

Abstract
Targeted cancer therapy requires the rapid and accurate identification of genetic abnormalities predictive of therapeutic response. We sought to develop a high-throughput genotyping platform that would allow prospective patient selection to the best available therapies, and that could readily and inexpensively be adopted by most clinical laboratories. We developed a highly sensitive multiplexed clinical assay that performs very well with nucleic acid derived from formalin fixation and paraffin embedding (FFPE) tissue, and tests for 120 previously described mutations in 13 cancer genes. Genetic profiling of 250 primary tumours was consistent with the documented oncogene mutational spectrum and identified rare events in some cancer types. The assay is currently being used for clinical testing of tumour samples and contributing to cancer patient management. This work therefore establishes a platform for real-time targeted genotyping that can be widely adopted. We expect that efforts like this one will play an increasingly important role in cancer management.
Keywords: cancer, genotyping, profiling, targeted therapies
→See accompanying article:
http://dx.doi.org/10.1002/emmm.201000071
Introduction
The clinical management of cancer patients has traditionally relied on chemotherapeutic choices that are mostly dictated by pathologic tumour histology and organ of origin. In recent years, major efforts to define the molecular causes of cancer have revealed a wide number of genetic aberrations (Davies et al, 2005; Ding et al, 2008; Greenman et al, 2007; Rikova et al, 2007; Sjoblom et al, 2006; Stephens et al, 2005; Thomas et al, 2007; Wood et al, 2007). A small subset of these defects, usually referred to as ‘drivers’, is frequently present across cancer types and appears to be essential for oncogenesis and tumour progression (Greenman et al, 2007). A new generation of drugs has been developed to selectively target such cancer-promoting pathways, (Druker et al, 2001; Hanahan & Weinberg, 2000; Weinstein, 2000) and hence treatment dictated by genetic markers is starting to complement the more conventional therapeutic approaches.
While the clinical benefit observed with some targeted agents is encouraging, it has become clear that for such strategies to be successful, it will be necessary to identify the patient population carrying the genetic abnormalities targeted by each drug (McDermott et al, 2007; Sos et al, 2009). In advanced non-small cell lung cancer (NSCLC), activating mutations in the region encoding the kinase domain of the epidermal growth factor receptor (EGFR) gene predict tumour sensitivity to the tyrosine kinase inhibitors (TKI) erlotinib and gefitinib (Lynch et al, 2004; Paez et al, 2004; Pao et al, 2004; Sordella et al, 2004). Since NSCLC patients harbouring EGFR mutations benefit from these specific inhibitors in the first-line setting compared to standard chemotherapy (Mok et al, 2009), and only a small fraction of NSCLCs harbour these mutations, prospective screening for EGFR mutations at the time of diagnosis is becoming common practice (Sharma et al, 2007). Equally important is the identification of mutations that render tumours resistant to therapy. Activating mutations in KRAS predict resistance to EGFR TKI treatment in NSCLC (Pao et al, 2005b). In metastatic colorectal cancer, mutations in KRAS, BRAF and PIK3CA are associated with resistance to treatment with monoclonal antibodies cetuximab and panitumumab, which target the extracellular domain of EGFR (Di Nicolantonio et al, 2008; Lievre et al, 2006; Sartore-Bianchi et al, 2009). Similarly, in breast cancer, oncogenic mutations in PIK3CA or low levels of PTEN expression, may confer resistance to treatment with trastuzumab, a monoclonal antibody that targets the HER2/NEU receptor (Berns et al, 2007).
As the repertoire of selective therapeutic compounds continues to expand, the need to evaluate larger numbers of genetic mutations will be a major challenge (Chin & Gray, 2008). In addition to the dilemma of selecting the most relevant abnormalities, the tissue samples themselves pose many obstacles, including minute specimens derived from small core biopsies, poor quality fragmented nucleic acid due to the formalin fixation and paraffin embedding (FFPE) required for histology-based diagnosis (Srinivasan et al, 2002), and heterogeneous tumour samples comprised of normal tissue and cancerous cells which dilute the mutant alleles of interest. Thus, a useful clinical assay will have to: (1) be multiplexed, to maximize information retrieval from limited tissue; (2) perform well with FFPE-derived material and (3) be sensitive enough to detect low-level mutations. Additionally, the turn-around-time for the entire specimen processing and mutation detection platform has to be quick, in order to integrate into the rapid pace of clinical decision making and impact patient management.
Taking all of these constraints into account, we developed a robust and highly sensitive tumour genotyping assay that is currently being used for real-time testing of tumours, and assisting physicians in directing their cancer patients to the most appropriate targeted therapies.
RESULTS
Assay design and validation
In order to develop a robust assay for clinical tumour genotyping, several high-throughput platforms were evaluated for the ability to detect low-level mutations in DNA extracted from FFPE tissues. The SNaPshot assay from Applied Biosystems consisting of a multiplexed PCR step followed by a single-base extension reaction that generates allele-specific fluorescently labelled probes (Fig 1) was ultimately selected given its low background noise, high sensitivity, and good performance with FFPE-derived DNA in a multiplexed setting. Moreover, genetic analysis using the SNaPshot methodology follows a simple workflow, with the only major instrumentation requirement being a capillary electrophoresis automated DNA sequencer. The SNaPshot system is particularly attractive because virtually all clinical laboratories already have at least one of these sequencers, hence avoiding additional capital expenses and facilitating rapid implementation by most clinical testing sites.
Figure 1. Schematic representation of SNaPshot genotyping.
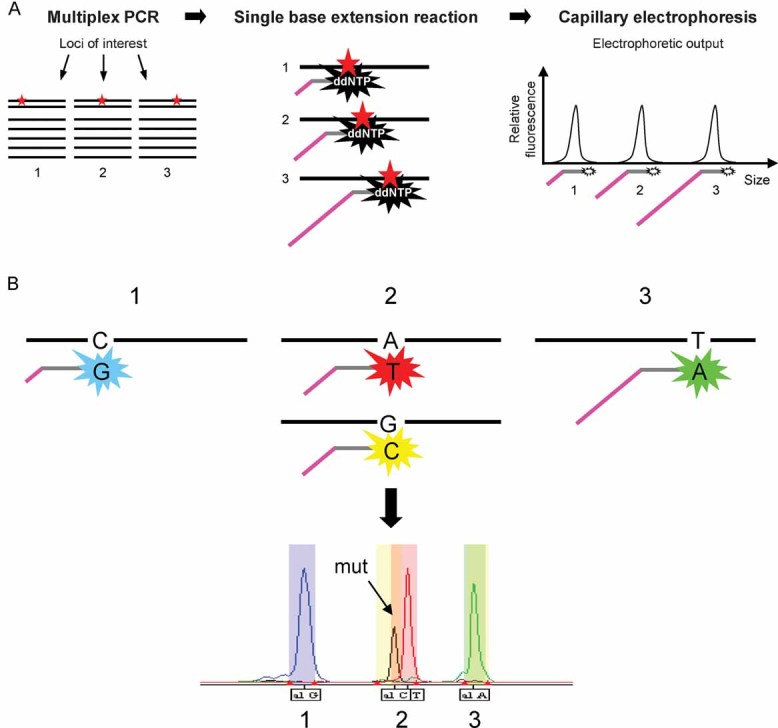
- The SNaPshot system follows a straightforward protocol and uses infrastructure already existent in most clinical laboratories. This method consists of a multiplexed PCR step, followed by a single-base extension sequencing reaction, in which allele-specific probes interrogate loci of interest and are fluorescently labelled using dideoxynucleotides. These probes are designed to have different sizes and are subsequently resolved by electrophoresis and analysed by an automated DNA sequencer. Thus, the identity of each locus is given by the position of its corresponding fluorescent peak in the spectrum, which is dictated by the length of the extension primer.
