

Abstract
Arterial hypertension is one of the most important health problems in industrialized cities. Blood pressure levels are influenced by renal salt handling and salt reabsorption in the kidney. In this Closeup, Castañeda-Bueno and Gamba discuss the work from Alessi and coworkers on the in vivo roles of the SPAK kinase in defining blood pressure levels.
Keywords: distal tubule, diuretic, hypertension, WNK
See related article by Rafiqi FH et al DOI 10.1002/emmm.200900058.
Arterial hypertension is a worldwide health problem, with up to 30% of the adult population affected with this asymptomatic disease that seriously increases the risk for cardiovascular fatal events such as acute myocardial infarction, end stage renal disease and stroke. Miscommunication between arterial pressure and salt excretion in the kidney underlies the aetiology of this disease. Accordingly, an imperceptible amount of salt retention over decades ends up increasing arterial pressure. Thus, the actual paradigm suggests that an inherited predisposition to retain salt in addition to high salt consumption results in increased prevalence of hypertension. Supporting this hypothesis is the fact that all Mendelian human disorders affecting blood pressure for which the diseased gene is known are associated with alterations of salt handling by the kidney.
» …all Mendelian human disorders affecting blood pressure for which the diseased gene is known are associated with alterations of salt handling by the kidney.«
One of the inherited diseases featuring arterial hypertension is pseudohypoaldosteronism type II (PHAII), caused by mutations in with-no-lysine kinases (WNK) WNK1 or WNK4. A deletion in the first intron of WNK1 or missense mutations on WNK4 results in a syndrome characterized by hyperkalemic metabolic acidosis with hypertension. PHAII is the mirror image of another inherited disease, Gitelman syndrome (hypokalemic metabolic alkalosis with arterial hypotension). The latter is a consequence of inactivating mutations in the renal thiazide-sensitive Na+:Cl− cotransporter (NCC), hence it has been suggested that mutant WNKs in PHAII could affect NCC activity thereby causing disease (reviewed in Gamba, 2009). Although WNKs are expressed in many different tissues, the clinical picture is due to altered ion handling in the distal nephron, in which NCC is exclusively expressed and where the final concentration of ions in urine is defined. This implies that PHAII phenotype is the consequence of the effects of mutated WNKs specifically in the distal nephron.
Vitari et al and Gagnon et al identified other serine/threonine kinases as targets for WNK1 and WNK4 (Gagnon et al, 2006; Vitari et al, 2005); the STE-20 related kinases known as STE20/SPS1-related proline/alanine-rich kinase (SPAK) and oxidative stress response-1 (OSR1). WNKs lie upstream of SPAK or OSR1 and regulate the SLC12A family of electroneutral cotransporters to which NCC belongs. Two other members of this family are the ubiquitously expressed Na+:K+:2Cl− cotransporter, NKCC1, and the kidney specific Na+:K+:2Cl− cotransporter, NKCC2. Interestingly, WNK1 and WNK4 can also modulate the activity of other ion transport channels in the distal nephron, including the apical sodium channel ENaC and the potassium channel, ROMK. The involvement of SPAK/OSR1 however, appears to be only required for WNKs modulation of NKCCs and NCC activity. While no Mendelian disease has been described in which SPAK/OSR1 are able to produce hypertension, a recent genome wide association study (Wang et al, 2009) found a strong correlation between intronic single nucleotide polymorphisms within the human SPAK gene and arterial hypertension, suggesting that SPAK may regulate blood pressure within the general population.
