

Abstract
The protein kinase Aurora-A is a major regulator of the cell cycle that orchestrates mitotic entry and is required for the assembly of a functional mitotic spindle. Overexpression of Aurora-A has been strongly linked with oncogenesis and this has led to considerable efforts at therapeutic targeting of the kinase activity of this protein. However, the exact mechanism by which Aurora-A promotes oncogenesis remains unclear. Here, we show that Aurora-A modulates the repair of DNA double-strand breaks (DSBs). Aurora-A expression inhibits RAD51 recruitment to DNA DSBs, decreases DSB repair by homologous recombination and sensitizes cancer cells to PARP inhibition. This impairment of RAD51 function requires inhibition of CHK1 by Polo-like kinase 1 (PLK1). These results identify a novel function of Aurora-A in modulating the response to DNA DSB that likely contributes to carcinogenesis and suggest a novel therapeutic approach to the treatment of cancers overexpressing this protein.
Keywords: Aurora-A, cancer, DNA repair, PARP inhibitors, RAD51
INTRODUCTION
Aurora-A is a centrosome-associated, cell cycle-regulated member of the Aurora serine/threonine protein kinase family which is important for mitosis (Bischoff & Plowman, 1999; Carmena & Earnshaw, 2003; Giet & Prigent, 1999; Nigg, 2001). The protein level and activity of Aurora-A peaks at G2 and during mitosis whereas expression is low in resting cells (Sasai et al, 2008; Zhou et al, 1998). A variety of Aurora-A substrates have been identified, of which the best characterized are p53, TPX2, Ajuba and D-TACC (Hirota et al, 2003; Meraldi et al, 2004). Aurora-A is essential for multiple processes during mitosis, including mitotic spindle formation and activation of cell cycle regulators such as PLK1 and CDK1 (Cazales et al, 2005; Seki et al, 2008). Perhaps unsurprisingly, targeting Aurora-A pharmacologically, by RNA interference or by genetic knockout, leads to aberrant mitosis and cell death (Manfredi et al, 2007; Sasai et al, 2008). The Aurora-A gene is located on human chromosome 20q13—a region that is amplified in a variety of human tumours (Kallioniemi et al, 1994). Aurora-A is overexpressed in a broad range of human tumours, including primary colorectal carcinoma, gliomas and breast, ovarian and pancreatic cancers (Bischoff et al, 1998; Gritsko et al, 2003; Zhou et al, 1998). Ectopic expression of Aurora-A induces, under certain conditions, abnormal spindle formation leading to polyploidy and has been reported to transform NIH3T3 and Rat1 fibroblasts (Bischoff et al, 1998; Meraldi et al, 2002; Zhou et al, 1998). Nevertheless, the exact mechanisms by which Aurora-A overexpression induces tumorigenicity remain unclear. A role of Aurora-A not directly linked to mitosis is its involvement in DNA-damage response (DDR). Aurora-A activity is tightly regulated during the response to genotoxic agents and is important for a normal DDR (Cazales et al, 2005; Krystyniak et al, 2006).
DNA damage is continuously generated by a variety of mechanisms including cell metabolism, exogenous genotoxic agents and the collapse of replication forks. Amongst the many types of DNA lesions, DNA double-strand breaks (DSBs) are especially lethal if left unrepaired. Deficiency in the DNA repair processes that normally deal with DSBs is associated with cancer susceptibility as illustrated by the tumour suppressor activity of breast and ovarian cancer susceptibility genes BRCA1 and BRCA2 which both are part of the DSB repair machinery (Kastan & Bartek, 2004). There are two mechanistically distinct pathways repairing DSBs, the non-homologous end-joining (NHEJ) and the homologous recombination (HR) pathways. The NHEJ pathway mediates the re-ligation of the two broken DNA ends, while HR involves the use of a homologous sequence as a template to faithfully repair the damaged region. Consequently HR is an essentially error-free repair pathway whereas NHEJ is mostly error-prone and generates mutations. HR requires a readily available homologous DNA sequence, a condition that is best fulfilled once the DNA has been replicated, in the S and G2 phases of the cell cycle. Indeed, HR is repressed in G1, and becomes activated during S and G2 phases, while NHEJ is constitutively active throughout the cell cycle (Mao et al, 2008b).
There is an intricate connection between the DDR and the cell cycle at multiple levels. First, as a response to a DSB is elicited, selection of the most appropriate DSB repair pathway occurs. This crucial step, which may have dramatic consequences on the maintenance of genome integrity, is considerably affected by cell cycle phase. Recent findings have illuminated the mechanistic basis for the cell cycle-dependent activation of DNA repair pathways. In both yeast and mammals, the G2 cyclin-dependent kinase CDK1 stimulates 5′–3′ resection of the DSB ends (Jazayeri et al, 2006). This modification is specifically required for HR, generates a substrate for the initiation of this process, and also triggers the full activation of the DNA damage checkpoint (Aylon et al, 2004; Huertas et al, 2008; Huertas & Jackson, 2009; Ira et al, 2004; Jazayeri et al, 2006). At a subsequent stage, once the HR machinery is fully active, the cell cycle is normally stalled by the activation of the DNA damage checkpoints. For the G2/M DNA damage checkpoint, the cell cycle arrest is mostly contributed by the regulation of CDC25 phosphatases and WEE1 either by the checkpoint kinases CHK1 and CHK2 or by the ATM/ATR kinases-dependent phosphorylation of PLK1. These two pathways converge to maintain an efficient inhibition of CDK1 and hence prevent cell cycle progression. Finally, during the DNA damage checkpoint recovery, the signal emanating from the mitotic kinase PLK1 becomes dominant and stimulates cell cycle progression. Interestingly, during this late phase of the DDR, but also during unperturbed cell cycle, Aurora-A has been identified as the upstream activator of PLK1 (Macurek et al, 2008; Seki et al, 2008). The resulting activation of CDK1 stems from two complementary, concomitant actions. Firstly, PLK1 and Aurora-A directly regulate CDC25 and WEE1. Secondly, PLK1 activation, by mediating a phosphorylation dependent degradation of Claspin, leads to the inactivation of CHK1 and hence alleviates the opposing effect of the checkpoint protein to the cell cycle (Mamely et al, 2006; Peschiaroli et al, 2006). In addition, the core machinery of HR is also targeted by the checkpoint recovery signalling. BRCA2 activity, which is required for RAD51 loading onto single-strand DNA (ssDNA), is also controlled by PLK1- and CDK-triggered phosphorylation (Esashi et al, 2005; Lee et al, 2004).
It appears, therefore, that a proper balance between the activity of cell cycle regulators and DNA repair proteins is crucial to produce an adapted response to DNA damage. One of the major regulators of the cell cycle, Aurora-A, is aberrantly expressed in cancer and its deregulation is a driving event in tumour formation (Greenman et al, 2007). This led us to investigate if the misexpression of Aurora-A observed in cancer could affect the DDR. We found that expression of high Aurora-A levels leads to the silencing of HR, the major error-free DSB repair pathway, in response to DNA damage. Aurora-A overexpression represses CHK1 kinase activity and the repression of HR requires the activation of PLK1. We propose that this function of Aurora-A could contribute to the oncogenic activity of Aurora-A and provide basis for novel therapeutic strategies.
