

Abstract
JunD regulates genes involved in antioxidant defence. We took advantage of the chronic oxidative stress resulting from junD deletion to examine the role of reactive oxygen species (ROS) in tumour development. In a model of mammary carcinogenesis, junD inactivation increased tumour incidence and revealed an associated reactive stroma. junD-inactivation in the stroma was sufficient to shorten tumour-free survival rate and enhance metastatic spread. ROS promoted conversion of fibroblasts into highly migrating myofibroblasts through accumulation of the hypoxia-inducible factor (HIF)-1α transcription factor and the CXCL12 chemokine. Accordingly, treatment with an antioxidant reduced the levels of HIF and CXCL12 and numerous myofibroblast features. CXCL12 accumulated in the stroma of HER2-human breast adenocarcinomas. Moreover, HER2 tumours exhibited a high proportion of myofibroblasts, which was significantly correlated to nodal metastases. Interestingly, this subset of tumours exhibited a significant nuclear exclusion of JunD and revealed an associated oxido-reduction signature, further demonstrating the relevance of our findings in human cancers. Collectively, our data uncover a new mechanism by which oxidative stress increases the migratory properties of stromal fibroblasts, which in turn potentiate tumour dissemination.
Keywords: AP-1, SDF-1, HIF-1, stroma, metastasis
INTRODUCTION
Carcinomas are highly complex tissues composed of neoplastic and stromal cells, including mesenchymal cells, fibroblasts or myofibroblasts, endothelial cells, pericytes and inflammatory cells (Bissell & Radisky, 2001; Mueller & Fusenig, 2004). In past decades, the major focus of cancer research has been the transformed cell itself. However, new clinical data have shown that the stroma contributes significantly to the development of a wide variety of tumours. Tissues exhibiting chronically inflamed stroma or those suffering from repetitive wound healings display a higher incidence of tumour formation (Joyce & Pollard, 2009; Tlsty & Coussens, 2006). Fibroblasts are the most common type of stromal cells in various human carcinomas, yet their specific contributions to tumour growth have only recently been clarified (Erez et al, 2010; Orimo et al, 2005). Stromal fibroblasts, named carcinoma-associated fibroblasts (CAFs), have been extracted from a number of invasive human breast carcinomas. CAFs are more competent in promoting growth of mammary carcinoma cells and enhancing tumour angiogenesis than fibroblasts derived from outside tumour masses (Olumi et al, 1999; Orimo et al, 2005). CAFs also mediate tumour-enhancing inflammation (Erez et al, 2010). CAFs isolated from the stroma of invasive human breast cancers include large populations of myofibroblasts (Eyden et al, 2009). Myofibroblasts are often referred to as activated fibroblasts that play key roles in wound repair (Hinz et al, 2007). The myofibroblastic properties of CAFs are believed to increase tumour growth and enhance vascular remodelling. Myofibroblasts are characterized by high de novo expression of smooth muscle α-actin (SM-α-actin), the actin isoform typically found in vascular smooth muscle cells, and possess greatly enhanced contractile ability. Recent work has shown that CAFs secrete elevated levels of CXCL12, also called stromal cell-derived factor 1 (SDF-1) (Orimo et al, 2005). CXCL12 is a homeostatic chemokine that mediates homing of stem cells to bone marrow by binding to its receptor (CXCR4) on circulating cells (Rossi & Zlotnik, 2000). CXCL12 not only stimulates carcinoma cell growth but also helps in recruiting endothelial progenitor cells to tumours, thereby furthering neo-angiogenesis (Orimo et al, 2005). The importance of this CXCL12–CXCR4 signalling pathway in the tumour microenvironment has already been addressed but, to our knowledge, its role in CAFs remains unexplored.
