 rumpshaker mice was similar to that of age-matched Plp1-transgenic mice. However, CD169+ cells appeared to be slightly more frequent in rumpshaker (Fig 6A). Only a small number of CtB foci, indicating local impairment of retrograde axonal transport (Fig 6B), were present in rumpshaker and in contrast to Plp1-transgenic mice at age P120, these foci occurred in similar numbers in both retinal and chiasmal portions.
Figure 6. Relatively few sites of axonal transport stasis are associated with diffuse, low level inflammation.
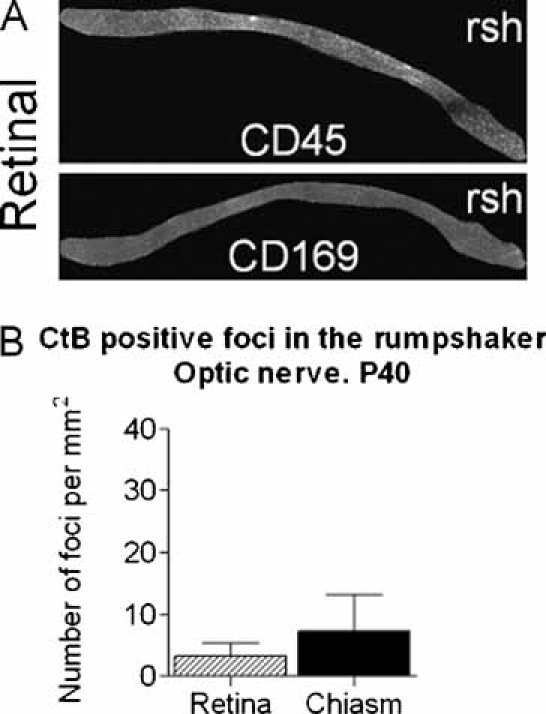
- Longitudinal sections of optic nerve (retinal end left) from the dysmyelinating rumpshaker mouse aged P40, immunostained for CD45 and CD169 (sialoadhesin). There is no gradient in the distribution of microglia/macrophages along the optic nerve and activated, sialoadhesin positive cells are present in low numbers compared to the P120 Plp1-transgenic nerve.
- Eighteen hours after injection of CtB into the rostral colliculus of P40 rumpshaker mouse, a relatively small number of CtB positive foci are present at similar densities in retinal and chiasmal portions of the nerve.
DISCUSSION
Axonal loss is widely accepted as the main determinant of neurological disability in MS (De Stefano et al, 1998). Axonal degeneration correlates with inflammatory demyelination in MS (Ferguson et al, 1997; Trapp et al, 1998), in EAE (Papadopoulos et al, 2006) and in aged mice in which demyelination is induced with cuprizone (Irvine & Blakemore, 2006). Whether axonal injury persists in MS patients once inflammation has resolved has been debated (Frischer et al, 2009; Trapp et al, 1998). To determine the relationship between axonal changes and myelin-related pathology we studied two different models of myelin disease. The Plp1-transgenic mouse optic nerve shows a temporo-spatially separated active demyelination with robust microglial/macrophage activation and chronic hypomyelination (due to dys- and demyelination) with low-grade inflammation. The C3H/101 rumpshaker optic nerve exhibits mild dysmyelination associated with modest cellular activation. Sites of axonal transport stasis were most prevalent at regions of active demyelination and inflammation, suggesting that the presence of inflammatory cells and/or damaged myelin are key factors.
Rostral to caudal progression of demyelination
The rostral to caudal progression of demyelination in the Plp1-transgenic mouse mirrors that of developmental myelination which commences at the optic nerve head approximately 7 days before it does so at the chiasm (Foran & Peterson, 1992). However, it seems unlikely that the small age difference between retinal and chiasmal oligodendrocytes explains the pattern of demyelination, which progresses over weeks in the Plp1-transgenic mouse. One possibility is that remyelination occurs concurrently with demyelination in the chiasmal portion of the nerve, but is less efficient in the retinal portion, manifesting as an apparent gradient of demyelination.