RESULTS AND DISCUSSION
Aurora-A overexpression impairs RAD51 focus formation
To assess the potential effect of Aurora-A on HR, we examined the formation of nuclear RAD51 foci after DNA damage. The localization of these foci after damage most likely represents the loading of the RAD51 DNA recombinase onto damaged DNA, an essential part of the HR process known to be controlled by other HR proteins, such as BRCA1 and BRCA2 (West, 2003). MCF10A cells were infected with lentiviral vectors expressing either high levels of both Aurora-A and GFP (from a bicistronic mRNA) or GFP alone. The levels of overexpressed Aurora-A in the infected MCF10A cell lines were comparable to the levels of Aurora-A found in a panel of cancer cell lines (Supporting Information Fig 1). In MCF10A cells expressing high levels of Aurora-A, RAD51 focus induction was impaired, suggesting a potentially repressive effect of Aurora-A on HR (Fig 1A, B). To address whether high Aurora-A levels altered the sensing of DNA lesions, we also examined the formation of serine 139 phosphorylated histone H2AX (γ-H2AX) nuclear foci (Bonner et al, 2008). High level Aurora-A expression did not affect γ-H2AX foci formation, suggesting that DNA damage sensing was not altered (Fig 1A, C). To exclude the possibility that these effects were specific to the cell model used, we also assessed RAD51 and γ-H2AX foci formation in mouse embryonic stem (ES) cells (Supporting Information Fig 2). ES cells transfected with a plasmid driving the expression of a myc-tagged Aurora-A showed impaired RAD51 foci formation upon X-ray treatment, while eliciting a normal γ-H2AX foci induction. As a control, ES cells lacking a functional HR pathway due to BRCA2 gene inactivation were used. A reduction of RAD51 foci formation was also observed (Farmer et al, 2005); (BRCA2−/−; Supporting Information Fig 2A).
Figure 1. Impairment of irradiation-induced RAD51 nuclear focus formation by Aurora-A overexpression.

MCF10A cells were infected with a lentivirus construct driving the expression of GFP (empty vector) or both GFP and Aurora-A (Aurora-A) and mixed in equal proportion with non-infected cells to provide an internal control. The mixed cells were exposed to 5-Gy X-ray, followed by a 24 h (A) or 4, 16 or 32 h (B and C) recovery.
- A. Immunofluorescence images of MCF10A cells stained for RAD51 (grey) and γ-H2AX (red). Green fluorescence indicates GFP expression by infected cells. DNA was counterstained with DAPI (blue). Arrowheads indicate empty vector infected or Aurora-A overexpressing cells.
- B, C. Quantification of RAD51 (B) and γ-H2AX (C) foci-positive nuclei. Immunofluorescence images of MCF10A were acquired as in (A). The cells were pre-treated with the Aurora-A inhibitor MLN8054 (Aurora-A + MLN8054) or vehicle, 1 h before irradiation. Nuclei containing more than three foci were scored as positive. The percentage of RAD51 (B) or γ-H2AX (C) positive nuclei of non-infected (GFP negative) or infected (GFP positive) cells was plotted. Error bars represent SEM. Results are representative of at least two independent experiments.
Given that Aurora-A is a kinase, we assessed whether catalytic activity was required for the modulation of RAD51 function. Pre-treatment of MCF10A cells overexpressing Aurora-A with a specific Aurora-A kinase inhibitor MLN8054, at concentrations that preferentially inhibit Aurora-A (Manfredi et al, 2007), restored normal RAD51 foci induction in response to ionizing radiation (Fig 1B). The specificity of this effect was confirmed by the observation that the Aurora-B kinase inhibitor ZM447439 (Ditchfield et al, 2003) was unable to restore the RAD51 response in cells expressing high levels of Aurora-A (Supporting Information Fig 3). The requirement of Aurora-A kinase activity for the inhibition of RAD51 foci formation was also confirmed by the use of a kinase inactive mutant of Aurora-A (Aurora-A D256A; Jiang et al, 2003; Supporting Information Fig 4).
The effect of Aurora-A on HR is not caused by cell cycle perturbation
The cellular choice between NHEJ and HR is regulated during the cell cycle and therefore, reduced activity of the HR pathways could conceivably result from modulation of the cell cycle by Aurora-A overexpression. To exclude this possibility, we monitored the cell cycle profile of irradiated MCF10A and ES cells by flow cytometry (Fig 2; Supporting Information Fig 2C). In these experimental systems, we did not observe the generation of polyploid cells upon overexpression of Aurora-A as described in other experimental settings (Meraldi et al, 2002; Supporting Information Fig 5). As expected, irradiation resulted in a cell cycle arrest in both cellular models. DNA profiles of MCF10A were performed at various times after irradiation. Up to 16 h after irradiation, the DNA profiles of Aurora-A overexpressing and normal cells were indistinguishable. As, at this time, a pronounced effect of Aurora-A overexpression on RAD51 foci formation is already clearly visible, we conclude that Aurora-A kinase activity modulates the HR independently of its function in cell cycle. However, at later time points, the presence of high levels of Aurora-A leads to cell cycle re-entry. Therefore, it is possible that, as the DNA damage checkpoint weakens, the kinase may overcome the arrest. Alternatively, this might reflect that a faster and therefore, less accurate repair pathway, such as NHEJ, operates in these conditions.
Figure 2. Effect of Aurora-A overexpression on the DNA damage-induced cell cycle arrest.

- MCF10A cells were infected with a lentivirus construct driving the expression of GFP (empty vector) or both GFP and Aurora-A (Aurora-A) and mixed in equal proportion with non-infected cells to provide an internal control (fluorescence images, top left panel). The mixed cells were exposed to 5-Gy X-ray, followed by a 16, 24 or 40 h recovery, or left untreated, as indicated. The DNA was counterstained by Hoechst 33342, and the DNA profiles of infected (GFP positive) and uninfected (GFP negative cells) were acquired separately by flow cytometry.
- The percentage of cells in each phase of the cell cycle was determined from the DNA profiles shown in (A) and plotted.
Aurora-A overexpression inhibits HR
As RAD51 activity is essential for HR, we assessed whether the impairment of RAD51 foci formation by Aurora-A overexpression could lead to a corresponding silencing of homology directed DNA repair itself. To address this, we used a previously validated synthetic DSB repair substrate stably introduced into 293 cells. This model is based on the induction of a DSB by the restriction enzyme I-SceI at a single chromosomal locus. When this lesion is repaired by a homology-directed mechanism, a functional blasticidin resistance gene is generated (Tutt et al, 2001; Fig 3A). Using either colony formation (Fig 3B, C) or GFP competition (Fig 3D, E) assays to estimate blasticidin resistance, fewer blasticidin resistant 293 cells were generated when Aurora-A was overexpressed, suggesting that HR is repressed by Aurora-A in this model. In line with the RAD51 foci formation assay, the kinase activity of Aurora-A is required for its effect on HR, as the overexpression of a kinase inactive mutant, Aurora-A S361*, did not affect the acquisition of blasticidin resistance in this assay (Fig 3B-E; Bibby et al, 2009). We also verified that the cell cycle profile of the 293 cells was unperturbed by Aurora-A overexpression in these experiments (Supporting Information Fig 6). Taken together, these results indicate that Aurora-A exerts a repressive effect on the DNA repair by HR.
