

»I was captivated by the idea of understanding and controlling the pluripotent state.«
In the 1960s it was shown that a rare type of tumour called a teratocarcinoma contains cells that are both pluripotent and self-renewing (Kleinsmith & Pierce, 1964). Pluripotent means the capacity of an individual cell to give rise to all other cell types of the body and the germline. This property is normally restricted to a brief window in early development. Self-renewal is the production of identical daughter cells while retaining the ability for differentiation. It is the defining feature of a stem cell. The study of teratocarcinoma stem cells led to particular culture conditions that allowed them to be propagated ex vivo without differentiation. In 1981 Martin Evans, Matt Kaufman and Gail Martin found that cells from early mouse embryos exposed to the same culture environment can suspend developmental progression and continue to multiply while remaining pluripotent (Evans & Kaufman, 1981; Martin, 1981). These are embryonic stem (ES) cells. I was completing my undergraduate studies in Oxford at that time and was fortunate to learn about this rather esoteric research from a wonderful teacher, Chris Graham. Chris also talked about parallel discoveries of growth factors and the provocative idea that they might regulate cell fate. I was captivated by the idea of understanding and controlling the pluripotent state and that has been my obsession ever since.
Finding the differentiation inhibiting activity
I went to Edinburgh to do a PhD on teratocarcinoma stem cells with Martin Hooper. There I was struck by an observation by a previous PhD student that the requirement of the stem cells for co-culture with a feeder layer could be replaced for a short time by conditioned medium. To me this meant only one thing; a growth factor that could block differentiation. This was not my main PhD project but Martin gave me the freedom to pursue this idea. I thought the factor might be insulin-like growth factor (IGF) but as growth factors were not available in catalogues in those days I had to use a cell culture supernatant as a source for them. This turned out to be very fortunate; I used buffalo rat liver (BRL) cells as a cell source. I added BRL cell medium to the ES cells and when I finally looked at them a week later my life changed. I brought Martin to the microscope and he just said ‘They're beautiful!’. And they were; for the first time totally undifferentiated colonies without feeders. The search for the responsible growth factor then started. To my surprise, I found that the activity that kept the stem cells undifferentiated separated away from IGF when I fractionated the BRL medium. We had a new factor in hand and I called it differentiation inhibiting activity (DIA) (Smith & Hooper, 1987).
I went back to Oxford to work with John Heath and purify this protein. We managed this in little over a year but then hit a brick wall; the protein was N-terminally blocked and very heavily glycosylated. Several sequencing attempts failed. We had started a collaboration with the biotechnology company Genetics Institute and I persuaded them to screen purified DIA in a panel of haematopoietic assays to see if we could get a clue to its possible identity. We were lucky; DIA was active in a particular proliferative assay that had just been used to clone the cytokine we now know as leukaemia inhibitory factor (LIF). We showed that DIA and LIF were identical (Smith et al, 1988). At the same time, Lindsay Williams and colleagues reported the reciprocal experiment of assaying LIF on ES cells (Williams et al, 1988). Peter Rathjen and I then found that feeder cells are a potent provider of LIF because they produce both soluble and cell-associated forms (Rathjen et al, 1990).
»The field has grown enormously and the biomedical opportunities are thrilling.«
In 1990 I was given the opportunity to start my own group in the new Centre for Genome Research in Edinburgh. There I had the privilege and pleasure of working alongside a great developmental biologist Rosa Beddington. In 1996 I became Director of the Institute and began to reorient the research focus of the Centre to stem cell biology, leading to the formation of the Institute for Stem Cell Research in 2000. Five years later it was time to move on and I came to Cambridge to help set up the Wellcome Trust Centre for Stem Cell Research where I am currently a Director. My ‘obsession’ has accompanied me throughout these changes. The field has grown enormously and the biomedical opportunities are thrilling, although my group still mainly studies the fundamental biology of pluripotent stem cells and the decision between self-renewal and commitment.