Surprisingly, axonal diameter frequency distributions at the chiasmal portion of the P120 nerve were not altered compared to WT. In the retinal portion of the nerve, where complete demyelination occurred earlier, a greater proportion of axons were less than 0.6 µm in diameter in the Plp-transgenic mouse compared to the WT (average 34.0% vs. 17.2%, respectively); however the overall frequency distributions were not significantly different. Whereas peripheral nervous system (PNS) axons and large diameter spinal cord axons are profoundly influenced by myelination (De Waegh et al, 1992; Tanaka et al, 2009), the subtle differences we observed may be due to the fact that the size of small diameter optic nerve axons is only minimally increased by myelination (Kirkpatrick et al, 2001; Perge et al, 2009).
Sites of transport impairment and swelling do not necessarily represent axonal transection
Some markers of axonal damage, such as focal accumulations of APP, represent sites of axonal transport stasis but may not necessarily indicate that the associated axon is transected (Ferguson et al, 1997). Thus, the damage may be reversible. For example, in EAE models, transient accumulations of APP have been correlated with iNOS expression by perivascular macrophages (Aboul-Enein et al, 2006) or with co-transfer of antibodies to neurofascin (Mathey et al, 2007). In these models, the extent of APP immunoreactivity correlates with clinical symptoms and subsides during remission, providing indirect evidence that the affected axons do not degenerate. Here, we took advantage of the anatomy of the optic pathway to show directly, using simultaneous injection of CtB into the eye and the colliculus that spheroids belong to intact axons in the demyelinating Plp1-transgenic mouse.
Similarly, in a mouse model of Alzheimer's disease, axons remain continuous along their entire length despite containing large focal swellings, indicative of a partial block of axonal transport (Adalbert et al, 2009). Thus, it appears that axonal transport has an in-built overcapacity, such that most axons can survive limited transport defects for some time.
Effectors of axonal transport stasis
Sites of transport impairment were noted in the chiasmal and optic tract portions of the visual pathway at P120, which at this age represent sites of active, inflammatory demyelination. Spheroids were noted in both myelinated internodes and in demyelinated axonal segments. In contrast, focal sites of transport stasis were much rarer in completely demyelinated regions where axons appeared structurally intact and were able, in the case of some axons, to conduct electrical impulses.
Activated microglia/macrophages probably contribute to axonal transport stasis at sites of active demyelination. For example, microglia/macrophage ‘activation’ has been associated with low levels of acute axonal injury in pre-demyelinating lesions in rats receiving focal injection of lipopolysaccharide (Marik et al, 2007), and inflammatory factors (e.g. nitric oxide and tumour-necrosis factor (TNF-α)) produced by this population, can possibly contribute directly to the dysregulation of molecular motors (De Vos et al, 2000; Stagi et al, 2006).
Alternatively, these effects could be indirect, for example triggered by oligodendrocytes that appear physically stressed in an inflammatory milieu (see Fig 2 Butt et al, (2004)). In this context, we note that axonal transport stasis can precede microglial activation and can occur in the absence of demyelination when axons are ensheathed by dysfunctional oligodendrocytes, such as in the myelinated Plp1 and Cnp1 knockout mice (Edgar et al, 2004, 2009; Lappe-Siefke et al, 2003). Thus, transport impairment in the Plp-transgenic mouse (which was sometimes observed within myelinated internodes) most likely indicates that axons are ensheathed by dysfunctional oligodendrocytes (Edgar & Nave, 2009; Nave & Trapp, 2008).
Good evidence for a functional link between demyelination, inflammation and axonal changes is provided by a comparison with shiverer mice, which lack compact myelin in the optic nerve. Indeed, shiverer mice show no signs of axonal loss/swelling, unless confronted additionally with the lack of PLP/DM20 from mutant oligodendrocytes (see Fig 1E (Griffiths et al, 1998)). This confirms that shiverer axons are in principle at risk for a similar axonal breakdown, but that it requires a ‘second hit’ on top of the lack of myelin. This could be either inflammation or deficiency of a critical oligodendroglial molecule. Taking these data together, we propose a model in which both neuroinflammation and oligodendrocyte dysfunction provide the highest risk for axonal loss, with demyelination surprisingly improving the chance of axon survival, at least in the short term.
The paper explained
PROBLEM
Axonal loss is widely accepted as the major cause of permanent neurological disability in multiple sclerosis (MS), an inflammatory demyelinating disorder of the central nervous system. However, the relationship between demyelination, inflammation and axonal damage in neuroinflammatory disorders is not yet fully understood.