Figure 3. HR deficiency in Aurora-A overexpressing cells.
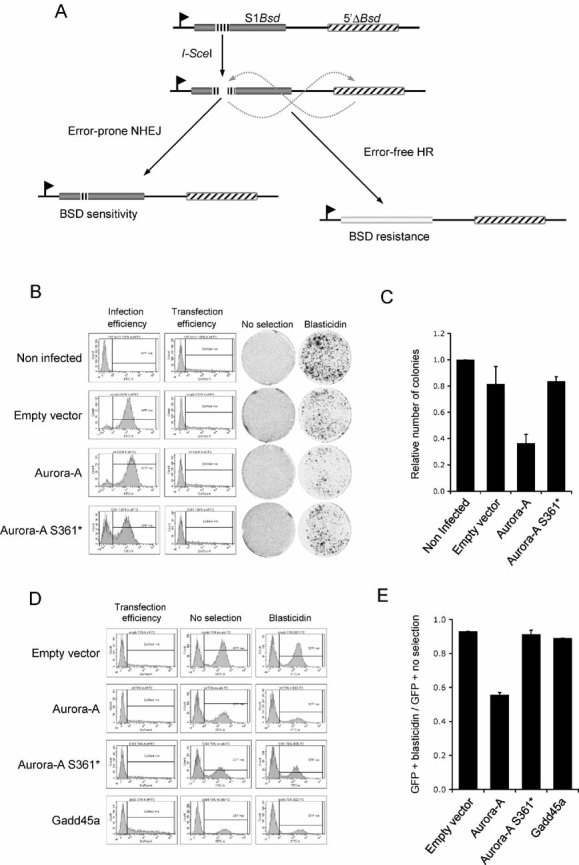
- Schematic representation of the HR repair substrate DR1Bsd. The single copy HR reporter construct chromosomally integrated into HEK 293 cells consists of two tandem copies of the blasticidin resistance gene (Bsd). The insertion of an I-SceI restriction site (black vertical dashes), which also encodes two in-frame stop codons, into the upstream copy of the blasticidin-resistance gene (S1Bsd; grey box) prevents its expression. The downstream copy of the gene (5′ΔBsd; black diagonal dashed box) is promoterless, and hence non-functional. Expression of the I-SceI restriction enzyme induces a DNA DSB at the I-SceI restriction site in S1Bsd. The repair of this lesion by NHEJ will mostly leave the S1Bsd gene non-functional, whereas HR mechanisms, using the 5′ΔBsd copy as a repair template, generates a functional gene and cell resistance to blasticidin.
- 293 cells bearing the HR repair substrate DR1Bsd were infected with a lentiviral vector expressing GFP alone (empty vector), or GFP along with Aurora-A (Aurora-A) or a kinase inactive Aurora-A mutant (Aurora-A S361*). Infection efficiency was monitored in flow cytometry by the measurement of GFP expression (infection efficiency). Non-infected cells were included as a control. The plasmid encoding the NLS-tagged I-SceI restriction enzyme was co-transfected with a DsRed expressing construct. The transfection efficiency was assessed by flow cytometry measurement of DsRed expression (transfection efficiency). Cells were then grown in normal medium (no selection) or in the presence of blasticidin (blasticidin) and stained with crystal violet as described in material and methods.
- Colonies formed in the presence of blasticidin were scored and plotted, after normalization by the platting efficiency (number of colonies formed in the absence of blasticidin) and the transfection efficiency. Results are representative of at least two independent experiments. Error bars represent SEM.
- 293 cells bearing the HR repair substrate DR1Bsd were infected with a lentivirus construct driving the expression of GFP (empty vector), GFP along with Aurora-A (Aurora-A), a kinase inactive Aurora-A mutant (Aurora-A S361*) or Gadd45a (Gadd45a) and mixed with non-infected cells to provide an internal control. The mixed cells were co-transfected with NLS-tagged I-SceI and DsRed expression plasmids. Transfection efficiency was assessed by flow cytometry measurement of DsRed expression (transfection efficiency). The GFP expression was analysed by flow cytometry in cells maintained in normal (no selection), or blasticidin-containing (blasticidin) medium.
- Ratio of GFP-positive cells after blasticidin selection versus non-selected cells as measured in (D). The average of two independent experiments is shown. Error bars represent SEM.
Aurora-A regulates HR through the PLK1/CHK1 pathway
The presence of DNA damage triggers a coordinated cellular response aimed at repairing damaged DNA while at the same time stalling the cell cycle so that damage is not eventually passed onto daughter cells. Unsurprisingly, cross-talk between repair and checkpoint pathways has been described (Harper & Elledge, 2007). Interestingly, the Polo-like kinase (PLK1) was recently shown to control the degradation of Claspin (Mamely et al, 2006) and hence the activity of the checkpoint protein CHK1, which is also required for HR (Sorensen et al, 2005). Aurora-A kinase has been identified as an upstream activator of PLK1 (Seki et al, 2008). Based on these observations we investigated whether high level Aurora-A expression could modulate HR activity through a PLK1-mediated regulation of the CHK1-Claspin complex.
We first determined the activity of PLK1 in either irradiated (5 Gy) or untreated MCF10A cells expressing endogenous or high levels of Aurora-A (Fig 4A). Using an antibody specific to the active threonine 210 phosphorylated form of PLK1 (p-Thr210-PLK1), we observed an increased PLK1 activity in cells expressing high Aurora-A levels. Moreover, the Aurora-A-mediated activation of PLK1 kinase activity was not affected by irradiation. We next tested the requirement of PLK1 activation for Aurora-A impairment of RAD51 focus formation by using a specific inhibitor of PLK1 kinase activity, BI2536 (Steegmaier et al, 2007). Inhibition of PLK1 efficiently abolished the effect of Aurora-A on RAD51 foci induction (Fig 4B), indicating that PLK1 activation is required for the impairment of RAD51 foci induction by Aurora-A.
Figure 4. Aurora-A activation of PLK1 is required for the impairment of RAD51 focus formation.