- Detailed view of the single-base extension reaction. The identity of the nucleotide(s) present at each locus is given by two parameters: the molecular weight and the colour of the fluorescently labelled ddNTPs added to the allele specific probes during the extension step. Thus, mutant and wild-type alleles can be distinguished based on the slightly different positions and on the distinct colours of their corresponding peaks. These two factors are used to establish the bins used for automatic data analysis (described in the Supporting Information).
We designed assays to detect recurrent mutations in some of the most important cancer genes, many of which activate cancer signalling pathways that are currently targeted by either Food and Drug Administration (FDA)-approved therapies or by agents in advanced stages of clinical development (Table 1). Our genotyping platform consists of eight multiplexed reactions that query 58 commonly mutated loci within 13 key cancer genes. Since multiple nucleotide variants have been described at most of these sites, the test can detect 120 previously described mutations (Supporting Information Table S1). We focused predominantly on oncogenes over tumour suppressors because aberrantly activated oncogenes are preferential targets for pharmacologic inhibition, and gain-of-function mutations in oncogenes are usually limited to a small set of codons. Accordingly, our assay captures 94–99% of the mutation frequency described for the BRAF, KRAS and JAK2 oncogenes, which are frequently mutated in very few hotspots. Representative spectra of all eight SNaPshot genotyping panels are depicted in Supporting Information Fig S1, which illustrates the good performance of the assay with both high-quality, commercially available genomic DNA (A) and total nucleic acid extracted from FFPE primary tumour tissue from patients (B).
Table 1.
Cancer genes included in the assay and available targeted cancer therapies
| Gene | SNaPshot coverage | Relevant drugs: launched (developer) | Relevant drugs in clinical testing (number of compounds)1 |
|---|---|---|---|
| APC | 15% | None | None |
| BRAF | 94% | Sorafenib (Bayer HealthCare Pharmaceuticals, Onyx Pharmaceuticals) | Raf inhibitors (4) |
| MEK inhibitors (12) | |||
| ERK inhibitor (1) | |||
| CTNNB1 | 74% | None | None |
| Gefitinib (AstraZeneca) | |||
| EGFR | 69% | Cetuximab (ImClone Systems, Merck Serono, Bristol-Myers Squibb) Erlotinib hydrochloride (Genentech, OSI Pharmaceuticals, Roche) Panitumumab (Amgen) Nimotuzumab (YM BioSciences, Biotech Pharmaceuticals, Oncoscience, Daiichi Sankyo) Lapatinib (GlaxoSmithKline) | EGFR inhibitors (26) |
| FLT3 | 22% | Sorafenib (Bayer HealthCare Pharmaceuticals, Onyx Pharmaceuticals) Sunitinib (Pfizer) | FLT3 inhibitors (10) |
| JAK2 | 99% | None | JAK2 inhibitors (4) |
| Imatinib mesylate (Novartis Oncology) | |||
| KIT | 24% | Sorafenib (Bayer HealthCare Pharmaceuticals, Onyx Pharmaceuticals) Sunitinib (Pfizer) | KIT inhibitors (11) |
| KRAS | 98% | None | Raf inhibitors (4) |
| MEK inhibitors (12) | |||
| ERK inhibitor (1) | |||
| NOTCH1 | 9% | None | Notch1/Gamma-Secretase inhibitors (3) |
| NRAS | 97% | None | Raf inhibitors (4) |
| MEK inhibitors (12) | |||
| ERK inhibitor (1) | |||
| PIK3CA | 76% | mTOR inhibitors: Sirolimus (Wyeth Pharmaceuticals) Everolimus (Novartis Pharmaceuticals) Temsirolimus (Wyeth Pharmaceuticals) | PI3K inhibitors (10) |
| PKB/AKT inhibitors (6) | |||
| mTOR inhibitors (13) | |||
| PTEN | 15% | Sirolimus (Wyeth Pharmaceuticals) Everolimus (Novartis Pharmaceuticals) Temsirolimus (Wyeth Pharmaceuticals) | mTOR inhibitors: |
| PI3K inhibitors (10) | |||
| PKB/AKT inhibitors (6) | |||
| mTOR inhibitors (13) | |||
| TP53 | 29% | None | None |
The numbers on the second column reflect the frequency of somatic mutations described for each gene (COSMIC database v42 release) that are captured by SNaPshot genotyping. The data on targeted agents was compiled using the Prous Science database (www.prous.com). Of note, many compounds have multiple targets or overlapping activities.
Cancer trials.
Assay validation was carried out with control DNA harbouring the mutations of interest, which included: primary tumour DNA, cancer cell line DNA and custom-designed synthetic oligonucleotides (Supporting Information Table S1). All SNaPshot assays identified the expected mutations. In addition, allele-specific assays that could be validated using genomic DNA were assessed for sensitivity, which ranged from 11.4 to 1.4% and was on average approximately 5% (Supporting Information Fig S2) an improvement over direct sequencing that is reported to have a sensitivity of about 20% (Hughes et al, 2006). Since allele-specific detection methods test a sequence change at one site, we would not anticipate the sensitivity of each assay to be affected by the mechanism that caused the mutation (point mutation vs. insertion or deletion). Our own experience with the SNaPshot system supports this hypothesis. The sensitivity data summarized in Supporting Information Fig S2 includes 44 assays (39 point mutations and 5 deletions) and the average sensitivity for the deletions (4.69%) was very similar to the average sensitivity for all assays (4.64%).
As an example of validation and sensitivity testing, Fig 2 illustrates SNaPshot analysis for two clinically relevant mutations, KRAS G12D and EGFR T790M, both of which confer resistance to anti-EGFR therapy. In each case, sensitivity was determined using DNA from a cancer cell line harbouring the mutation of interest, serially diluted with commercially available wild-type DNA. The A427 lung carcinoma cell line was used to detect the highly prevalent KRAS G12D mutation (Fig 2A) (Bamford et al, 2004) and the NCI-H1975 lung adenocarcinoma cell line was used to identify the EGFR T790M mutation (Fig 2B), which represents the most commonly described mechanism of acquired resistance to EGFR TKIs in lung cancer (Ladanyi & Pao, 2008; Pao et al, 2005a). In both instances, assay sensitivity was approximately 3% and data quality was very comparable to traditional Sanger sequencing analysis (panels on the right). A detailed illustration of the process used to calculate assay sensitivity for these two cases is shown in Supporting Information Fig S3. Of note, the use of fluorescently labelled probes in the SNaPshot assay enables allele recognition to be contingent on two parameters: slightly different masses and distinct colour readouts. These features facilitate the ability to distinguish low-level mutations from background noise. Finally, while 75% of the assays (33 out of 44) shown in Supporting Information Fig S2 were highly sensitive detecting levels of mutant allele of ≤5%, when analysing samples of unknown genotype we typically use a mutant allele cut-off of 10%, which in our experience is a conservative value that allows us to confidently call a mutation (detailed scoring guidelines are provided as Supporting Information). Additional sensitivity data and examples of assay validation using synthetic oligonucleotide probes are illustrated in Supporting Information Figs S4 and S5.