RESULT
Using a mouse model of the leukodystrophy, Pelizaeus-Merzbacher disease, we show that fast transport within optic nerve axons is focally impaired at sites of damage, where active, inflammatory demyelination takes place and also at sites where axons remain ensheathed by mutant oligodendrocytes. Most interestingly, where demyelination is complete, axons appear preserved.
IMPACT
Our results challenge the field to re-think the mechanisms of glial support to axons as they suggest that, at least in the short-term, neuroinflammation and associated changes are more deleterious for axonal health than overt demyelination per se.
MATERIALS AND METHODS
Mice
The mice used in this study (line #72) carry a cassette harbouring three copies of a Plp1 genomic transgene (Readhead et al, 1994). These were backcrossed for more than six generations to the C57BL/6N (Charles River) background. WT and homozygous mice were examined at P20, P30, P40, P60 and P120. Only males were used for quantitative analysis, except for the electrophysiological studies, when only females were used. See Supporting Information for genotyping methods. For axonal transport assays (see below), homozygous transgenic mice were compared to age and sex-matched littermates. For all other analyses, comparisons were between age and sex-matched littermates or closely related individuals from the same in-bred colony.
Rumpshaker (rsh/y) and WT (+/y) male offspring were also examined. These were distinguished on the basis of the neurological phenotype of rumpshaker. All animal studies were approved by the Ethical Committee of the University of Glasgow and licensed by the UK Home Office.
Tissue collection and preparation
For electron microscopy, tissues were prepared as described previously (Griffiths et al, 1981). For immunohistochemistry, frozen tissue sections were used. Details of the fixatives employed can be found in Supporting Information.
Antibodies and immunohistochemistry
Antibodies to CD3 (1:100), CD45 (1:600), CD169 (sialoadhesin; 1:100), B-cells (CD45R/B220; 1:100) and MBP (1:500) were obtained from Serotec Ltd (Oxford, UK) SMI-32, which recognizes non-phosphorylated neurofilaments (1:1000) was obtained from Sternberger Monoclonals, Inc. (Lutherville, MD). Secondary antibodies were conjugated with FITC or Texas Red (Cambridge Biosciences). In all cases (except for B cells) sections were permeabilized with methanol (−20 °C), blocked in 10% goat serum in PBS and incubated overnight in primary antibody in blocking solution. Secondary antibodies in blocking solution were applied for 1 h at room temperature (RT). For the B-cell marker, frozen sections were blocked and the primary antibody was applied for 1 h at RT. Sections were then fixed in 4% paraformaldehyde and the secondary antibody applied, as above.
Digital images
Fluorescence images were taken using an Olympus IX70 microscope, QIcam Fast digital camera (QImaging Surrey, BC, Canada) and Image-Pro Plus 6 software (Media Cybernetics, Silver Spring, MD). Images were processed using Adobe Photoshop. No significant electronic manipulations were made except to images of longitudinal optic nerve sections in which tissue debris around the nerve was digitally removed.
Collicular injections for retrograde transport
To study retrograde axonal transport, WT and Plp1-transgenic homozygous littermates at P40 and P120, and WT and rumpshaker littermates at P40, were used. Fluorescein-conjugated CtB subunit was injected into the right rostral colliculus and its accumulation in the retina was measured 18 h later (Edgar et al, 2004). The total area occupied by fluorescent signal and the mean density of the signal were determined using Image-Pro Plus 6 software; data are expressed as area of fluorescence per mm2 of retina and average mean density (intensity) of signal.
Intraocular injections for anterograde transport
Some mice were simultaneously injected with FITC–CtB and with 1 µl of 1% TRITC–CtB in physiological saline (Invitrogen Ltd., Paisley, UK) into the left eye.
TMB labelling and visualization of CtB–HRP by electronmicroscopy
Twenty-four hours after injection of peroxidase-conjugated CtB (0.5% CtB/1% peroxidase in sterile PBS; Sigma–Aldrich Company Ltd.) into the colliculus of P120 homozygous #72 mice, the animals were perfused with 1% paraformaldehyde/1% glutaraldehyde/0.1 M PB. Nerves were treated with 0.07% solution of the chromogen TMB and stabilized with 0.7% 3,3/-diaminobenzidine (DAB) tetrahydrochloride/0.33% Cobalt chloride as described by van der Want et al, (1997). Resin embedded tissue was mounted on blank Araldite stubbs and 70 nm section cut and stained with Reynold's lead citrate for EM viewing.