The AP-1 (activator protein-1) transcription factor plays a critical role in regulating environmental stress responses (Mechta-Grigoriou et al, 2001). Recently, we discovered a new function of JunD, a member of the AP-1 family, in controlling oxidative stress and angiogenic switch (Gerald et al, 2004; Laurent et al, 2008). JunD protects cells against oxidative stress by regulating genes involved in antioxidant defence and H2O2 production. Subsequently, inactivation of junD leads to a persistent accumulation of reactive oxygen species (ROS) in cells and tissues. Thus, junD-deficient mice and junD−/− derived-fibroblasts constitute good models for investigating the physiological consequences of chronic oxidative stress. Using these systems, we uncovered a molecular mechanism linking oxidative stress to angiogenesis and ageing (Gerald et al, 2004; Laurent et al, 2008). Accumulation of H2O2 reduces the activity of hypoxia-inducible factor (HIF)-prolyl-hydroxylases (PHDs), which signal HIF-α subunits for proteosomal degradation. In consequence, HIF-α proteins accumulate and enhance transcription of specific target genes such as VEGF-A (Pouyssegur & Mechta-Grigoriou, 2006).
In this paper, we take advantage of the persistent oxidative stress due to junD inactivation to examine the role of ROS in tumour development. In a model of ras-mediated mammary carcinogenesis, junD deletion increases tumour growth and revealed extensively modified stroma. Moreover, dissemination of junD+/+ neoplastic cells was enhanced when grafted into junD−/− mice. Since JunD expression is detected in stromal fibroblasts, these data suggest that inactivation of junD in these cells, and consecutive oxidative stress, may affect the fibroblastic properties and potentiate tumour spread. Using junD-deficient fibroblasts, we demonstrated that oxidative stress promotes conversion of fibroblasts into myofibroblasts in an HIF-1α and CXCL12-dependent pathway. Conversely, long-term antioxidant treatment partially reverses myofibroblast differentiation. In agreement with our observations on mice, HER2-amplified tumours exhibit the highest expression levels of CXCL12 in the stroma and the highest correlated proportion of myofibroblasts, when compared to aggressive basal-like breast cancers (BLC) or to good prognosis luminal-A (Lum-A) breast carcinomas. Interestingly, HER2-subtype of tumours display a molecular signature characteristic of stress–response and a nuclear exclusion of the JunD protein. Since breast tumours which overexpress HER2 exhibit one of the poorest prognosis of all molecular classes of breast carcinomas and show a high rate of axillary lymph node metastases (Bartlett et al, 2007), these observations underline the role of oxidative stress and myofibroblasts in cancer metastases.
RESULTS
junD inactivation results in a reactive stroma and promotes tumour metastasis
We have previously shown that inactivation of the junD gene leads to constitutive oxidative stress (Gerald et al, 2004; Laurent et al, 2008). To further investigate the role of such stress in tumour development, we crossed junD−/− mice with an MMTV-v-Ha-ras transgenic strain; a model for breast cancer (Sinn et al, 1987). Tumour-free survival rates were significantly lower in junD-deficient females compared to ras junD+/+ (referred to as ras) ones (Fig 1A). Moreover, the number and volume of tumours were higher in ras junD−/− mice compared to control animals (Fig 1B and C). To better understand the underlying mechanism, we compared histological properties of ras junD−/− and ras tumours when they reached the same size (Fig 1D; Fig S1). Inactivation of junD generally affected characteristics of the tumours, as well as features of the associated stroma. When compared to ras tumours (Fig 1Da,c; Fig S1Aa), deletion of junD in ras-mediated tumours resulted in an increased proportion of polycystic carcinomas (Fig 1Db; Fig S1Ac) and massive fibrosis (Fig 1Dd), indicated by the accumulation of various forms of collagen. Moreover, the number and the size of blood vessels increased in ras junD−/− tumours compared to ras (Fig 1De,f), confirming our previous results that junD deletion increased angiogenesis in vivo (Gerald et al, 2004; Laurent et al, 2008). We quantified each cell type composing the tumours by specific immunohistochemistry staining. Epithelial cells, fibroblasts, myofibroblasts, macrophages and haematopoietic cells were specifically stained using E-cadherin, vimentin, SM-α-actin, F4/80 and CD45-specific antibodies, respectively (Fig 1D and E; Fig S1B). In both genotypes, epithelial cells remained highly differentiated, as evaluated by expression of E-cadherin at cellular surface (Fig 1Dg,h; Fig S1B), further indicating that junD inactivation did not promote massive epithelial to mesenchymal transition (EMT). In contrast, the tumour surrounding stroma was quantitatively and qualitatively modified by junD deletion. The proportion of CAF was significantly higher in ras junD−/− tumours compared to ras ones (Fig 1Di,j and E; Fig S1B). Moreover, ras junD−/− fibroblasts expressed higher levels of vimentin, a type III intermediate filament, than controls (Fig 1Di,j; Fig S1B) and accumulated podoplanin, a glycoprotein characteristic of reactive stroma (Fig 1Dm,n). Furthermore, ras junD−/− tumours exhibited significant increase in myofibroblasts content, evaluated by the number of SM-α-actin-positive fibroblasts (Fig 1Do,p). Finally, the stroma of ras junD−/− tumours overproduced the CXCL12 chemokine (Fig 1Dq,r; Fig S1D) and exhibited increased recruitment of inflammatory cells (Fig 1Dk,l). Staining with specific markers for macrophages (Fig 1Ds,t and E) and haematopoietic cells (data not shown) indicated that both cell types were recruited more efficiently in ras junD−/− tumours compared to ras. All these observations reveal highly vascularized tumours with reactive stroma in junD-deficient ras-mediated tumours, features that are not seen in ras tumours alone strongly arguing that JunD is involved in these processes.