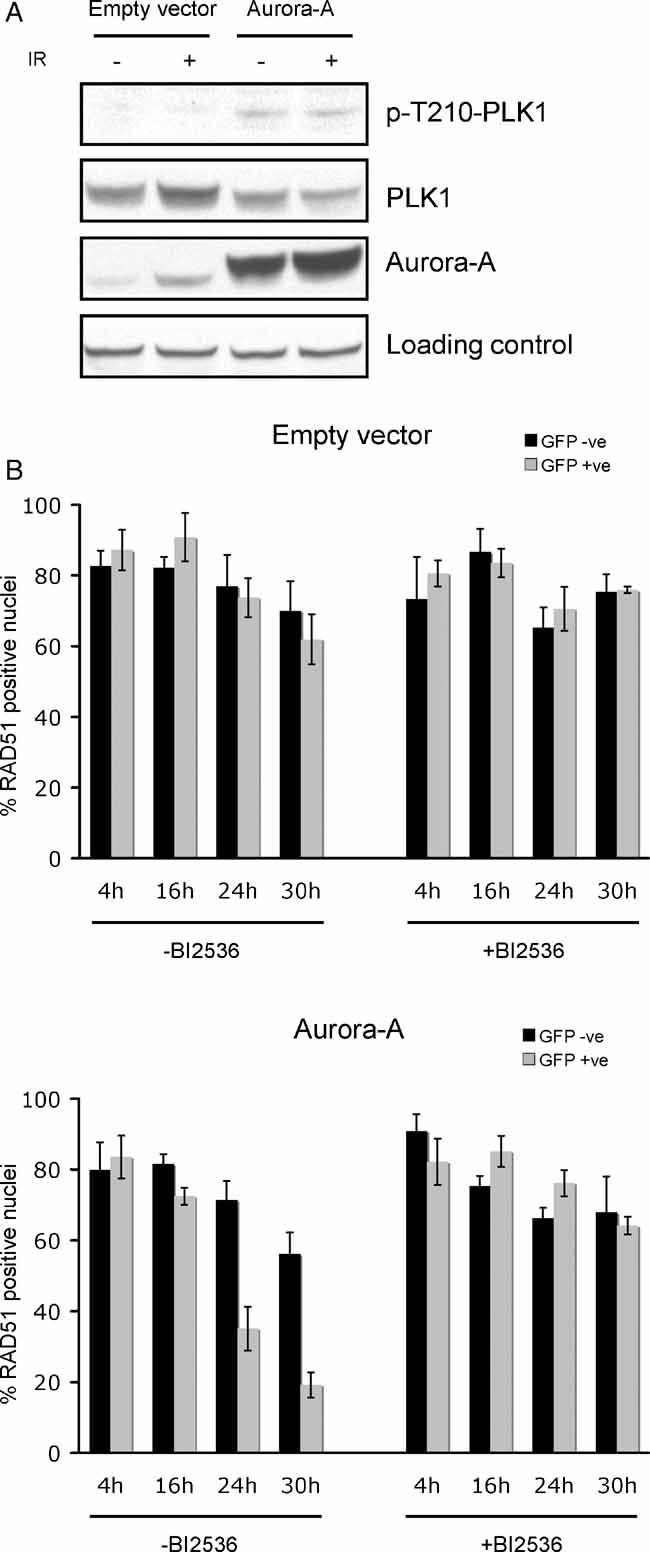
- Immunoblot analysis of total PLK1, threonine 210 phosphorylated PLK1 (p-T210-PLK1) and Aurora-A in non-irradiated or irradiated (5 Gy) MCF10A cells infected with a lentiviral construct expressing GFP alone (empty vector) or GFP and Aurora-A (Aurora-A). Ezrin was used as loading control.
- PLK1 activity is required for the inhibition of RAD51 nuclear focus formation by Aurora-A. MCF10A cells were infected with a lentivirus construct driving the expression of GFP (empty vector) or both GFP and Aurora-A (Aurora-A) and mixed in equal proportion with non-infected cells to provide an internal control. The mixed cells were pre-treated with the PLK1 kinase inhibitor BI2536 (+BI2536) or with vehicle (−BI2536), 1 h before a 5-Gy X-ray treatment, followed by a 4, 16, 24 or 30 h recovery, as indicated. RAD51 foci were then detected by immunofluorescence and RAD51 foci-positive nuclei were scored in uninfected (GFP negative) and infected (GFP positive) cells as described in Fig 1B. Shown is the average percentage of RAD51 foci positive cells from two independent experiments. Error bars indicate SEM.
To address whether the constitutive activation of PLK1 by Aurora-A could affect CHK1 activity, we immunoprecipitated CHK1 from MCF10A cells with varying levels of Aurora-A expression, and assessed the ability of CHK1 to phosphorylate the substrate myelin binding protein (MBP) in vitro. Both, basal and DNA-damage-induced CHK1 activities were decreased in MCF10A cells overexpressing Aurora-A (Fig 5A, B). Similar samples were analysed by immunoblot in order to directly monitor the phosphorylation status of CHK1. Two major activating phosphorylations of CHK1, on serine 317 and serine 345 (p-Ser317-CHK1 and p-Ser345-CHK1, respectively) were decreased in cells expressing high levels of Aurora-A (Fig 5C). The effect of Aurora-A was most likely specific to CHK1, as the activity of CHK2, when monitored by its autophosphorylation on the threonine 68 (p-T68-CHK2) residue, was not affected (Fig 5C). Finally, both basal and radiation-induced Claspin levels were also decreased by the presence of high Aurora-A levels (Fig 5C). Taken together, these results suggest that Aurora-A modulates CHK1 activity through PLK1-triggered Claspin degradation.
Figure 5. Aurora-A overexpression correlates with decreased CHK1 activity.
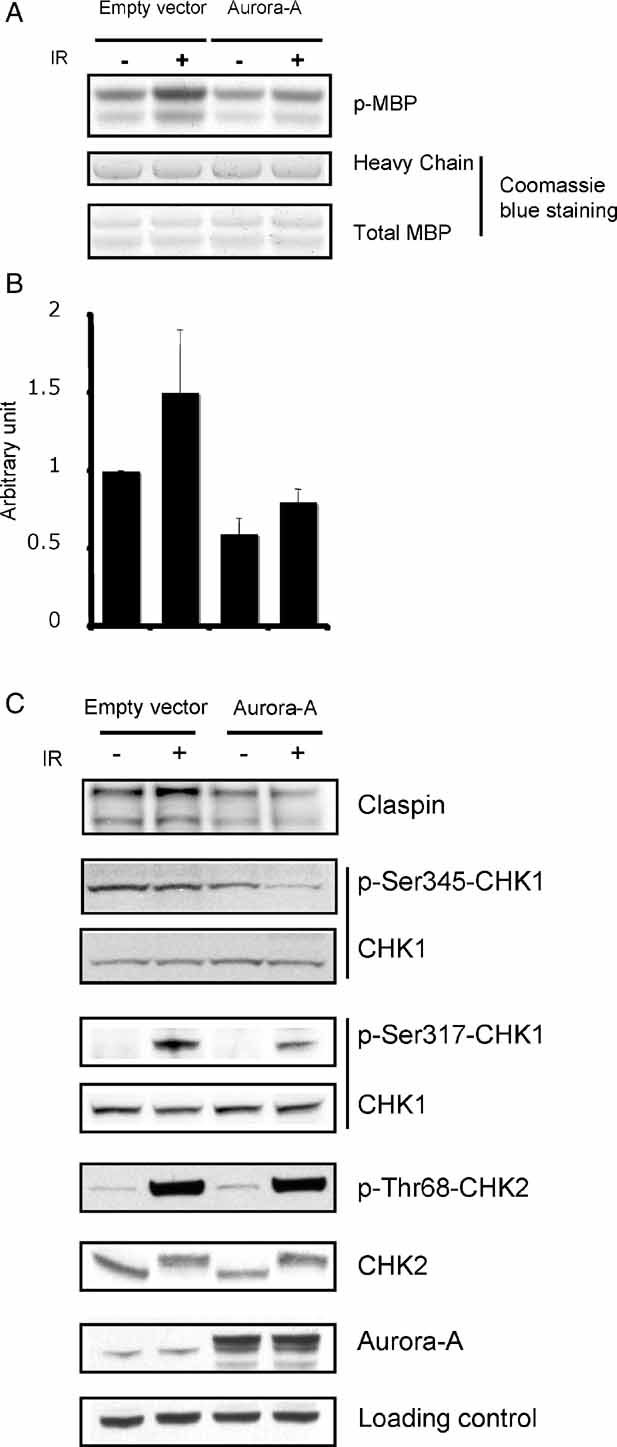
- MCF10A cells infected with a lentiviral construct expressing Aurora-A (Aurora-A) or the corresponding empty vector (empty vector) were treated by 5-Gy X-ray, followed by 24 h recovery. Cell lysates were prepared and immunoprecipitated CHK1 kinase activity was measured by in vitro kinase assay using MBP as a substrate. Equal levels of total MBP and IgG heavy chain were monitored by Coomassie blue staining.