Axonal morphometry and quantification of axonal changes
Axon diameter, density and the myelination status were assessed from electron micrographs (initial magnification ×6700) of randomly selected fields at the chiasmal end of optic nerve from P20, P40, P60 and P120 mice. EM negatives were scanned for analysis using Image-Pro Plus 6 software. Optic nerve areas were measured from light micrographs. Axon densities and nerve area measurements were used to derive total numbers. Axon diameter frequency distributions and the percentage of myelinated axons were also determined. The myelin volume (Mv) and the axonal volume (Av) were determined by a point counting method (Williams, 1977). Myelination status was also ascertained for the retinal end of the optic nerve and the optic tract from Plp-transgenic mice.
Cell counts
Cell counts were made in the retinal and chiasmal portions of the optic nerve and in the optic tract. Ten micrometer thick transverse sections, 90 µm apart, were immunostained for CD45, counterstained with DAPI then photographed at ×40 magnification. At least six sections per region were quantified. The densities of nucleated CD45+ cells were counted within an area of interest using Image-Pro Plus 6 software (Al-Saktawi et al, 2003). Optic nerve and tract transverse areas were measured and the average number of cells per 10 µm thick section was calculated.
Quantification of CtB in retinal and chiasmal portions of the optic nerve
Longitudinal sections (10 µm thick) of the optic nerve from animals injected with CtB were imaged using a confocal microscope (Leica SP2 AOBS, Leica Microsystems Semiconductor GmbH, Wetzlar, Germany) at ×63 magnification. Images were taken through the entire depth of the tissue, in retinal and chiasmal portions, at 0.5 µm steps. The image stack was flattened and the numbers of CtB+ foci, greater than 10 µm2 were counted within an AOI, using Image-Pro Plus 6 software. Foci < 10 µm2 could be observed in WT nerves and therefore were excluded from analysis. At least nine images per nerve were taken from each animal. Lipofucsin granules, which were visible in both red and green channels, were excluded from the analysis.
Electrophysiology
WT and homozygous Plp1-transgenic female mice were killed by cervical dislocation. The optic nerves were dissected and placed in an interface perfusion chamber (Medical Systems Corp., Greenvale, NY), maintained at 37°C and superfused with artificial cerebrospinal fluid (aCSF; see Supporting Information) and aerated by humidified gas mixture of 95% O2/5% CO2. The CAP was recorded following supramaximal stimulation, as described previously (Allen et al, 2006).
Statistical analyses
A two-sample Kolmogorov–Smirnov test was used to compare axonal diameter frequency distributions. Calculations were made using Microsoft Office Excel and significance was assessed by reference to published tables (Siegal & Castellan, 1988). All other statistical analyses were made using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA). Comparisons between the morphometry of WT and homozygous mice were made using a grouped t-test; comparisons along the length of the optic pathway used ANOVA. The comparisons between CtB transport in pairs of WT and transgenic littermates were performed with a paired t-test, as were comparisons between CtB foci at retinal and chiasmal portions of the optic nerve. Data are expressed as Mean ± SEM (N was at least 3) and significance in bar charts indicated as <0.05 (*), <0.01 (**) and <0.001 (***).
Acknowledgments
The authors thank Marie Ward and Gemma Thomson for technical assistance, Professor Rudolf Martini for advice, Professor V. Hugh Perry for helpful comments on the manuscript and Professor Dominic Mellor for assistance with statistics. The work was funded by a Multiple Sclerosis Society (Scotland) Junior Fellowship to J.M.E. and I.R.G. and grants from DFG (SFB-TR43) and BMBF (Leukonet) to K.A.N.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Authors contributions
J.E., I.R.G., K.-A.N. conceived and designed the experiments and wrote the manuscript. J.E., I.R.G., M.M., P.M., A.M.B., S.T., L.P. and F.G. performed experiments. J.E., I.R.G. and A.M.B. analysed the data.