Understanding pluripotency
A pluripotent stem cell can be considered as a blank state or tabula rasa. Our laboratory and others have shown that emergence of the pluripotent state within the inner cell mass (ICM) of the pre-implantation embryo is orchestrated by two transcriptional organizers, Oct4 and Nanog (Mitsui et al, 2003; Nichols et al, 1998; Silva et al, 2009). These factors confer on a subset of ICM cells, the epiblast, potency to generate embryonic lineages. Epiblast cells persist in this naïve state for only 24 h in the mouse embryo but, as described above, they may be immortalized in vitro as ES cells. A pillar of our research strategy has been to simplify the complex culture environment originally employed for ES cells so as to define the essential requirements for maintaining pluripotency. Having found that co-culture could be replaced by LIF, we then showed that LIF activates the transcription factor Stat3 (Niwa et al, 1998) and feeds into the core pluripotency network via induction of Kruppel-like transcription factors (Fig 1) (Hall et al, 2009), see also (Niwa et al, 2009). More recently, we realized that ES cell commitment is triggered by the mitogen activated protein kinase (Erk) cascade (Kunath et al, 2007). This has allowed replacement of serum with highly selective small molecule inhibitors (Ying et al, 2008). Chemical inhibition or genetic ablation of the Erk cascade suppresses ES cell differentiation and maximizes self-renewal in the presence of LIF. If Erk signalling is blocked, LIF can largely be replaced by partial inhibition of glycogen synthase kinase-3, which acts principally by stabilizing intracellular β-catenin. An interest of our current research is to define the convergence between Stat3 and β-catenin and to resolve the non-overlapping effects of Stat3.
Figure 1. LIF reinforces the pluripotent transcription factor hub.
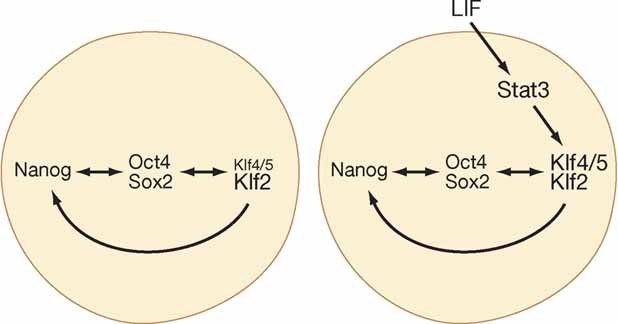
Oct4/Sox along with Nanog and Klfs comprise an autoregulatory hub that radiates to sustain the pluripotency gene regulatory network. When this network is confronted by activated Erk, the hub must be reinforced to prevent differentiation. LIF achieves this by boosting the expression of Klf4 and Klf5.
»Our next challenge is to elucidate how pluripotent cells exit from the ground state.«
Ability to withstand loss of Erk signalling is unusual for non-transformed cells. We have proposed that ES cells represent a ground state for mammalian cells, meaning a basal self-replicating unit that is developmentally and epigenetically un-programmed and has minimal requirements for extrinsic stimuli to support survival and proliferation (Silva & Smith, 2008). This ground state is, however, highly sensitive to the environment, in particular stimuli that activate Erk. We hypothesize that the underlying molecular network of ground state pluripotency may be conserved among eutherian mammals. Indeed, Erk inhibition enables very efficient derivation of ES cells from all strains of mice tested (Nichols et al, 2009a) and also from rats (Buehr et al, 2008; Li et al, 2008), which was not previously possible. Blockade of Erk signalling in the developing mouse embryo reveals that the entire ICM can acquire pluripotency (Nichols et al, 2009b). These findings lead to a reinterpretation of the nature of ES cells, suggesting that they are most likely identical to early epiblast cells in the blastocyst rather than an artefact of in vitro manipulation. Our next challenge is to elucidate how pluripotent cells exit from the ground state and select somatic lineage or germline differentiation paths (Fig 2). ES cells appear homogenous when Erk signalling is suppressed (Wray et al, 2010), consistent with the idea of being anchored in a ground state (Smith, 2009). When this pathway is active, however, the stem cells become heterogeneous in gene expression even though differentiation can largely be prevented by LIF. We surmise that Erk signalling perturbs the ground state creating a destabilized transition state. Cells in this critical state are poised between commitment and a return to ground state. The questions we are now seeking to answer are what are the molecular events underlying the transition and is exit from the ground state separable from or coincident with lineage specification.
Figure 2. From pluripotency to lineage-specific differentiation.