Figure 2. Sensitivity assessment revealed the ability to detect low-level mutations.
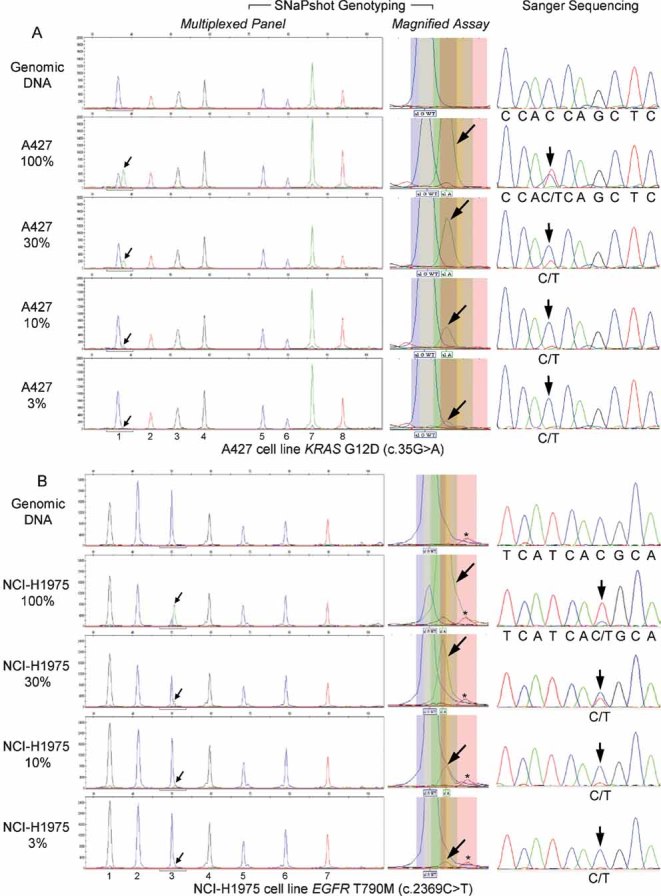
- The A427 lung carcinoma cell line was used to detect the KRAS G12D mutation (nucleotide change 35G > A). Sensitivity was ∼3% and the SNaPshot panel includes the following assays: (1) KRAS 35; (2) EGFR 2236_50del R; (3) PTEN 517; (4) TP53 733; (5) FLT3 2503; (6) PIK3CA 3139; (7) NOTCH1 4724 and (8) NOTCH1 4802.
- The NCI-H1975 lung adenocarcinoma cell line was used to identify the EGFR T790M mutation (nucleotide change 2369C > T). Assay sensitivity was ∼3% and the SNaPshot panel tests for: (1) KRAS 34; (2) EGFR 2235_49del F; (3) EGFR 2369; (4) NRAS 181; (5) PIK3CA 1633; (6) CTNNB1 94 and (7) CTNNB1 121. As can be appreciated in the middle section, decreasing levels of ‘green’ mutant signal (arrows), absent from wild-type DNA (top panel), can be easily distinguished from the nearby ‘red’ background peak (asterisk), which is also found in the assay run on the normal control (top panel). Of note, the EGFR c.2369C assay was designed in the reverse orientation, thus the observed alleles are G (blue) for the wild-type and A (green) for the mutant.
Tumour genotyping
We profiled 250 primary cancer samples representative of major human malignancies, and detected a total of 100 mutations in 86 (34%) of the cases (Supporting Information Table S2). Of note, the majority of these tumour samples (96%) were derived from FFPE tissue. The most frequently mutated gene was KRAS, across multiple tumour types, followed by EGFR, which was detected in lung adenocarcinomas (Table 2 and Fig 3). Consistent with previous reports (Subramanian & Govindan, 2008), KRAS mutations in lung cancer were strongly associated with a history of smoking (89% of KRAS mutations were found in patients that smoked >10 packs/year), while the reverse was true for EGFR, with 73% of EGFR-mutant tumours originating from patients who had never smoked.
Table 2.
Somatic mutations detected by SNaPshot genotyping of primary tumours
| Tumour type | Total no. of cases | Mutations (no. of cases) |
|---|---|---|
| Breast | 33 | KRAS G12V + PIK3CA E545K (1)a |
| PIK3CA H1047L (1) | ||
| PIK3CA H1047R (2) | ||
| TP53 R175H (1) | ||
| TP53 R248Q (1) | ||
| Chronic myeloproliferative disorder | 10 | JAK2 V617F (4) |
| Colorectal | 30 | APC R1114X (1) |
| BRAF V600E (1) | ||
| KRAS G12C (1) | ||
| KRAS G12D (2) | ||
| KRAS G12S (1) | ||
| KRAS G12V (2) | ||
| KRAS G12V + PIK3CA E545K (1) | ||
| KRAS G13D (1) | ||
| KRAS G13D + PIK3CA R88Q (1)a | ||
| KRAS G13D + TP53 R273H (1)a | ||
| NRAS G12D (2)a | ||
| NRAS Q61H + TP53 R175H (1)a | ||
| PI3KCA E545K (1) | ||
| TP53 R175H (1) | ||
| Lung | 87 | CTNNB1 S37F + EGFR E746_A750del (1)a |
| EGFR E746_A750del (6) | ||
| EGFR E746_A750del + EGFR T790M + TP53 R175H (1)a | ||
| EGFR L858R (4) | ||
| EGFR L858R + EGFR T790M (1) | ||
| KRAS G12A (2) | ||
| KRAS G12C (10) | ||
| KRAS G12D (1) | ||
| KRAS G12D + TP53 R248Q (1)a | ||
| KRAS G12V (3) | ||
| KRAS G13D (1) | ||
| NRAS Q61L + TP53 R248P (1)a | ||
| PIK3CA E542K (1) | ||
| TP53 R248Q (1) | ||
| TP53 R273L (1) | ||
| Melanoma | 11 | BRAF V600E (4) |
| BRAF V600M (1) | ||
| NRAS Q61L (1) | ||
| NRAS Q61R (1) | ||
| Pancreatic | 23 | KRAS G12D (2) |
| KRAS G12D + TP53 R175H (1)a | ||
| KRAS G12R (2) | ||
| KRAS G12V (5) | ||
| KRAS G12V + TP53 R248Q (1)a | ||
| Prostate | 20 | CTNNB1 S33C (1) |
| CTNNB1 S37Y + PIK3CA E542K (1)a | ||
| KRAS G13R (1)a | ||
| Other | 36 | BRAF V600E (1)a, unknown primary, presumed breast |
| KRAS G12D (1), cervical | ||
| TP53 R306X (1)a, thyroid Hurthle cell carcinoma |
Mutations or combination of mutations that are rare or not-previously described in the corresponding tumour type.