For more information
NINDS Pelizaeus-Merzbacher Disease Information Page
http://www.ninds.nih.gov/disorders/pelizaeus_merzbacher/pelizaeus_merzbacher.htm
Multiple Sclerosis Society UK
http://www.mssociety.org.uk/index.html
Proteolipid protein gene information
Authors' websites
Julia M. Edgar
http://www.gla.ac.uk/faculties/vet/research/staff/researchinterests/?sid=128
Klaus-Armin Nave
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Aboul-Enein F, Weiser P, Höftberger R, Lassmann H, Bradl M. Transient axonal injury in the absence of demyelination: a correlate of clinical disease in acute experimental autoimmune encephalomyelitis. Acta Neuropathol (Berl) 2006;111:539–547. doi: 10.1007/s00401-006-0047-y. [DOI] [PubMed] [Google Scholar]
- Adalbert R, Nogradi A, Babetto E, Janeckova L, Walker SA, Kerschensteiner M, Misgeld T, Coleman MP. Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain. 2009;132:402–416. doi: 10.1093/brain/awn312. [DOI] [PubMed] [Google Scholar]
- Al-Saktawi K, McLaughlin M, Klugmann M, Schneider A, Barrie JA, McCulloch MC, Montague P, Kirkham D, Nave K-A, Griffiths IR. Genetic background determines phenotypic severity of the Plp rumpshaker mutation. J Neurosci Res. 2003;72:12–24. doi: 10.1002/jnr.10561. [DOI] [PubMed] [Google Scholar]
- Allen L, Anderson S, Wender R, Meakin P, Ransom BR, Ray DE, Brown AM. Fructose supports energy metabolism of some, but not all, axons in adult mouse optic nerve. J Neurophysiol. 2006;95:1917–1925. doi: 10.1152/jn.00637.2005. [DOI] [PubMed] [Google Scholar]
- Anderson TJ, Klugmann M, Thomson CE, Schneider A, Readhead C, Nave K-A, Griffiths IR. Distinct phenotypes associated with increasing dosage of the Plp gene: implications for CMT1A due to Pmp22 gene duplication. Ann N Y Acad Sci. 1999;883:234–246. [PubMed] [Google Scholar]
- Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–171. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- Brown AM, Tekkok SB, Ransom BR. Glycogen regulation and functional role in mouse white matter. J Physiol. 2003;549:501–512. doi: 10.1113/jphysiol.2003.042416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt AM, Pugh M, Hubbard P, James G. Functions of optic nerve glia: axoglial signalling in physiology and pathology. Eye (Lond) 2004;18:1110–1121. doi: 10.1038/sj.eye.6701595. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Matthews PM, Fu LQ, Narayanan S, Stanley J, Francis GS, Antel JP, Arnold DL. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis—results of a longitudinal magnetic resonance spectroscopy study. Brain. 1998;121:1469–1477. doi: 10.1093/brain/121.8.1469. [DOI] [PubMed] [Google Scholar]
- De Vos K, Severin F, Van Herreweghe F, Vancompernolle K, Goossens V, Hyman A, Grooten J. Tumor necrosis factor induces hyperphosphorylation of kinesin light chain and inhibits kinesin-mediated transport of mitochondria. J Cell Biol. 2000;149:1207–1214. doi: 10.1083/jcb.149.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waegh SM, Lee VMY, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Werner HB, McCulloch MC, Barrie JA, Brown A, Faichney AB, Snaidero N, Nave KA, Griffiths IR. Early ultrastructural defects of axons and axon-glia junctions in mice lacking expression of Cnp1. Glia. 2009;57:1815–1824. doi: 10.1002/glia.20893. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler J, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, et al. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol. 2004;166:121–131. doi: 10.1083/jcb.200312012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, Nave KA. The role of CNS glia in preserving axon function. Curr Opin Neurobiol. 2009;19:498–504. doi: 10.1016/j.conb.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120:393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- Foran DR, Peterson AC. Myelin acquisition in the central nervous system of the mouse revealed by an MBP-Lac Z transgene. J Neurosci. 1992;12:4890–4897. doi: 10.1523/JNEUROSCI.12-12-04890.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischer JM, Bramow S, Dal Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Lassmann H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132:1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths IR, Duncan ID, McCulloch M. Shaking pup: a disorder of central myelination in the spaniel dog. II. Ultrastructural observations on the white matter of cervical spinal cord. J Neurocytol. 1981;10:847–858. doi: 10.1007/BF01262657. [DOI] [PubMed] [Google Scholar]