Figure 1. junD inactivation promotes appearance of a reactive stroma and tumour progression.
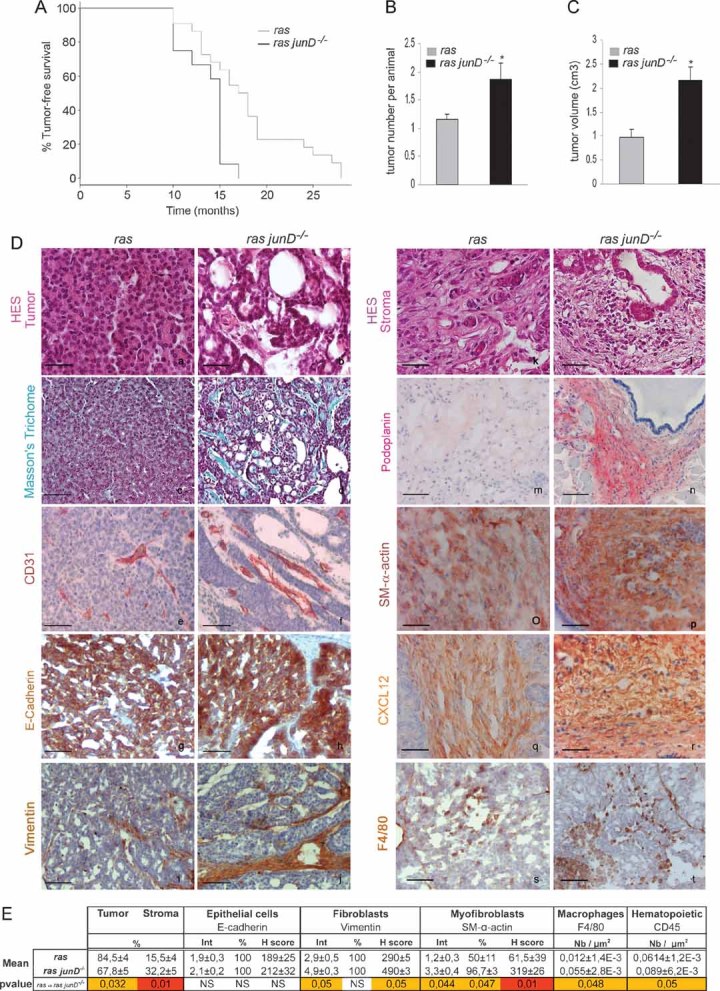
- Kaplan–Meyer tumour-free survival curve of ras junD+/+ (referred to as ras) animals (n = 12) and ras junD−/−mice (n = 12) (p = 0.0092, log-rank test).
- Number of tumours per animal in ras (n = 12) and ras junD−/− (n = 12) mice.
- Tumour volumes in ras (n = 10) and ras junD−/− (n = 9) mice.
- Sections and histological analysis of epithelial tumours (a–h) and immediate adjacent stroma (i–t) from ras or ras junD−/− animals. Sections have been coloured with HES (haematoxylin-eosin-saffranin) (a,b,k,l), Masson's trichrome (c,d) or immunostained with specific antibodies, as indicated (e–j,m–t).