Figure 3. Distribution of somatic mutations in primary human cancers.
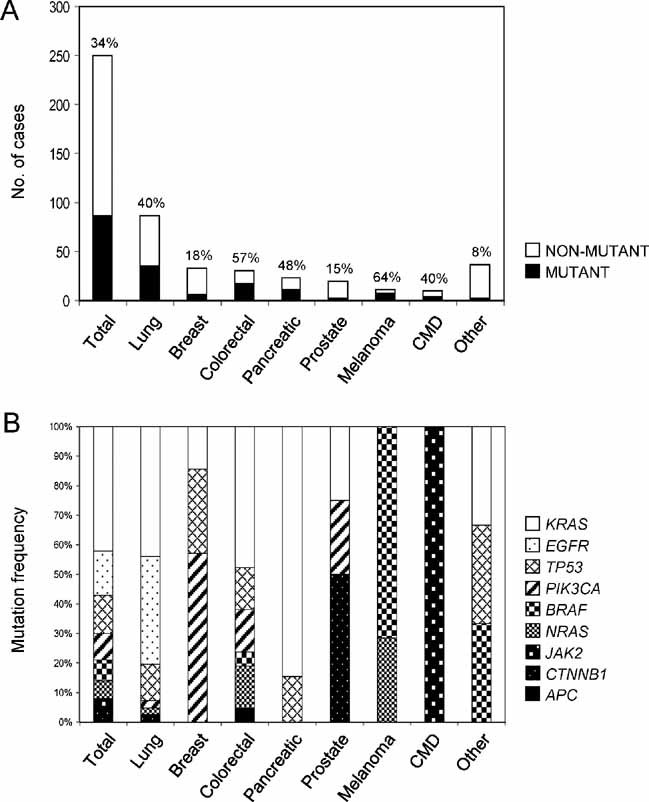
- their mutational status and
- the mutation frequency of individual genes.
The specificity of SNaPshot genotyping was evaluated by analysis of primary tumour samples and matching normal tissue from the same individual. Figure 4 includes examples of adenocarcinomas of the lung (Fig 4A) and pancreas (Fig 4B), and of malignant melanoma (Fig 4C), and depicts the most prevalent activating mutations in our data set for EGFR (L858R), KRAS (G12V) and BRAF (V600E), respectively. The mutant allele (arrow) is only detected in the tumour specimen and not in the matching normal tissue, demonstrating the specificity of the test.
Figure 4. Profiling of primary tumours and matching normal tissue established assay specificity.
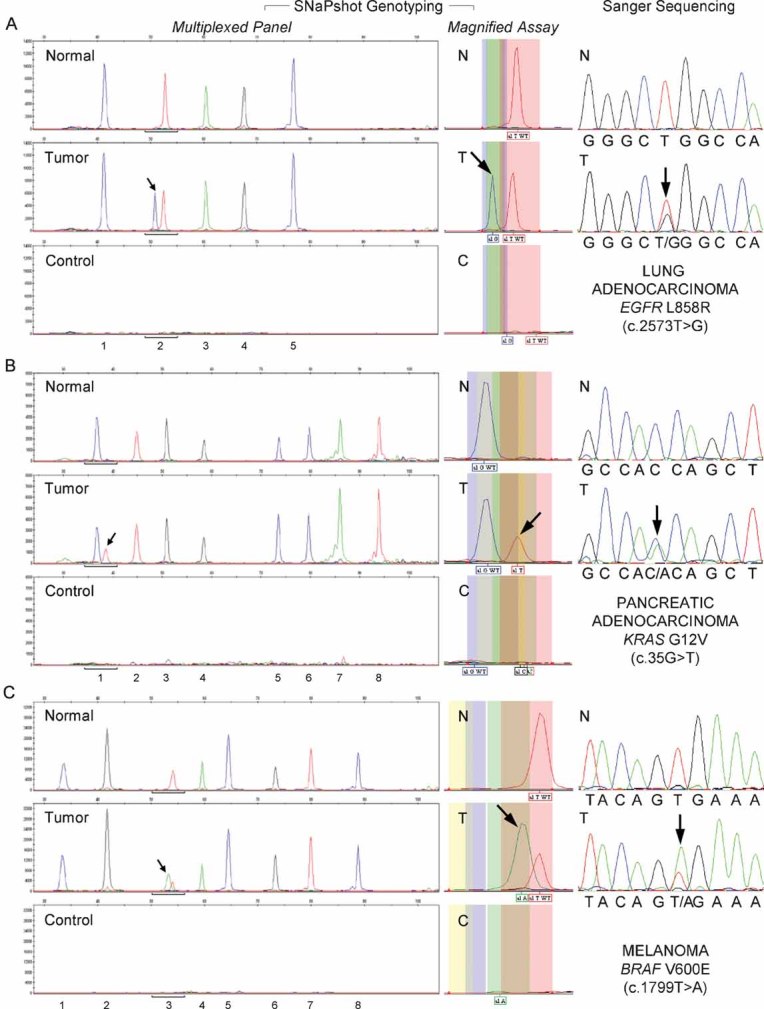
- Detection of the EGFR L858R (c.2573T > G) mutation in a case of lung adenocarcinoma. Assays: (1) EGFR 2236_50del F; (2) EGFR 2573; (3) CTNNB1 133; (4) PIK3CA 1624 and (5) NRAS 35.
- Identification of the KRAS G12V (c.35G > T) mutation in a pancreatic adenocarcinoma. Assays: (1) KRAS 35; (2) EGFR 2236_50del R; (3) PTEN 517; (4) TP53 733; (5) FLT3 2503; (6) PIK3CA 3139; (7) NOTCH1 4724 and (8) NOTCH1 4802.
- Detection of the BRAF V600E (c.1799T > A) mutation in melanoma. Assays: (1) EGFR 2235_49del R; (2) NRAS 38; (3) BRAF 1799; (4) NRAS 182; (5) PIK3CA 263; (6) TP53 742; (7) CTNNB1 95 and (8) CTNNB1 122.
In general, our genotyping results were consistent with the documented mutational prevalence for oncogenes, but we observed lower than expected mutational frequencies for tumour suppressors (Supporting Information Table S3). Slight discrepancies between our observations and the reported mutation frequencies for oncogenes included lower than expected mutation prevalences for beta-catenin (CTNNB1) and BRAF in pancreatic and colorectal tumours, respectively; and higher than the reported frequencies for NRAS in colorectal cancer. Surprisingly, the incidence of NRAS mutations in the colorectal cancer population tested was threefold higher than previously described. Interestingly, we also identified a number of mutations and combination of mutations (marked by “a” in Table 2) that are rare or not previously described in the respective tumour types. Some of these less common events are illustrated in Supporting Information Fig S6 and include the co-occurrence of activating mutations in KRAS and PIK3CA in breast cancer, which were proposed to be mutually exclusive events based on cell line studies (Hollestelle et al, 2007), and of beta-catenin and EGFR mutations in a rarely recognized case of foetal-type lung adenocarcinoma (Nakatani et al, 2002).