- Griffiths IR, Klugmann M, Anderson TJ, Yool D, Thomson CE, Schwab MH, Schneider A, Zimmermann F, McCulloch MC, Nadon NL, et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- Ip CW, Kroner A, Crocker PR, Nave K-A, Martini R. Sialoadhesin deficiency ameliorates myelin degeneration and axonopathic changes in the CNS of PLP overexpressing mice. Neurobiol Dis. 2006;25:105–111. doi: 10.1016/j.nbd.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Irvine KA, Blakemore WF. Age increases axon loss associated with primary demyelination in cuprizone-induced demyelination in C57BL/6 mice. J Neuroimmunol. 2006;175:69–76. doi: 10.1016/j.jneuroim.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick LL, Witt AS, Payne HR, Shine HD, Brady ST. Changes in microtubule stability and density in myelin-deficient shiverer mouse CNS axons. J Neurosci. 2001;21:2288–2297. doi: 10.1523/JNEUROSCI.21-07-02288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave K-A. Disruption of the CNP gene uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Marik C, Felts PA, Bauer J, Lassmann H, Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130:2800–2815. doi: 10.1093/brain/awm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathey EK, Derfuss T, Storch MK, Williams KR, Hales K, Woolley DR, Al Hayani A, Davies SN, Rasband MN, Olsson T, et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med. 2007;204:2363–2372. doi: 10.1084/jem.20071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- Papadopoulos D, Pham-Dinh D, Reynolds R. Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp Neurol. 2006;197:373–385. doi: 10.1016/j.expneurol.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Perge JA, Koch K, Miller R, Sterling P, Balasubramanian V. How the optic nerve allocates space, energy capacity, and information. J Neurosci. 2009;29:7917–7928. doi: 10.1523/JNEUROSCI.5200-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Readhead C, Schneider A, Griffiths IR, Nave K-A. Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron. 1994;12:583–595. doi: 10.1016/0896-6273(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Schneider A, Montague P, Griffiths IR, Fanarraga ML, Kennedy PGE, Brophy PJ, Nave K-A. Uncoupling of hypomyelination and glial cell death by a mutation in the proteolipid protein gene. Nature. 1992;358:758–761. doi: 10.1038/358758a0. [DOI] [PubMed] [Google Scholar]
- Scolding N, Franklin R. Axon loss in multiple sclerosis. Lancet. 1998;352:340–341. doi: 10.1016/S0140-6736(05)60463-1. [DOI] [PubMed] [Google Scholar]
- Siegal S, Castellan NJ., Jr . Two independent samples. In: Anker JD, editor. Nonparametric Statistics for the Behavioural Sciences. New York: McGraw and Hill; 1988. pp. 102–167. [Google Scholar]
- Stagi M, Gorlovoy P, Larionov S, Takahashi K, Neumann H. Unloading kinesin transported cargoes from the tubulin track via the inflammatory c-Jun N-terminal kinase pathway. FASEB J. 2006;20:2573–2575. doi: 10.1096/fj.06-6679fje. [DOI] [PubMed] [Google Scholar]
- Stys PK, Hubatsch DA, Leppanen LL. Effects of K+ channel blockers on the anoxic response of CNS myelinated axons. Neuroreport. 1998;9:447–453. doi: 10.1097/00001756-199802160-00017. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Ma J, Tanaka KF, Takao K, Komada M, Tanda K, Suzuki A, Ishibashi T, Baba H, Isa T, et al. Mice with altered myelin proteolipid protein gene expression display cognitive deficits accompanied by abnormal neuron-glia interactions and decreased conduction velocities. J Neurosci. 2009;29:8363–8371. doi: 10.1523/JNEUROSCI.3216-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- van der Want JJ, Klooster J, Cardozo BN, de Weerd H, Liem RS. Tract-tracing in the nervous system of vertebrates using horseradish peroxidase and its conjugates: tracers, chromogens and stabilization for light and electron microscopy. Brain Res Brain Res Protoc. 1997;1:269–279. doi: 10.1016/s1385-299x(96)00042-6. [DOI] [PubMed] [Google Scholar]
- Williams MA. Stereological techniques. In: Glauert AM, editor. Practical Methods in Electron Microscopy. Vol. 6. Quantitative Methods in Biology. Amsterdam: North Holland; 1977. pp. 5–84. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.