- Percentage of epithelial and fibroblastic compartments in the tumours has been evaluated using E-cadherin and Vimentin-specific staining, respectively. Are also indicated intensity (Int), percentage of positive cells (%) and H scoring (Int × %) for E-cadherin, Vimentin and SM-α-actin-staining as well as the number of F4/80- or CD45-positive cells per µm2. n represents the number of animals analysed per genotype; n represents the number of tumours analysed per genotype. Data are means ± SEM. *p < 0.05 by student's test. Scale bars = 20 µm in (Da,b,k,l) and 40 µm in (Dc–j,Dm–t).
To further define the function of JunD, we monitored its pattern of expression in ras-derived tumours. JunD expression was detected in neoplastic epithelial cells (Fig S1Ca,e) and in stromal fibroblasts (Fig S1Cb,f), suggesting that its deletion can directly impact both compartments. In order to explore the role of JunD only in stromal fibroblasts, we performed transplant experiments by injecting B16F10, a transformed cell line of the same immunotype as our immunocompetent wt and junD−/− mice. Resulting tumours developed in either a wt or a junD−/− host. Although junD-deficient mice showed earlier tumour onset than wt animals (Fig 2A), both types of tumours reached the same mean volume (data not shown) and did not display obvious accumulation of inflammatory cells (Fig S1E). In contrast, tumours developed in junD-deficient environment accumulated significantly both SM-α-actin and CXCL-12 (Fig 2B and C). In addition, stromal inactivation of junD notably increased the incidence and size of metastases in lungs (Fig 2D and E). To confirm the role of CXCL12 in junD-mediated tumourigenesis, we have treated daily grafted junD−/− mice by specific CXCL12 siRNA (Fig 2F). Interestingly, silencing of CXCL12 decreased significantly tumour size and prevented lung metastases. Taken together, these results indicate that inactivation of junD in the tumour environment is sufficient to modify tumour properties, increase the content in SM-α-actin-expressing cells and promote tumour growth and spread, in a CXCL12-dependent manner.
Figure 2. junD inactivation in the stroma potentiates tumour metastasis.
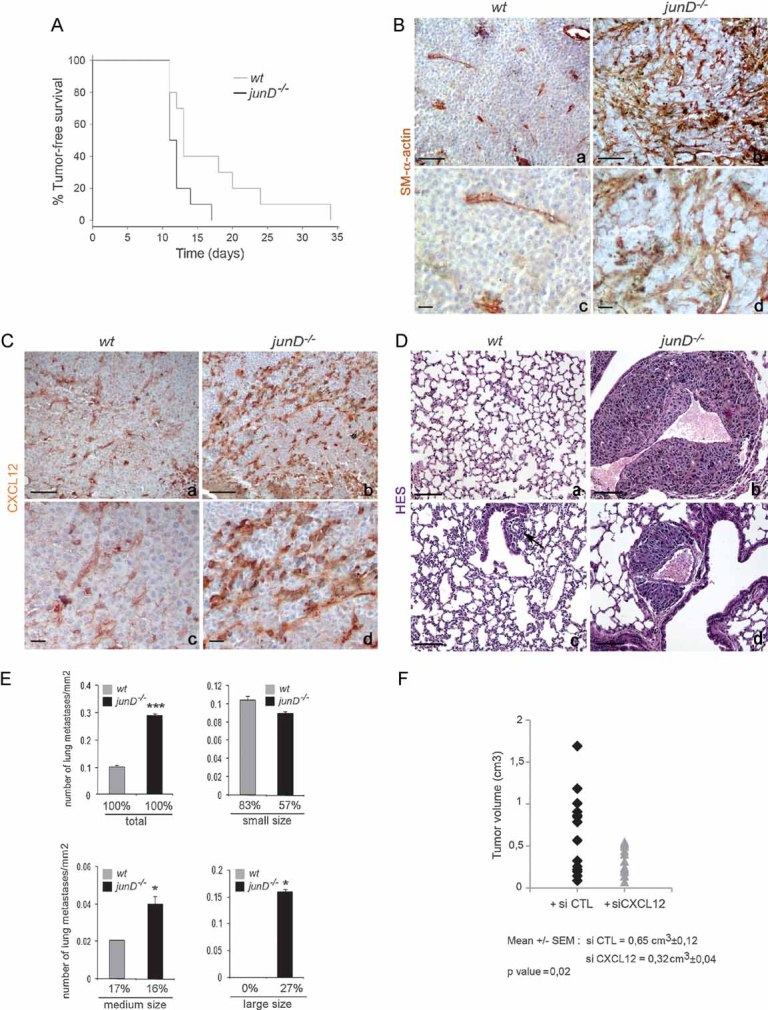
- A. Kaplan–Meyer tumour-free survival curve of wt (n = 10) and junD−/− mice (n = 10) in graft experiments using B16F10 cells (p = 0.041, log-rank test).