- Quantification of CHK1-induced MBP phosphorylation. Kinase assay signals from (A) were quantified. The average of two independent experiments was plotted. Error bars indicate SEM.
- The amount of Claspin, total CHK1, Serine 317-phosphorylated CHK1 (p-Ser317-CHK1), Serine 345-phosphorylated CHK1 (p-Ser345-CHK1), total CHK2, threonine 68-phosphorylated CHK2 (p-T68-CHK2) and Aurora-A were determined in the cell lysates used in (A) by immunoblotting. Ezrin was used as loading control.
Increased radiosensitivity of MCF10A cells overexpressing Aurora-A
Deficient HR causes sensitivity to ionizing radiation. Therefore, we examined the effect of high level Aurora-A expression on the radiosensitivity of normal human mammary epithelial cells (MCF10A) using a clonogenic survival assay (Fig 6A). In this assay, MCF10A cells were infected with lentiviral vectors expressing either high levels of both Aurora-A and GFP (from a bicistronic mRNA; Aurora-A) or GFP alone (empty vector; Fig 6C). The ability of these cells to form colonies after irradiation was assessed. As expected, we observed a dose-dependent decrease of the colony formation. Moreover, the overexpression of Aurora-A substantially aggravates the effect of irradiation, when compared to cells expressing GFP alone. To confirm these results, we performed a GFP competition assay in which empty vector-, or Aurora-A-infected cells were mixed with non-infected cells used as an internal control (Fig 6B). As in the colony formation assay, Aurora-A overexpression sensitizes MCF10A cells to irradiation in a dose-dependent manner. This suggests that Aurora-A expression modulates radiosensitivity and provided further evidence that high levels of Aurora-A might interfere with HR.
Figure 6. Aurora-A overexpression enhances cell sensitivity to irradiation and to PARP inhibition.
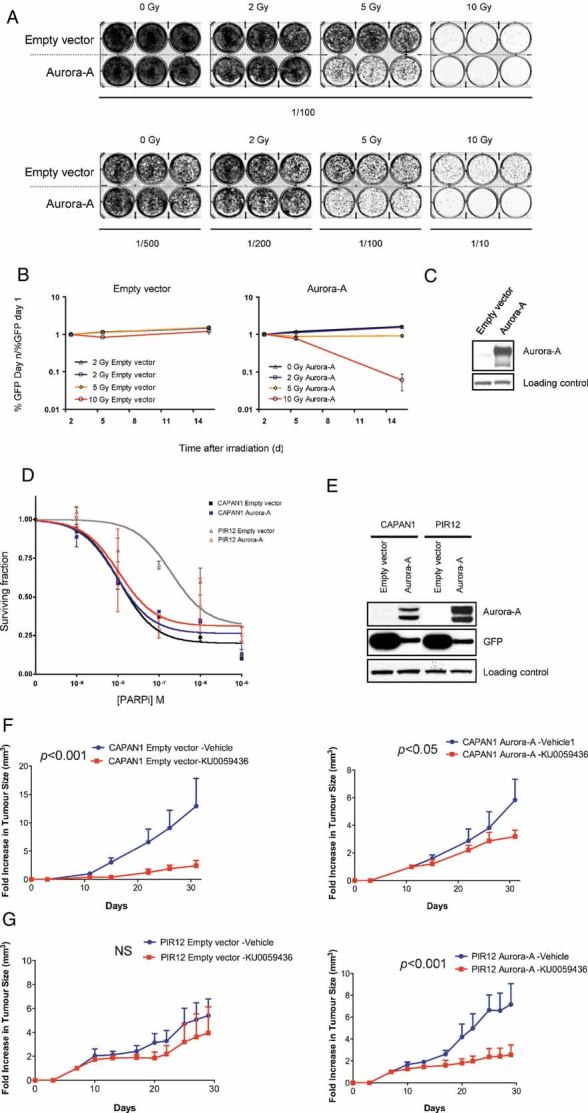
- A. MCF10A cells were infected with a lentivirus construct driving the expression of GFP (empty vector) or both GFP and Aurora-A (Aurora-A), and then exposed to 0-, 2-, 5- or 10-Gy X-ray. After a 7 days recovery, cells were re-plated at the indicated dilutions, left to grow for 7 days and stained with crystal violet.
- B. MCF10A cells were infected with a lentivirus construct driving the expression of GFP (empty vector) or both GFP and Aurora-A (Aurora-A) and mixed in equal proportion with non-infected cells to provide an internal control. The mixed cells were exposed to 0-, 2-, 5- or 10-Gy X-Ray, as indicated. GFP expression was determined by flow cytometry at the indicated time and the proportion of infected, GFP-positive cells in the live cells population was plotted. Error bars represent SEM.
- C. Cell lysates were prepared from infected MCF10A cells used in (B) and the expression of Aurora-A was determined by immunoblotting. Ezrin was used as loading control.
- D. CAPAN1 and PIR12 cells were infected with a lentivirus construct driving the expression of GFP (empty vector) or both GFP and Aurora-A (Aurora-A), and exposed to the PARP inhibitor KU00589482 or to vehicle for sixty days. Cell viability was measured as described in Material and Methods. The mean and SD of a representative experiment with internal triplicate are shown.
- E. Cell lysates were prepared from CAPAN-1 and PIR12 cells used in (D). The expression of Aurora-A and GFP were determined by immunoblotting. Ezrin was used as loading control.
- F, G. CAPAN1 and CAPAN1 cells overexpressing Aurora-A (F) and PIR12 and PIR12 overexpressing Aurora-A (G) were injected into the lateral flanks of athymic nude mice. The results are expressed as fold increase in tumour volume, ±SEM. p-values were calculated by two-way ANOVA.