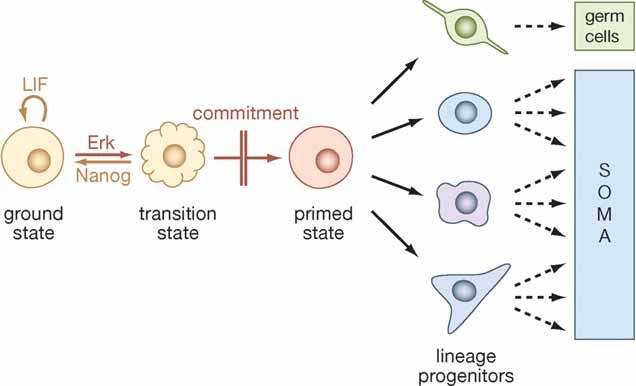
In this model ES cells must first exit the ground state to generate effector cells that are enabled for lineage specification and differentiation. The primed state can be considered as a metastable entity in which multiple lineage-affiliated programmes compete directly. An alternative model envisions that ES cells transit directly to lineage restricted progenitors such that loss of ES cell identity is inseparable from lineage choice.
A ground state human pluripotent stem cell?
In rodents, the epigenetic conversion of unipotent primordial germ cells into pluripotent embryonic germ (EG) stem cells (Matsui et al, 1992; Resnick et al, 1992) and the generation of induced pluripotent stem (iPS) cells by molecular reprogramming (Takahashi & Yamanaka, 2006) are also facilitated by inhibition of Erk signalling (Silva et al, 2008) and unpublished. However, human ‘ES’ and iPS cells are neither responsive to LIF nor sustained by Erk inhibition. On the contrary, they depend on Erk signalling for continued proliferation. A possible explanation for this divergence is that the human ‘ES’ cells we currently have available may represent a later stage of epiblast development (Brons et al, 2007; Smith, 2001; Tesar et al, 2007). A central unanswered question therefore is whether a ground state equivalent to rodent ES cells exists for all mammals? Contrary to the widespread view that after Shinya Yamanaka's iPS cell breakthrough human embryo research is no longer needed, I contend that it remains essential. It is remarkable how little is known about development of the human blastocyst and in particular of the epiblast. However, the variable quality of supernumerary human embryos and the lack of functional measures of blastocyst maturation make this research problematic. We are therefore also examining the development of pluripotency and establishment of ES cells in other mammalian species including non-human primates.
If it becomes possible to generate and define a ‘ground state’ human pluripotent stem cell, this would enable objective classification of human-induced pluripotency. Currently the high degree of variability observed among human ‘ES’ cells and iPS cells presents a major obstacle to developing standardized operating procedures essential for large scale bioindustrial and clinical applications.
The medical possibilities of pluripotent cells
I expect that for the next 10 years the major biomedical applications of pluripotent stem cells will be in disease modelling, drug discovery and toxicology screening (Fig 3). Nonetheless, researchers will remain entranced by the concept of direct stem cell therapies. Indeed clinical trials are already planned using cells differentiated from human pluripotent cells. Perhaps the most promising area in the short term is retinal disease such as age-related macular degeneration and retinitis pigmentosum, major causes of vision impairment and blindness. This prospect arises from the partly serendipitous observation that some human ES cells can consistently and efficiently differentiate into retinal cells (Osakada et al, 2008). Experiments in rat models suggest that relatively small numbers of transplanted cells may be able to repair the damaged region of the retina in patients with macular degeneration. If these trials are successful they will encourage other efforts, notably to develop beta cell replacement therapy for type I diabetes and potentially bring life-changing benefits to a large number of patients. I am confident that through insightful basic and pre-clinical studies and responsible approaches in the clinic, pluripotent stem cell research will usher in regenerative medicine in various forms.
Figure 3. Biomedical potential of pluripotent stem cells.