- B, C. Representative immunohistochemistry of tumours from injected wt (a,c) and junD−/− mice (b,d) using SM-α-actin and CXCL12-specific antibodies.
- D. Typical HES views of lungs from injected mice. Sections show large (b) and medium (d) sizes of metastatic nodules in junD−/− mice compared to wt animals (a,c).
- E. Number of total, small-sized (<10 cells), medium-sized (10 cells < × < 50 cells) and large-sized (>50 cells) metastasis in wt and junD−/− mice. Numbers below indicate the percentage of large-, medium- and small-sized metastasis in the respective populations.
- F. Tumour volumes in junD−/− mice treated daily either with control siRNA (black) or with specific CXCL12-directed siRNA. Data are means ± SEM. *p < 0.05 and ***p < 0.005 by student's test. N represents the number of tumours analysed per genotype. Scale bars = 40 µm.
junD-deficient fibroblasts exhibit features of CAFs
Since junD expression has been detected in tumour-associated fibroblasts, we next investigated whether inactivation of junD, followed by oxidative stress, was sufficient to alter the properties of fibroblasts. We investigated the gene expression profile and morphology of immortalized junD−/− fibroblasts in a tumour-free context and compared them with the already reported characteristics of CAFs (Fig 3). We first identified a subset of 1934 genes that were significantly up-regulated (p < 0.05) in junD-deficient fibroblasts compared to wt cells. We next compared this list (referred to as junD−/−) with two partially overlapping lists of CAF-specific genes (Allinen et al, 2004; Farmer et al, 2009), set of genes that is also up-regulated in desmoid-type fibromatosis, further underscoring their tumour-associated fibroblastic molecular signature (West et al, 2005) (Fig 3A). The Allinen' and Farmer's lists were composed of 201 and 161 genes, respectively. Among these genes, 44 from Allinen's list and 17 from Farmer's were up-regulated in junD−/− versus wt fibroblasts (Table S1). This is significantly more than would be expected by chance (namely, p = 10−20 for Allinen versus junD−/− and p = 10−4 for Farmer versus junD−/−, using Fisher Exact test). Genes encoding extracellular matrix proteins (including collagens I, III, IV, fibronectin, sparc), components of the cytoskeleton (myosin) and matrix metalloproteases (MMP2, MMP14) were up-regulated in the three lists (Fig 3A). These data argue that expression profiles of junD−/− fibroblasts are related to the expression signature of CAFs. junD inactivation is thus sufficient to confer CAF properties, even in a tumour-free context.
Figure 3. junD−/− fibroblasts exhibit features of carcinoma-associated myofibroblasts.