Aurora-A overexpression confers sensitivity to PARP inhibition
PARP inhibition generates DNA lesions that are normally repaired by HR, and accordingly cells with compromised HR are highly sensitive to PARP inhibition (Ashworth, 2008). It has been previously shown that the CAPAN1 pancreatic tumour cell line, which carries the protein-truncating c.6174delT frameshift mutation (Goggins et al, 1996), is highly sensitive to PARP inhibitors such as KU0058948 (Edwards et al, 2008; McCabe et al, 2005). We have previously generated a CAPAN1-derived PARP inhibitor-resistant cell line, PIR12, which carries a functional BRCA2 gene and is thus HR competent (Edwards et al, 2008). We used this model to explore the effect of high level of Aurora-A expression on PARP inhibitor sensitivity. Ectopic expression of Aurora-A sensitized the normally HR-competent PIR12 cell line to the potent PARP inhibitor KU0058948 (Farmer et al, 2005; Fig 6D, E). Conversely, high level Aurora-A expression was unable to increase PARP inhibitor sensitivity in the isogenic, HR deficient, CAPAN1 cell line. Finally, to assess the in vivo selectivity of PARP inhibition towards Aurora-A overexpressing tumours, we performed xenograft studies in nude mice using CAPAN1 (Fig 6F) and PIR12 (Fig 6G) cells expressing normal or high levels of Aurora-A. Mice were treated with the PARP inhibitor KU0058948. Similarly to our in vitro data (Fig 6D), the CAPAN1 tumours were sensitive to the PARP inhibitor, regardless of Aurora-A status (p < 0.001 and <0.05, respectively). The PIR12 tumours, however, were resensitized to PARP inhibition by overexpression of Aurora-A (p < 0.001). Our data showing radiosensitization of Aurora-A overexpressing cells combined with the finding that PARP inhibitor sensitivity was increased by high Aurora-A expression, further suggest that HR is suppressed and that these cells may be sensitized to agents targeting deficiencies in this DNA repair pathway.
CONCLUSION
Here, we show that Aurora-A overexpressing cells have suppressed HR. Our results suggest a signalling cascade emanating from Aurora-A, where activated PLK1 represses CHK1 activity through Claspin regulation, resulting in inhibition of RAD51 function and decreased HR (Fig 7). HR activity has been recently reported to be repressed before and upon mitotic entry (Mao et al, 2008b). This repression of HR has conceivably evolved to prevent annealing of DNA between sister chromatids as they are scheduled to segregate into daughter cells. Interestingly, the repression of HR that occurs upon mitotic entry parallels the normal expression and activation pattern of Aurora-A. Repression of HR upon entry to mitosis might be part of the normal function of Aurora-A.
Figure 7. Model for the regulation of the DNA damage response by Aurora-A.

- During the S and G2 phases of the cell cycle, HR is the preferred pathway to repair DSBs. Activated CHK1 stimulates repair by phosphorylating the RAD51 recombinase, while stalling the cell cycle by keeping CDK1 inactive. CDK1 inactivation is mediated by a number of pathways, including inactivation of CDC25 phosphatases and Wee1 kinase by CHK1.
- High levels of Aurora-A result in constitutive activation of the PLK1 kinase. Claspin is degraded upon phosphorylation by PLK1, an event normally restricted to the G2 checkpoint recovery phase. The inactivation of CHK1 that ensues weakens the response of HR protein, such as RAD51 accumulation to the DSBs. Consequently, DNA is repaired by the constitutively operative NHEJ pathway. The shortening of the cell cycle arrest observed in Aurora-A overexpressing cells after irradiation might be a result of both the activation of a faster DNA repair process, such as the NHEJ pathway and the stimulation of CDK1.
We did not observe persistence of γ-H2AX signal in cells overexpressing Aurora-A, suggesting that the repair might be fulfilled by an alternative error-prone mechanism. A series of reports have demonstrated that in the absence of functional HR, error-prone repair mechanisms predominate (Moynahan et al, 2001; Tutt et al, 2001). Amongst the possible alternative mechanisms to HR, NHEJ is a good candidate as the repaired DSBs in HR deficient cells are characterized by deletions flanked by short tandem repeats (Edwards et al, 2008) which is reminiscent of the repair performed by NHEJ mechanism. The use of NHEJ, which is a faster process compared to HR, is also suggested by the shorter cell cycle arrest observed in Aurora-A overexpressing cells (Fig 2A, B; Mao et al, 2008a).
Our results suggest that overexpression of Aurora-A results in a repression of the error-free HR pathway, presumably favouring a lower-fidelity process, such as NHEJ. This might consequently increase genomic instability and account, at least in part, for the oncogenic activity of Aurora-A. Finally, several small molecule inhibitors of PARP have shown therapeutic efficacy against tumours deficient in HR in pre- and early clinical studies (Bryant et al, 2005; Farmer et al, 2005; Fong, 2009). Our results suggest that Aurora-A overexpression decreases the repair of DSB by HR pathways and confers sensitivity to PARP inhibition. This latter observation may have important implications for the development of cancer therapeutics aimed at targeting tumour cells with Aurora-A overexpression. Moreover, previous studies have reported a lack of correlation between the sensitivity of cells treated with Aurora kinase inhibitors and Aurora-A levels (Chan et al, 2007; Soncini et al, 2006) suggesting that methods other than targeting Aurora-A are required. This raises the possibility of extending the therapeutic utility of PARP inhibition to tumours with high levels of Aurora-A expression.
MATERIALS AND METHODS
Inhibitors
Aurora-B inhibitor ZM447439 (Ditchfield et al, 2003) and PLK1 inhibitor BI2536 (Steegmaier et al, 2007) were obtained from Tocris Bioscience and AxonMedchem, respectively. Aurora-A inhibitor MLN8054 (Manfredi et al, 2007) was synthesized in-house. The PARP inhibitor KU0058948 was provided by KuDOS Pharmaceuticals.
Cell culture and transfection
HEK 293 and the packaging line 293T were grown in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% foetal calf serum (FCS), 2.4 mM l-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. MCF10A cells were grown in DMEM/F12 (1:1) medium (Gibco) supplemented with 5% horse serum, epidermal growth factor (20 ng/ml) (Peprotech), hydroxycortisone (0.5 µg/ml), cholera toxin (100 ng/ml), insulin (10 µg/ml) (Sigma–Aldrich), penicillin (60 µg/ml) and streptomycin (100 µg/ml). CAPAN1 and PIR12 cells were grown in DMEM (Gibco) supplemented with 20% FCS, 2.4 mM l-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin. ES cells were grown on mitomycin-inactivated mouse embryonic fibroblasts (MEF) as feeder cells, or on a gelatin-coated substrate in DMEM (Sigma) supplemented with 15% FCS, 2 mM l-glutamine (Sigma), MEM-non-essential amino acids (Gibco), 50 µM 2-mercaptoethanol (Gibco) and 100 U/ml (when grown on feeder MEFs) or 200 U/ml (when grown on gelatin-coated substrate) LIF (Esgro, Chemical International).
The paper explained
PROBLEM
The Aurora-A gene is frequently upregulated in a variety of cancers, including breast and colon cancer. The clinical application of inhibitors of Aurora-A kinase activity is now being evaluated in clinical trials. However, there is a need for a better understanding of the oncogenic properties of Aurora-A to identify the relevant patient population for treatment.
RESULTS
We demonstrate that deregulation of Aurora-A in normal and cancer cell lines affects DNA damage repair. Aurora-A represses the HR repair pathway and confers increased cellular sensitivity to PARP inhibitors (known to target cancer cells with deficient DNA repair pathways) in vitro and in vivo.