NKCC2 and NCC are the major transport pathways for salt reabsorption in the apical membrane of the thick ascending limb of Henle's loop and distal convoluted tubule of the nephron, respectively (Figure 1). These are the only epithelial cells in which these transporters are expressed. They together are responsible for the reabsorption of 20–25% of the glomerular filtrate. Particularly important is the location of NCC after a highly specialized region of the nephron known as macula densa in which sensing the intraluminal chloride concentration through NKCC2 modulates the glomerular filtration rate. Thus, any change in salt reabsorption beyond this point can no longer be compensated by tubuloglomerular feedback and will affect final salt excretion. NKCC2 and NCC are the targets of the most commonly prescribed diuretic drugs worldwide such as loop diuretics (furosemide) or thiazide-type diuretics, respectively. Because of their inhibitory effect on NCC, thiazides are currently recommended as the first line pharmacologic therapy for the treatment of arterial hypertension (Chobanian et al, 2003). It has been demonstrated that NCC and NKCC2 activity is increased by phosphorylation of certain conserved threonine/serine residues in their amino terminal domain and that SPAK/OSR1 are the kinases that drive such effect. Upon phosphorylation, the NCC or NKCC2 containing vesicles are trafficked to the plasma membrane (Richardson & Alessi, 2008). Recent evidence suggests that WNK4–SPAK modulation of NCC is regulated by the prohypertensive peptide hormone angiotensin II (reviewed in Gamba, 2009). Thus, regulation of NKCC2/NCC by the interaction between WNKs and SPAK/OSR1 could play a major role in salt reabsorption and thus regulation of arterial blood pressure. As the cotransporters are only expressed in the distal nephron, this would explain the specificity of the PHAII phenotype.
Figure 1. Major salt reabsorption pathways modulated by SPAK in the kidney.
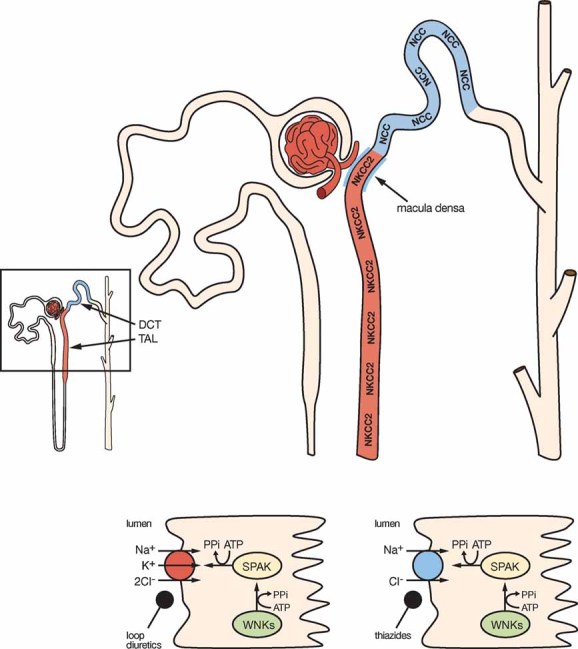
The nephron is the functional unit of the kidney (left lower corner). NKCC2 and NCC are key proteins involved in transport pathways driving salt reabsorption in the apical membrane of the thick ascending limb of Henle's loop (TAL; in red) or distal convoluted tubule (DCT; in blue). Rafiqi et al demonstrate that SPAK is expressed in these segments of the nephron and that absence of SPAK activity is associated with reduced NKCC2 and NCC expression and activity resulting in reduction of arterial blood pressure. These observations support the hypothesis that WNKs effect on NKCC2 or NCC is mediated by SPAK.