Within the subset of events captured by our panel, our observations were consistent with previous findings from genome wide studies (Supporting Information Fig S7). The most common mutations observed in colorectal cancer were C:G to T:A transitions, previously shown to be abundant in this tumour type and a possible effect of dietary carcinogens (Sjoblom et al, 2006). Moreover, consistent with previous reports, we identified C:G to A:T transversions (34%) and C:G to T:A transitions (24%) as the most frequent mutation classes in lung cancer (Ding et al, 2008). C:G to A:T transversions have been associated with smoking and are thought to be induced by tobacco smoke carcinogens (Slebos et al, 1991). All C:G to A:T transversions detected in our lung cancer population were found in smokers (Fig S7B), which is likely in part due to the pattern of KRAS mutations commonly seen in smokers. Finally, we identified a higher proportion of mutations in smokers than in never-smokers for lung (49% vs. 28%) and pancreatic (67% vs. 13%) cancers, in agreement with previously observed correlations between smoking and the number of genetic changes in these tumour types (Blackford et al, 2009; Ding et al, 2008).
Clinical application of genetic profiling
Out of all primary tumours examined, 62 cases were genotyped as part of what has now become routine clinical testing at our institution (Supporting Information Table S2). Exon 19 of the EGFR gene is a hotspot for in-frame deletions, often found in lung cancer and that have been associated with response to EGFR TKI therapy (Lynch et al, 2004; Mok et al, 2009; Paez et al, 2004; Pao et al, 2004). Although the SNaPshot assay tests for the two most common deletions in the EGFR intracellular domain, due to the therapeutic implications of this region, mutational profiling of clinical cases was complemented by a PCR-based sizing assay designed to capture all deletions (or insertions) in EGFR exon 19. For most cases (98%) there was concordance between SNaPshot and the exon 19 sizing data, however, the second approach identified one additional deletion in EGFR which was not captured by SNaPshot genotyping (Supporting Information Table S2).
In our early experience implementing this assay in a clinical setting, approximately two to three weeks are required from the time of test requisition until genotyping report finalization. We thus refer to this as a ‘real-time’ assay, as oncologists ordering the test will have access to their patients' tumour mutational profiling data in time to influence clinical decision making. In these initial analyses, we have already observed that the SNaPshot results have substantially impacted therapeutic decisions. For lung cancer patients, detection of activating mutations in EGFR will identify patients most appropriate for first-line treatment with EGFR TKI therapy (Kobayashi et al, 2005; Lynch et al, 2004; Paez et al, 2004; Pao et al, 2004; Zhu et al, 2008). Conversely, tumours harbouring KRAS mutations are associated with lack of responsiveness to EGFR TKI treatment, and such patients are advised to pursue other therapeutic options (Pao et al, 2005b). Although mutational analysis for these two genes is already widely viewed as the modern standard of care, our genotyping effort uncovered a few additional events, less commonly tested for, that also influenced clinical decisions. Supporting Information Fig S8A illustrates the case of a breast cancer patient with metastatic disease that had progressed through all previous therapy regimens. Identification of the PIK3CA H1047L activating mutation in her tumour prompted enrolment in a clinical trial of a new PIK3CA inhibitor. Supporting Information Fig S8B represents the case of a lung cancer patient with an activating mutation in EGFR that had previously responded to anti-EGFR therapy, but who recently relapsed. Re-biopsy and genotyping of the recurrence revealed the presence of the EGFR T790M mutation, which confers resistance to first-generation EGFR TKIs (Pao et al, 2005a). This finding prompted subsequent therapy with an irreversible EGFR TKI, which also targets the newly acquired T790M EGFR mutant (Riely, 2008). Supporting Information Fig S8C is an example of how SNaPshot genotyping can offer some insight into tumour heterogeneity. Here, profiling of bilateral tumour masses in a patient with lung cancer revealed two distinct genotypes. Our results supported the clinical suspicion that this was not metastatic disease, but rather two synchronous early stage primary tumours. This interpretation provided a better prognosis for the patient, and affected the consideration for pursuing aggressive surgical therapy and adjuvant chemotherapy, directly impacting the management of her disease.
To further investigate sample heterogeneity within the primary tumours evaluated for clinical testing, we re-examined all mutant cases and compared the levels of mutant alleles identified by SNaPshot genotyping with the extent of stromal contamination in each original tumour specimen. As shown in Supporting Information Table S4 the extent of stromal contamination (column 2), and the levels of mutant alleles (column 3) are distinct for different tumour specimens, which is most likely reflecting our inability to accurately predict stromal contamination in a tridimensional tumour specimen, based on the histological evaluation of a single tumour section. In addition, some of these discrepancies may be due to tumour heterogeneity and the presence of activating mutations within variable subsets of tumour cell populations. Concerns with tumour heterogeneity underscore the importance of using highly sensitive mutation detection methods. This matter has been widely appreciated, particularly for mutations that confer resistance to targeted therapeutics where the detection of minor resistant clones, either in the primary tumour or during the course of treatment, is critical to predict response (Maheswaran et al, 2008; Marchetti et al, 2009; Yung et al, 2009). By contrast, the clinical implications of identifying low levels of drug-sensitizing mutations are currently unknown. To begin to address this issue, we examined whether patients with low abundance EGFR sensitizing mutations responded to EGFR TKIs. Within this small cohort, we identified two patients (NA09-129 and NA09-184) with low levels (<20%) of EGFR exon 19 deletions both of whom achieved a clinical response to EGFR TKI therapy (Supporting Information Table S4). While further studies will be required to properly examine this matter, our preliminary observations suggest that the use of targeted agents may be helpful even in cases where the sensitizing mutations are restricted to smaller clones of the tumour cell population. Importantly, our findings indicate that highly sensitive detection methods will be fundamental in identifying these patients.
DISCUSSION
The conventional approach of treating cancer according to histological parameters and tissue of origin is increasingly accommodating molecular genetic information derived on a case-by-case basis. As a step towards personalized cancer medicine, our goal was to develop a high-throughput genetic profiling platform to rapidly query resection or biopsy specimens for relevant genetic changes in a time and cost-effective manner, and to help direct the administration of available targeted therapies. To maximize the clinical utility of our assay, we: (1) focused predominantly on genes targeted by FDA-approved therapies or by therapeutics in clinical trials; and (2) sought to develop a clinical test that could be easily adopted by many laboratories.