IMPACT
The data shed light on the molecular mechanisms underlying Aurora-A tumorigenic properties and we propose using PARP inhibitors as a new therapeutic approach to target tumour cells with upregulated Aurora-A.
Transfection of 293T packaging cells, 293 and ES cells was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Plasmids and lentiviruses
A human myc-tagged Aurora-A cDNA was PCR amplified using the following primers containing a PmeI restriction site: AurA F 5′-GTTTAAACATGGAGCAGAAGCTG-3′, AurA R 5′-GTTTAAACCTAAGACTGTTTGCT-3′ and pCMV-myc-Aurora-A as a template. The amplification product was inserted into PCR2.1 (Invitrogen). The cDNA was released by PmeI digestion before insertion into PmeI linearized pWPI lentiviral expression plasmid (addgene). Mutations in Aurora-A cDNA were performed by site directed mutagenesis. The human Gadd45a cDNA was a kind gift from Richard Bayliss. The cDNA was released from the pET30 plasmid by digestion with EcoRI and NcoI, treated with Klenow DNA polymerase to generate blunt ends and inserted into the PmeI linearized pWPI.
The lentivirus production in 293T packaging cells was performed following the guidelines provided by Didier Trono's laboratory (available online http://tronolab.epfl.ch/). Briefly, 293T cells were co-transfected as described above with the pWPI expression plasmids, the packaging plasmid psPAX2 and the envelope vector pMD2.G. Transfection medium was changed after 16 h, and the lentivirus-containing medium was subsequently harvested every 24 h for 3 days and frozen at −80°C. Infectivity of the viral solution was titrated on 293T cells. MCF10A, CAPAN1, PIR12 and HEK293 were infected by adding viral particles to the growing medium for at least 24 h.
Immunofluorescence
Cells grown on a glass coverslip were fixed in PBS–4% paraformaldehyde for 1 h, permeabilized in PBS–0.5% Triton X-100 for 15 min, blocked in PBS–2%FCS–1%BSA, before incubation with rabbit-anti-RAD51 (Santa Cruz) and mouse-anti-γ-H2AX (Upstate) antibodies. Secondary antibodies were Alexa Fluor® 555-conjugated goat anti-rabbit IgG (H + L) and Alexa Fluor® 633-conjugated goat anti-mouse IgG (H + L) (Invitrogen). DNA was conterstained with 4′,6-diamidino-2-phenylindole (DAPI). Image acquisition was performed on a Leica SP2 confocal scanning microscope (Leica Microsystems, Milton Keynes, Bucks, UK).
Flow cytometry
Flow cytometry data acquisition was performed on a BD LSR II flow cytometer (BD). Analysis was carried out using the FacsDiva software (BD). For all flow cytometry analysis, live cells were gated based on a FSC-A versus SSC-A dot plot. GFP was detected using the FITC-A channel. DNA profiling was performed by propidium iodide (PI) staining of cold ethanol fixed-cells after RNAse treatment (ES cells) using the PE-Cy5 channel, or by live staining with Hoechst 33342 (Pacific blue channel). In both cases, single cells were selected for analysis based on SSC-W versus SSC-A and PE-Cy5-W versus PE-Cy5-A (PI staining) or Pacific blue-W versus Pacific blue-A (Hoechst 33342 staining) plot gating.
Immunoblotting, immunoprecipitation and kinase assay
Proteins were extracted in lysis buffer (50 mM HEPES pH 7.4, 250 mM NaCl, 0.5% Igepal CA-630 (Sigma), protease inhibitor (Complete, Roche), 0.1 mM Na3VO4, 1 mM NaF, 10 mM β-glycerophosphate). The following antibodies were used for immunoblotting: Rabbit anti-Aurora-A, anti-p-Ser317-CHK1, anti-p-Ser345-CHK1 and anti-p-Thr68-CHK2 (Cell Signalling), rabbit anti-Claspin (Abcam), mouse anti-Aurora-A and mouse anti-GFP (clone JL-8) (BD Bioscience), goat anti-CHK2, mouse anti-CHK1 (Santa Cruz), mouse anti-p-T210-PLK1 (BD Pharmingen) and mouse anti-PLK1 (Upstate). The anti-Ezrin antibody used for loading control was a kind gift from Prof. Clare Isacke.
For immunoprecipitation, 500 µg of protein was diluted 1:10 in binding buffer [PBS supplemented with protease inhibitor (Complete, Roche), 0.1 mM Na3VO4, 1 mM NaF, 10 mM β-glycerophosphate] and incubated with 2 µg of mouse anti-CHK1 antibody (Santa Cruz) overnight at 4°C. Immunocomplexes were pulled-down by addition of Protein A-AgarosePlus (Santa Cruz) and washed three times in binding buffer, resuspended in 1× kinase assay buffer (50 mM Tris pH 7.5, 10 mM NaCl, 2.5 mM MgCl2, 1 mM DTT). The kinase reaction was performed in the presence of 0.5 mg/ml MBP, 10 µM cold ATP, 2 µCi γ-33P-ATP (3000 Ci/mmol, 10 mCi/ml, PerkinElmer), at 25°C for 30 min. Proteins were then resolved in SDS–PAGE before autoradiography. Band density was measured using the ImageJ software. The gel was subsequently rehydrated and Coomassie stained (SimplyBlue SafeStain, Invitrogen).
Homologous recombination reporter assay
293 cells bearing the stably integrated DNA repair reporter construct (see Fig 3; Tutt et al, 2001) were infected with lentiviruses expressing GFP alone or GFP along with Aurora-A (wild type or mutants) or Gadd45a. Cells were subsequently co-transfected with a plasmid expressing an NLS-tagged I-SceI restriction enzyme and a DsRed expressing vector. After 24 h, the transfection efficiency was assessed by flow cytometry quantification of DsRed positive cells. For colony formation assay, cells were plated in six-well plates in the presence of 5 µg/ml of blasticidin (Invitrogen; 1.2 × 106 cells) or without (3 × 105 cells) antibiotic. After 21 days, cells were fixed and stained with crystal violet. Colonies were counted in a ColCount (Oxford Optronics). For the GFP competition assay, GFP positive cells were quantified by flow cytometry at various time during the selection process.
PARP inhibitor sensitivity assay
Exponentially growing cells were seeded in 24-well plates and exposed continuously to the PARP inhibitor KU0058948 (gift from KuDOS/AstraZeneca) at concentrations ranging from 10−9 to 10−5 M, or DMSO. Medium and inhibitor were replaced every 4 days. PARP inhibitor- or DMSO-treated cells were split equally when appropriate. After 60 days the survival of KU0058948 versus DMSO-treated cells was measured using a luminescent cell viability assay (CellTiter-Glo, Promega).