»SPAK is indeed a kidney kinase that modulates the activity of the salt transporters NKCC2 and NCC.«
In this issue of EMBO Molecular Medicine, Rafiqi et al provide an interesting piece of information that helps to integrate in vivo several observations done using in vitro systems (Rafiqi et al, 2010). It was critical to elucidate the roles of SPAK and/or OSR1 in regulating blood pressure in vivo and assess if these are associated with functional properties of the kidney. The authors generated mice knock-in models in which the threonine residues of the T-loop in SPAK (T243A) or OSR1 (T185A) were substituted by alanines. It was previously demonstrated that phosphorylation of these residues by WNKs triggers activation of SPAK/OSR1. SPAK234A/234A mice were viable and grew normally while OSR1185A/185A mice died at embryonic day 17.5, suggesting a crucial role for OSR1 during development. SPAK expression is not affected by the T243A mutation but as expected its kinase activity is ablated. Interestingly, along the nephron SPAK is only expressed in the thick ascending limb and the distal convoluted tubule where NKCC2 and NCC are also expressed. Radiotelemetry analysis of arterial blood pressure revealed hypotension in adult SPAK234A/234A mice when compared with wild-type mice. Consistent with reduction in salt reabsorption as the mechanism of hypotension, blood pressure was normalized after exposing the SPAK234A/234A mice to high salt diet during a 14-day period. Renal function tests showed that SPAK234A/234A mice actually developed a Gitelman-like syndrome with hypomagnesemia and hypocalciuria. The characteristic hypokalemia of Gitelman's disease was not observed in normal sodium diet, but became apparent with low sodium diet. These physiological observations strongly suggest that absence of SPAK activity results in decreased activity of NKCC2 and/or NCC. Indeed, the expression of NCC in the kidney of SPAK234A/234A mice was reduced by 30% and phosphorylation at residues T53, T58, and S89 (indicative of cotransporter's activity) was reduced by about 80%. Similar observations were done using specific antibodies against NKCC2. Because the mice phenotype resembles Gitelman's rather than Bartter's disease (that is due to inactivating mutations of NKCC2), it appears as though SPAK inactivation has a strong effect on NCC function. Interestingly, aldosterone levels were similar in SPAK234A/234A and wild-type mice in both, normal or low sodium diet. This is intriguing because low blood pressure could be corrected by high salt intake, implying that reduced blood pressure was associated with volume contraction, a physiological situation in which one would expect increased aldosterone secretion. In this regard, up-regulation of the three subunits of the epithelial sodium channel ENaC was observed, which suggests a compensatory mechanism in which ENaC drives sodium reabsorption in the collecting duct located beyond the distal convoluted tubule. ENaC up-regulation, however, would also be expected to be secondary to aldosterone effects in collecting duct. Finally, by switching from high to low salt diet, the authors observed that SPAK234A/234A mice reach salt and potassium balance at slower rates than control mice.
The observations presented in this issue of EMBO Molecular Medicine show that SPAK is indeed a kinase expressed in the kidney that modulates the activity of the salt transporters NKCC2 and NCC. Absence of SPAK activity is associated with arterial hypotension. As discussed, intronic SNPs that result in increased expression of SPAK in the kidney (Wang et al, 2009) are associated with increased blood pressure and SPAK is a target of WNKs and modulates NKCC2/NCC activity. It is reasonable to conclude that the WNKs–SPAK–NKCC/NCC pathway plays a critical role to define the baseline activity of NKCC2 and NCC and thus, salt reabsorption rates. These observations establish a molecular mechanism for blood pressure regulation that can be exploited for the development of new therapeutic agents for arterial hypertension. Importantly, as the WNKs–SPAK–NCC pathway is likely only active in the kidney distal convoluted tubule, a compound targeting it would have diuretic, antihypertensive properties without additional secondary effects.
Supplementary material
See related article by Rafiqi FH et al http://dx.doi.org/10.1002/emmm.200900058.
References
- Chobanian AV, et al. JAMA. 2003;289:2560–2571. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- Gagnon KB, et al. Am J Physiol Cell Physiol. 2006;290:C134–C142. doi: 10.1152/ajpcell.00037.2005. [DOI] [PubMed] [Google Scholar]
- Gamba G. Am J Physiol Renal Physiol. 2009;297:F838–F848. doi: 10.1152/ajprenal.00159.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafiqi FH, et al. EMBO Mol Med. 2010;2:63–75. doi: 10.1002/emmm.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C, et al. J Cell Sci. 2008;121:3293–3304. doi: 10.1242/jcs.029223. [DOI] [PubMed] [Google Scholar]
- Vitari AC, et al. Biochem J. 2005;391:17–24. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. Proc Natl Acad Sci USA. 2009;106:226–231. [Google Scholar]