The tumour genotyping assay described here is currently being applied for real-time testing of patient samples, and uses expertise and infrastructure already present in most clinical settings. We found it to be highly sensitive and specific, and to perform very well with nucleic acid extracted from FFPE tissue, which is a practical requirement for broad implementation by pathology departments. The system is modular, so as more targeted drugs become available and more predictors of response are identified, new assays can be designed and introduced to existing panels. In addition to the high quality of the data, a major advantage of using SNaPshot technology for tumour genotyping is the lack of need for an upfront investment in high-tech instrumentation not commonly available even in modern clinical laboratories. For example, the minimum cost of equipment for some of the current multiplexed allele-targeted technologies and next-generation sequencing platforms, approaches several hundreds of thousands of dollars. Due to the multiplexing features of the SNaPshot technique, the tissue requirements and cost-per-assay are also low. Our data suggest that signal detection by the SNaPshot system, which combines capillary electrophoresis with the ability to identify four possible fluorescently labelled extension products, offers added advantages when compared to other methods. For instance, while in array-based single fluorophore-detecting technologies one assay tests for the presence or absence of a single mutant allele, each SNaPshot assay queries all three possible mutant variants at once. Also, in contrast to mass spectrometry-based methods (which rely on molecular weight for allele recognition), since the SNaPshot method identifies different nucleotide variants at the same locus not only by their mass but also by their colour, an additional parameter is available to help distinguish between wild-type, mutant, and background signals. This point is illustrated in Supporting Information Fig S9 which provides a direct comparison between SNaPshot and Sequenom MassArray assays. Finally, while the use of next-generation sequencing may ultimately replace current platforms for tumour genotyping, it will likely be several years before the technology and computational infrastructure become affordable and are mature enough for validation and generalized clinical use. For a more detailed discussion of the costs, workflow and tissue requirements of the SNaPshot platform and its comparison with other technologies please refer to Supporting Information.
The main limitation of the SNaPshot method appears to be the number of reactions that can be multiplexed together (plex level), which appears to be optimal below 10. While other allele-specific platforms usually use much higher plex levels for SNP-detection, when employed for rare mutation profiling, their plex level is also lower than 10 (Thomas et al, 2007). Since there is a limit to the number of assays that can be performed on scarce tumour biopsies, as with other targeted sequencing strategies, SNaPshot genotyping is best suited to test for genes affected by point mutations, insertions or deletions at only a few hotspots. Our assay design has a better coverage for oncogenes than for tumour suppressors (Table 1), as the latter tend to be mutated at many more sites than the former. Accordingly, genotyping of primary tumours was overall consistent with reported data for the oncogenes, but captured lower mutation frequencies than what has been documented for most tumour suppressors (Supporting Information Table S3). Slight discrepancies between our observations and the reported mutation frequencies for oncogenes included both lower (CTNNB1 and BRAF) and higher (NRAS) than expected mutation levels within specific cancer types. This variability most likely reflects differences in tumour sub-populations and sample sizes rather than the performance of the assay.
In this era of genomic medicine, one of the most debated questions regarding tumour profiling is which cancer genes and mutations should be tested. If time and cost were not an issue and if tissue quality and quantity were not a limiting factor, most would agree that more information is usually better. Despite the challenges of interpreting highly complex data sets, a complete molecular picture of each tumour should provide the best resource to make informed treatment decisions and establish meaningful correlations between response to therapy and specific genetic signatures. Tumour profiling has advanced significantly in the past decade and will continue to evolve. However, if we want to improve cancer prospects today and until next-generation sequencing options become economically viable and rapid enough to address the time constraints of clinical decision making, highly multiplexed allele-specific platforms like the one presented here will be invaluable clinical resources. We selected the cancer mutations most likely to have immediate clinical impact, either because they are targeted by FDA-approved drugs or by therapeutic agents in clinical trial.
Ideally, the clinical application of targeted mutational profiling will be complemented by additional approaches to provide a more comprehensive picture of each individual cancer, which would include alterations in gene copy number, karyotype information and chromosomal rearrangements such as translocations and large insertions or deletions. Such analyses will require the application of additional technologies and in some cases the use of different materials. For instance, the EGFRvIII mutation commonly observed in human glioblastoma, results in a constitutively active receptor with an in-frame truncation within its extracellular ligand-binding domain, and is of relevance for targeted therapy (Pedersen et al, 2001). While the mutation mechanisms leading to EGFRvIII are diverse, ranging from large deletions of genomic DNA to point mutations that affect splicing, the end product is a single mRNA splice variant lacking exons 2–7, making it an ideal candidate for detection by allele-specific assays like the one described herein. Such a test would require slight adaptation of the protocol to include a cDNA synthesis step, and possibly the use of a better quality tissue source than the highly fragmented nucleic acid extracted from archived FFPE tumour tissue that was employed in the present study.
It is suspected that deregulation of a core of common signalling pathways is one of the major underlying causes driving human carcinogenesis (Jones et al, 2008). Therefore, we decided to apply the same broad genotyping platform to all tumours, rather than restricting the analysis to focused mutational panels for specific cancer types. We hope that this approach will identify novel treatment opportunities for a broader set of malignancies. To that extent, our study uncovered a number of mutations and mutation co-occurrences that had not been previously appreciated in the tumour types tested (Table 2). We suspect that clinical application of mutation profiling programs such as this one, across multiple malignancies, will swiftly impact disease management not only of common cancers but also of rare tumours, which have to date received less comprehensive attention. Moreover, while some current examples have established a convincing foundation for using specific mutations as predictors of response to selective agents (e.g. EGFR kinase mutations predict tumour sensitivity to gefitinib and erlotinib, and KRAS mutations confer resistance to EGFR TKIs) (Mok et al, 2009; Pao et al, 2005b) the future landscape of cancer therapy is likely to be less simple. As novel therapeutic approaches contemplate the use of multiple agents and of multi-targeted drugs, the correlations between specific mutational genotypes and sensitivity or resistance to treatment will probably be more complex. Genetic profiling strategies will be essential to dissect these intricate connections and will likely play an increasingly important role in cancer management. Going forward, determining the optimal application of novel agents will require carefully designed clinical trials which integrate tumour molecular analysis in both up-front patient selection and retrospective correlative analyses.
Finally, most genes included in our panel are targeted by currently available drugs making them ideal candidates for genetic profiling however, with the exception of KRAS and EGFR, the full clinical implications of many of the mutations tested by our assay are still under investigation. Clinical questions of interest include not only the efficacy of novel agents in clinical trials, but also the viability of using a given drug to fight different cancer types with similar genetic abnormalities, and the optimal treatment of heterogeneous tumours harbouring different levels of the target mutation. The lack of established treatment algorithms based on results of genetic profiling needs to be clearly conveyed to patients undergoing testing, which in our case is accomplished by a consent form and counselling session with background information about the test. Thus, before giving permission to have their tumour tested, patients understand that molecular profiling may (or may not) provide information that could help them and their doctor decide which therapies could be most or least successful in treating their tumour, as either part of standard therapy or as part of research studies that may be of interest to them. The costs of testing are billed to the patient's insurance company and issues related to reimbursement are addressed by hospital-wide policies in the same way as with any other clinical test or procedure.
Targeted cancer therapy is revolutionizing clinical oncology and driving efforts to integrate tumour molecular analysis in clinical decision making. The EGFR story in NSCLC has demonstrated that genotype-driven treatment choices affect patient outcomes. Robust and practical genotyping strategies such as the one described here will be instrumental in moving forward the optimal application of targeted therapies. Such approaches will undoubtedly see increasing application in the selection of patients for early-stage clinical trials, providing the potential for better response rates and improved interpretation of trial results. Tumour genetic analysis therefore holds great promise to make personalized cancer care a reality.