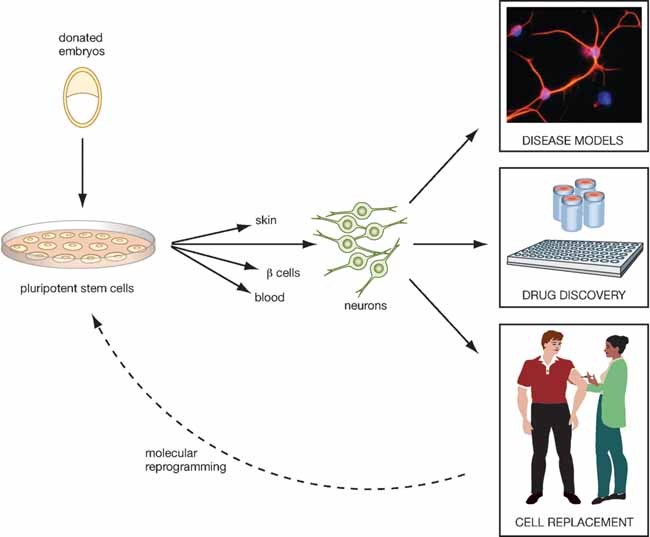
As a renewable source for all types of differentiated cell, human pluripotent stem cells provide unprecedented opportunities for studying human cellular pathogenesis and for developing efficacious pharmaceuticals. In the longer term it is hoped that stem cell differentiation can provide supplies of cells for transplantation therapy in conditions such as retinal diseases, type I diabetes and Parkinson's.
 |
 |
Acknowledgments
The author declares that he has no conflict of interest.
References
- Brons IG, et al. Nature. 2007;448:191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- Buehr M, et al. Cell. 2008;135:1287–1298. doi: 10.1016/j.cell.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Evans MJ, et al. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- Hall J, et al. Cell Stem Cell. 2009;5:597–609. doi: 10.1016/j.stem.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Kleinsmith LJ, et al. Cancer Res. 1964;24:1544–1552. [PubMed] [Google Scholar]
- Kunath T, et al. Development. 2007;134:2895–2902. doi: 10.1242/dev.02880. [DOI] [PubMed] [Google Scholar]
- Li P, et al. Cell. 2008;135:1299–1310. doi: 10.1016/j.cell.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GR. Proc Natl Acad Sci USA. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, et al. Cell. 1992;70:841–847. doi: 10.1016/0092-8674(92)90317-6. [DOI] [PubMed] [Google Scholar]
- Mitsui K, et al. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- Nichols J, et al. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- Nichols J, et al. Nat Med. 2009a doi: 10.1038/nm.1996. [Google Scholar]
- Nichols J, et al. Development. 2009b;136:3215–3222. doi: 10.1242/dev.038893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, et al. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, et al. Nature. 2009;460:118–122. doi: 10.1038/nature08113. [DOI] [PubMed] [Google Scholar]
- Osakada F, et al. Nat Biotechnol. 2008;26:215–224. doi: 10.1038/nbt1384. [DOI] [PubMed] [Google Scholar]
- Rathjen PD, et al. Cell. 1990;62:1105–1114. doi: 10.1016/0092-8674(90)90387-t. [DOI] [PubMed] [Google Scholar]
- Resnick JL, et al. Nature. 1992;359:550–551. doi: 10.1038/359550a0. [DOI] [PubMed] [Google Scholar]
- Silva J, et al. Cell. 2008;132:532–536. doi: 10.1016/j.cell.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, et al. PLoS Biol. 2008;6:e253. doi: 10.1371/journal.pbio.0060253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, et al. Cell. 2009;138:722–737. doi: 10.1016/j.cell.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG. Ann Rev Cell Dev Biol. 2001;17:435–462. doi: 10.1146/annurev.cellbio.17.1.435. [DOI] [PubMed] [Google Scholar]
- Smith A. EMBO Mol Med. 2009;1:251–254. doi: 10.1002/emmm.200900035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG, et al. Dev Biol. 1987;121:1–9. doi: 10.1016/0012-1606(87)90132-1. [DOI] [PubMed] [Google Scholar]
- Smith AG, et al. Nature. 1988;336:688–690. doi: 10.1038/336688a0. [DOI] [PubMed] [Google Scholar]
- Takahashi K, et al. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tesar PJ, et al. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- Williams RL, et al. Nature. 1988;336:684–687. doi: 10.1038/336684a0. [DOI] [PubMed] [Google Scholar]
- Wray J, et al. Biochem Soc Trans. 2010;15:814–818. [Google Scholar]
- Ying QL, et al. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]