Xenografts
CAPAN1 and PIR12 cells (4 × 106) infected with empty lentiviral vector (empty vector) or a vector expressing Aurora-A (Aurora-A) were mixed 1:1 in matrigel (BD Biosciences) and then injected subcutaneously into the lateral flank of 6- to 8-week-old female athymic nude mice (12 animals per cohort, 24 in total). The mice were allowed to recover for 2 days and then treated with KU0058948 or vehicle alone. PARP inhibitor (or vehicle) was administered by intraperitoneal injection at a dose of 15 mg/kg in 10% 2-hydroxypropyl-b-cyclodextrin (HBC; Farmer et al, 2005) for five consecutive days followed by 2 days of no treatment after which the same treatment cycle was continued until the end of the study. Tumour volumes were measured every 2–4 days from the initiation of tumour growth. Tumours were measured every 2–4 days after the initiation of drug dosing. The results are expressed as fold increase in tumour volume, ±SEM. p-values were calculated by two-way ANOVA.
Acknowledgments
We thank G. Smith, N. Martin and M. O'Connor (KuDOS Pharmaceuticals) for providing KU0058948 and V. Bavetsias and B. Atrash for synthesizing MLN8054. We thank Professor Steve Jackson for technical advice and insightful comments on this work. We thank members of the Cancer Drug Target Discovery Team and P. Workman for comments on the manuscript, D. Robertson for his assistance with confocal microscopy and members of the Gene Function Team for technical advice. This study was supported by Breakthrough Breast Cancer and Cancer Research UK [CRUK] grant number C309/A8274.
Supporting information is available at EMBO Molecular Medicine online.
Alan Ashworth may benefit financially from the development of PARP inhibitors through The Institute of Cancer Research ‘rewards to inventors’ scheme as a result of patents held jointly between The Institute of Cancer Research and KuDOS/AstraZeneca.
Author contributions
TS and SL designed the experiments. TS performed most of the experiments. DM and CT performed in vitro experiments. AMC performed the xenograft study. CJL and AA provided reagents and critical comments. TS and SL analysed the experiments and wrote the manuscript.
For more information
The Breakthrough Research Centre:
http://www.breakthroughresearch.org.uk/breakthrough_research_centre/index.html
The Cancer Therapeutics:
http://www.icr.ac.uk/research/research_sections/cancer_therapeutics/index.shtml
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–3790. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004;23:4868–4875. doi: 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibby RA, Tang C, Faisal A, Drosopoulos K, Lubbe S, Houlston R, Bayliss R, Linardopoulos S. A cancer-associated aurora A mutant is mislocalized and misregulated due to loss of interaction with TPX2. J Biol Chem. 2009;284:33177–33184. doi: 10.1074/jbc.M109.032722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff JR, Plowman GD. The Aurora/Ipl1p kinase family: regulators of chromosome segregation and cytokinesis. Trends Cell Biol. 1999;9:454–459. doi: 10.1016/s0962-8924(99)01658-x. [DOI] [PubMed] [Google Scholar]
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–854. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- Cazales M, Schmitt E, Montembault E, Dozier C, Prigent C, Ducommun B. CDC25B phosphorylation by Aurora-A occurs at the G2/M transition and is inhibited by DNA damage. Cell Cycle. 2005;4:1233–1238. doi: 10.4161/cc.4.9.1964. [DOI] [PubMed] [Google Scholar]
- Chan F, Sun C, Perumal M, Nguyen QD, Bavetsias V, McDonald E, Martins V, Wilsher NE, Raynaud FI, Valenti M, et al. Mechanism of action of the Aurora kinase inhibitor CCT129202 and in vivo quantification of biological activity. Mol Cancer Ther. 2007;6:3147–3157. doi: 10.1158/1535-7163.MCT-07-2156. [DOI] [PubMed] [Google Scholar]
- Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi: 10.1038/nature03404. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Swaisland H, Lau A, O'Conner MJ, Ashworth A, et al. A synthetic lethal cancer therapy: poly(ADP)-ribose polymerase (PARP) inhibition is well-tolerated and shows antitumour activity in BRCA1 and BRCA2 mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- Giet R, Prigent C. Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine-threonine kinases. J Cell Sci. 1999;112:3591–3601. doi: 10.1242/jcs.112.21.3591. [DOI] [PubMed] [Google Scholar]
- Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, Yeo CJ, Jackson CE, Lynch HT, Hruban RH, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56:5360–5364. [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, Fiorica JV, Nicosia SV, Cheng JQ. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003;9:1420–1426. [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, Hatakeyama K, Saya H. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–598. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Jackson SP. Human CtIP mediates cell-cycle control of DNA-end resection and double-strand-break repair. J Biol Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Zhang Y, Lees E, Seghezzi W. AuroraA overexpression overrides the mitotic spindle checkpoint triggered by nocodazole, a microtubule destabilizer. Oncogene. 2003;22:8293–8301. doi: 10.1038/sj.onc.1206873. [DOI] [PubMed] [Google Scholar]
- Kallioniemi A, Kallioniemi OP, Piper J, Tanner M, Stokke T, Chen L, Smith HS, Pinkel D, Gray JW, Waldman FM. Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization. Proc Natl Acad Sci USA. 1994;91:2156–2160. doi: 10.1073/pnas.91.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Krystyniak A, Garcia-Echeverria C, Prigent C, Ferrari S. Inhibition of Aurora A in response to DNA damage. Oncogene. 2006;25:338–348. doi: 10.1038/sj.onc.1209056. [DOI] [PubMed] [Google Scholar]
- Lee M, Daniels MJ, Venkitaraman AR. Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene. 2004;23:865–872. doi: 10.1038/sj.onc.1207223. [DOI] [PubMed] [Google Scholar]
- Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455:119–123. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, Perrakis A, Medema RH, Freire R. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr Biol. 2006;16:1950–1955. doi: 10.1016/j.cub.2006.08.026. [DOI] [PubMed] [Google Scholar]
- Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, Galvin KM, Hoar KM, Huck JJ, LeRoy PJ, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci USA. 2007;104:4106–4111. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 2008a;7:1765–1771. doi: 10.1016/j.dnarep.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008b;7:2902–2906. doi: 10.4161/cc.7.18.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe N, Lord CJ, Tutt AN, Martin NM, Smith GC, Ashworth A. BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of Poly (ADP-Ribose) polymerase: an issue of potency. Cancer Biol Ther. 2005;4:934–936. doi: 10.4161/cbt.4.9.2141. [DOI] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J. 2002;21:483–492. doi: 10.1093/emboj/21.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr Opin Genet Dev. 2004;14:29–36. doi: 10.1016/j.gde.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell. 2006;23:319–329. doi: 10.1016/j.molcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Sasai K, Parant JM, Brandt ME, Carter J, Adams HP, Stass SA, Killary AM, Katayama H, Sen S. Targeted disruption of Aurora A causes abnormal mitotic spindle assembly, chromosome misalignment and embryonic lethality. Oncogene. 2008;27:4122–4127. doi: 10.1038/onc.2008.47. [DOI] [PubMed] [Google Scholar]
- Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320:1655–1658. doi: 10.1126/science.1157425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soncini C, Carpinelli P, Gianellini L, Fancelli D, Vianello P, Rusconi L, Storici P, Zugnoni P, Pesenti E, Croci V, et al. PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res. 2006;12:4080–4089. doi: 10.1158/1078-0432.CCR-05-1964. [DOI] [PubMed] [Google Scholar]
- Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, Griffin C, Thacker J, Ashworth A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20:4704–4716. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, Brinkley BR, Sen S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.