MATERIALS AND METHODS
Specimen collection
We tested 250 primary cancer samples spanning 26 human malignancies, which included: lung cancer (n = 87), breast cancer (n = 33), colorectal cancer (n = 30), pancreatic cancer (n = 23), prostate cancer (n = 20), melanoma (n = 11), chronic myeloproliferative disease (n = 10), cholangiocarcinoma (n = 6), gastric cancer (n = 4), ovarian cancer (n = 3), salivary gland cancer (n = 3) and thyroid cancer (n = 3) among others. Sixty-two of these primary tumour samples were evaluated for official clinical testing, and included 52 lung adenocarcinomas, most of them small core biopsies with very limited tissue. For haematopoietic malignancies, spare DNA that had been previously extracted from patient blood for clinical testing was obtained from the Massachusetts General Hospital (MGH) Molecular Diagnostics Laboratory. For solid tumours, formalin-fixed paraffin-embedded (FFPE) tumour blocks were obtained from the MGH archives. All samples were collected with institutional review board approval. Histological examination of haematoxylin and eosin-stained slides derived from FFPE samples was performed by a pathologist (AJI) and assessed for the presence of tumour. Available tumour tissue was manually macrodissected from serial 5 µm unstained sections, or cored from the paraffin block using a 1.5 mm dermal punch. Total nucleic acid was extracted from FFPE material using a modified FormaPure System (Agencourt Bioscience Corporation, Beverly, MA) on a custom Beckman Coulter Biomek NXP workstation. Blood-derived DNA was extracted using the QIAamp Blood kit (Qiagen, Inc., Valencia, CA).
Assay design
We evaluated the COSMIC (Bamford et al, 2004) database and PubMed to select a panel of genes and loci previously reported to be frequently affected by somatic mutation in human cancer. We chose 13 cancer genes and designed 58 assays to test for individual mutational events, which included: 1 insertion, 3 deletions and 52 substitutions (Supporting Information Table S1). Genomic position and sequencing information for all mutation sites were collected using the RefSeq gene sequences obtained using the human genome browser from the University of California Santa Cruz (UCSC), NCBI build 36.1. Primers for multiplexed PCR amplification were designed using Primer 3 software. Since FFPE tissue can be highly fragmented and of poor quality, design parameters restricted amplicon length to a maximum of 200 nt. All amplification primers (Supporting Information Table S5A) include a 10 nt long 5′ anchor tail (5′-ACGTTGGATG-3′) and the final PCR products range in length between 75 and 187 nt. The extension primer probes (Supporting Information Table S5B) were designed manually, according to the ABI PRISM SNaPshot Multiplex Kit protocol recommendations (Life Technologies/Applied Biosystems, Foster City, CA) and using primer analysis tools available through the Primer 3 and Integrated DNA Technologies (IDT, Coralville, IA) web interfaces. Optimal conditions for multiplexed assays were determined empirically and are summarized in Supporting Information Table S6.
The paper explained
PROBLEM
Cancer cells harbour genetic abnormalities that give them a survival and proliferative advantage. These are also the cancer's Achilles heel, as the tumour becomes highly dependent on them to grow. ‘Smart drugs’ are being developed to target specific genetic abnormalities and only the tumours harbouring the targeted mutations will respond to each drug. One of the main challenges facing the oncology community today is how to efficiently match each tumour with the right therapy, and give all cancer patients the best shot at fighting their disease.
RESULTS
We developed and extensively validated a simple and flexible multiplexed tumour genotyping assay that detects common mutations in some of the most important cancer genes (including EGFR, KRAS, NRAS, BRAF and PIK3CA), many of which activate cancer signalling pathways targeted by currently available ‘smart therapies’. Our analysis included the genetic profiling of 250 primary tumours, was consistent with the documented oncogene mutational spectrum and demonstrated that the SNaPshot system is fast, highly sensitive and very robust.
IMPACT
Over the past year we have consistently profiled lung cancers presented to the MGH. The test was recently expanded to colorectal malignancies and will soon be offered to all incoming cancer patients. We shared our protocols with other laboratories that have now also quickly implemented and adapted this system to their particular needs, which has further convinced us of its broad applicability and its potential to personalize cancer care and impact therapeutic decisions in a large scale.
As part of the design rationale, we included assays covering four adjacent loci that are commonly mutated in the therapeutically relevant KRAS and NRAS oncogenes (for both genes we are targeting nucleotide positions: 34G, 35G, 37G and 38G). Due to the close proximity of these sites, to avoid compromising assay sensitivity due to primer competition, we decided to assay each of them in an independent panel. In addition, due to the extreme sequence similarity between KRAS and NRAS, to avoid non-specific results, we segregated the assays for the two genes into individual multiplexed reactions. We thus started with eight panels, which were populated with the 58 assays outlined in Supporting Information Table S1. Many of these genes and assays are clinically relevant. In addition, since the costs of running the assay (regarding tumour material and the actual price per assay) are mainly dictated by the number of panels, we decided to also include a set of common mutations affecting critical cancer genes for which a therapeutic agent is still currently unavailable. We hope that the addition of these less ‘clinically relevant’ mutations will still be useful in a clinical setting, as we may find them to correlate with a better or worse prognosis or to influence response to specific therapies, and thus contribute to better cancer care in the future.
SNaPshot genotyping
The Applied Biosystems (ABI) Prism® SNaPshot® Multiplex system was originally developed to detect single nucleotide polymorphisms (SNPs) (Lindblad-Toh et al, 2000) (Fig 1). Multiplexed PCR was performed in a volume of 10 µl, containing 0.5 U of Platinum Taq polymerase (Invitrogen, Carlsbad, CA, USA), 30 nmol of MgCl2, 3 nmol of dNTPs (Invitrogen), amplification primers (IDT) as specified in Supporting Information Table S6A, and ideally either 20 ng of genomic DNA or 60 ng of total nucleic acid. When the amount of tissue was limiting, multiplexed PCR was performed with as low as 5 ng of total nucleic acid. Thermocycling was performed at 95°C for 8 min, followed by 45 cycles of 95°C for 20 s, 58°C for 30 s and 72°C for 1 min, and one last cycle of 72°C for 3 min. Excess primers and unincorporated dNTPs were inactivated using 3.3 U of shrimp alkaline phosphatase (USB, Cleveland, OH) and 2.7 U of exonuclease I (USB) for 60 min at 37°C, followed by 15 min at 75°C for enzyme inactivation. The primer extension reaction was performed in a volume of 10 µl, containing 3 µl of PCR product, 2.5 µl of SNaPshot Multiplex Ready Reaction mix, and the appropriate cocktail of PAGE-purified extension primers (IDT; Supporting Information Table S6B). Cycling conditions were 96°C for 30 s, followed by 25 cycles of 96°C for 10 s, 50°C for 5 s and 60°C for 30 s. After treatment with 2 U of shrimp alkaline phosphatase, 0.5 µl of labelled extension products were mixed with Hi–Di Formamide and 0.2 µl of GeneScan-120LIZ size standard (Life Technologies/Applied Biosystems) to a final volume of 10 µl. Following denaturation at 95°C for 5 min, the extension products were resolved by running on 36 cm long capillaries in an automatic sequencer (ABI PRISM 3730 DNA Analyser, Life Technologies/Applied Biosystems), according to the SNaPshot default settings established by ABI. Data analysis was performed with GeneMapper Analysis Software version 4.0 (Life Technologies/Applied Biosystems) using the automatic calling parameters described in the Supporting Information.
Acknowledgments
We thank J. M. Batten for identifying and providing spare DNA for the chronic myeloproliferative disease cases and the Aid for Cancer Research and anonymous donors for financial support. DDS thanks J. Teague at The Wellcome Trust Sanger Institute for providing the ftp link to the COSMIC spreadsheet summaries of all gene mutations, and S. Santagata for helpful comments on the manuscript.
Supporting information is available at EMBO Molecular Medicine online.
Authors DAH, TJL and JS have partial patent rights to EGFR mutation testing. DDS and AJI submitted a patent application for the SNaPshot genotyping methods described herein, which are the subject of licensing discussions.
Author contributions
DDS, AJI, DRB and LWE planned the experiments. DDS designed and directly supervised assay development. DNL and DAH devised the concept. SSD, SA, KV, GK, SLB and HS performed the experiments. ELK, JWC, SJI, UM, JS, LVS, JAE and TJL contributed samples and clinical correlations. DDS wrote the manuscript. AJI, LVS, LWE, JAE, DRB, SJI, JWC, SA and DNL edited the manuscript.
For more information
Wellcome Trust Sanger Institute COSMIC database, gene files and Cancer Cell Line Project:
http://www.sanger.ac.uk/genetics/CGP/cosmic/
ftp://ftp.sanger.ac.uk/pub/CGP/cosmic/data_export/genes/
http://www.sanger.ac.uk/genetics/CGP/CellLines/
UCSC genome browser:
Primer design and analysis tools:
http://biotools.idtdna.com/SciTools/SciTools.aspx
Massachusetts General Hospital Translational Research Laboratory:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, et al. The COSMIC (catalogue of somatic mutations in cancer) database and website. Br J Cancer. 2004;91:355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Blackford A, Parmigiani G, Kensler TW, Wolfgang C, Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Eshleman JR, et al. Genetic mutations associated with cigarette smoking in pancreatic cancer. Cancer Res. 2009;69:3681–3688. doi: 10.1158/0008-5472.CAN-09-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Gray JW. Translating insights from the cancer genome into clinical practice. Nature. 2008;452:553–563. doi: 10.1038/nature06914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Hunter C, Smith R, Stephens P, Greenman C, Bignell G, Teague J, Butler A, Edkins S, Stevens C, et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005;65:7591–7595. doi: 10.1158/0008-5472.CAN-05-1855. [DOI] [PubMed] [Google Scholar]
- Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW, Schutte M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol Cancer Res. 2007;5:195–201. doi: 10.1158/1541-7786.MCR-06-0263. [DOI] [PubMed] [Google Scholar]
- Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J, Baccarani M, Cortes J, Cross NC, Druker BJ, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108:28–37. doi: 10.1182/blood-2006-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod Pathol. 2008;21:S16–S22. doi: 10.1038/modpathol.3801018. [DOI] [PubMed] [Google Scholar]
- Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Winchester E, Daly MJ, Wang DG, Hirschhorn JN, Laviolette JP, Ardlie K, Reich DE, Robinson E, Sklar P, et al. Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat Genet. 2000;24:381–386. doi: 10.1038/74215. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, Inserra E, Diederichs S, Iafrate AJ, Bell DW, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359:366–377. doi: 10.1056/NEJMoa0800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti A, Milella M, Felicioni L, Cappuzzo F, Irtelli L, Del Grammastro M, Sciarrotta M, Malatesta S, Nuzzo C, Finocchiaro G, et al. Clinical implications of KRAS mutations in lung cancer patients treated with tyrosine kinase inhibitors: an important role for mutations in minor clones. Neoplasia. 2009;11:1084–1092. doi: 10.1593/neo.09814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci USA. 2007;104:19936–19941. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Masudo K, Miyagi Y, Inayama Y, Kawano N, Tanaka Y, Kato K, Ito T, Kitamura H, Nagashima Y, et al. Aberrant nuclear localization and gene mutation of beta-catenin in low-grade adenocarcinoma of fetal lung type: up-regulation of the Wnt signaling pathway may be a common denominator for the development of tumors that form morules. Mod Pathol. 2002;15:617–624. doi: 10.1038/modpathol.3880575. [DOI] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005a;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005b;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen MW, Meltorn M, Damstrup L, Poulsen HS. The type III epidermal growth factor receptor mutation. Biological significance and potential target for anti-cancer therapy. Ann Oncol. 2001;12:745–760. doi: 10.1023/a:1011177318162. [DOI] [PubMed] [Google Scholar]
- Riely GJ. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. J Thorac Oncol. 2008;3:S146–S149. doi: 10.1097/JTO.0b013e318174e96e. [DOI] [PubMed] [Google Scholar]
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69:1851–1857. doi: 10.1158/0008-5472.CAN-08-2466. [DOI] [PubMed] [Google Scholar]
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Slebos RJ, Hruban RH, Dalesio O, Mooi WJ, Offerhaus GJ, Rodenhuis S. Relationship between K-ras oncogene activation and smoking in adenocarcinoma of the human lung. J Natl Cancer Inst. 1991;83:1024–1027. doi: 10.1093/jnci/83.14.1024. [DOI] [PubMed] [Google Scholar]
- Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Sos ML, Michel K, Zander T, Weiss J, Frommolt P, Peifer M, Li D, Ullrich R, Koker M, Fischer F, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009;119:1727–1740. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan M, Sedmak D, Jewell S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am J Pathol. 2002;161:1961–1971. doi: 10.1016/S0002-9440(10)64472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens P, Edkins S, Davies H, Greenman C, Cox C, Hunter C, Bignell G, Teague J, Smith R, Stevens C, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005;37:590–592. doi: 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- Subramanian J, Govindan R. Molecular genetics of lung cancer in people who have never smoked. Lancet Oncol. 2008;9:676–682. doi: 10.1016/S1470-2045(08)70174-8. [DOI] [PubMed] [Google Scholar]
- Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–351. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- Weinstein IB. Disorders in cell circuitry during multistage carcinogenesis: the role of homeostasis. Carcinogenesis. 2000;21:857–864. doi: 10.1093/carcin/21.5.857. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Yung TK, Chan KC, Mok TS, Tong J, To KF, Lo YM. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin Cancer Res. 2009;15:2076–2084. doi: 10.1158/1078-0432.CCR-08-2622. [DOI] [PubMed] [Google Scholar]
- Zhu JQ, Zhong WZ, Zhang GC, Li R, Zhang XC, Guo AL, Zhang YF, An SJ, Mok TS, Wu YL. Better survival with EGFR exon 19 than exon 21 mutations in gefitinib-treated non-small cell lung cancer patients is due to differential inhibition of downstream signals. Cancer Lett. 2008;265:307–317. doi: 10.1016/j.canlet.2008.02.064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.