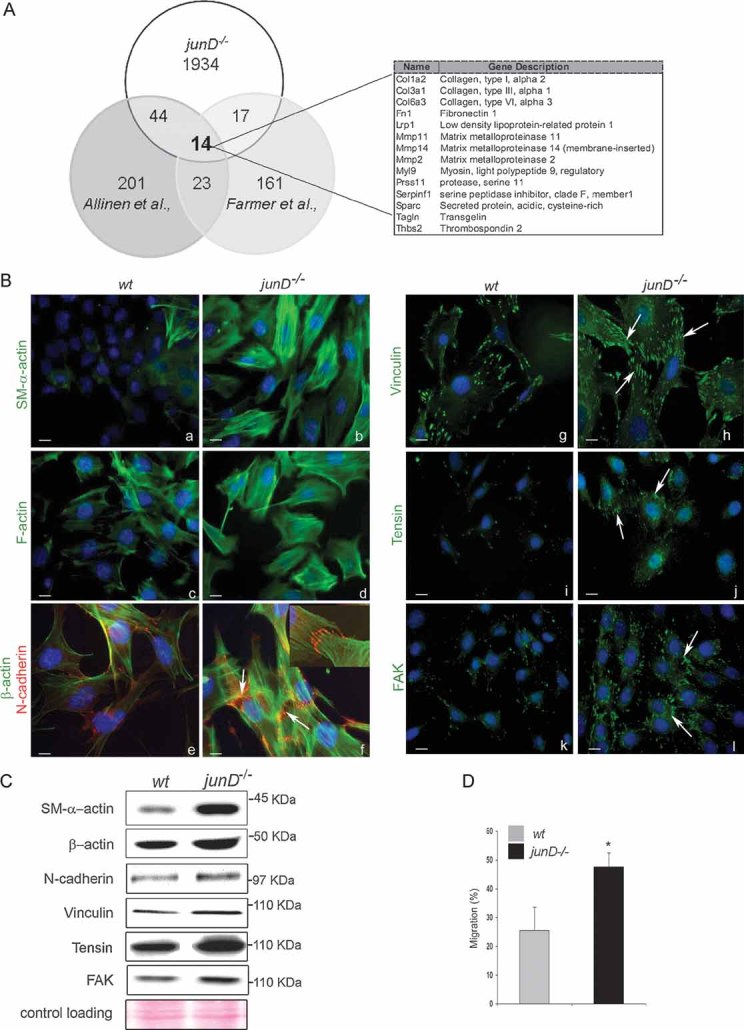
- Venn's diagram showing the number of common up-regulated genes in CAF (from Allinen's and Farmer's studies) and junD−/− fibroblasts. On the right panel, the 14 genes common to the three lists are listed.
- Representative immunofluorescence staining from wt and junD−/− cells using specific antibodies, as indicated. Arrows indicate typical staining. Inserted section in (f) shows a higher magnification (100×) image of a representative AJ co-stained with SM-α-actin (in green) and N-cadherin (in red).
- Western blots of whole cell extracts from wt and junD−/− fibroblasts. Ponceau colouration was used as an internal control for each protein loading; a representative gel is shown.
- Migration assay of wt compare with junD−/− fibroblasts. *p < 0.05 by student test. Scale bars = 10 µm (Ba–d,g–l) and 5 µm in (Be,f).
Since CAFs contain a high proportion of myofibroblasts, we analysed whether junD deletion caused fibroblasts to adopt myofibroblastic features. Compared to wt cells, junD−/− fibroblasts exhibited significant accumulation of SM-α-actin (Fig 3Ba,b), increased assembly of F-actin containing stress fibres (Fig 3Bc,d) and recruitment of SM-α-actin into those stress fibres (Fig S2A). Moreover, just as in differentiated myofibroblasts, junD−/− fibroblasts differed from wt cells in having a significantly higher number of adherens junctions (AJ) (Fig 3Be,f) and an enhanced assembly of mature focal adhesions (FA), characterized by an increase in vinculin, tensin and focal adhesion kinase (FAK) content (Fig 3Bg-l, Fig S2B for quantitative analyses). SM-α-actin protein (Fig 3C) and mRNA (Fig S2C) were increased in junD−/− cells compared to wt. In contrast, total amounts of all other tested proteins remained similar between the two cell types (Fig 3C), further suggesting that inactivation of junD modulated the polymerization of F-actin, the recruitment of N-cadherin to AJ and the association of vinculin or tensin to FA but had only a marginal effect on their total levels. Finally, cellular migration assessed by transwell assays was increased in junD−/− fibroblasts as compared to wt cells (Fig 3D). Hence, these data show that inactivation of junD in fibroblasts converts them into myofibroblasts and increases their cellular migration potential.
CXCL12 plays a key role in acquisition of myofibroblast properties
Since expression of SM-α-actin in myofibroblasts is coordinately regulated by transforming growth factor β1 (TGF-β1) (Hinz et al, 2007; Ronnov-Jessen et al, 1995), we analysed the role of TGF-β1 in acquisition of junD−/− myofibroblast properties. Treatment of junD−/− cells with an inhibitory drug targeting the TGF-β1 pathway did not alter their myofibroblast properties (such as accumulation of SM-α-actin containing stress fibres), despite clearly decreasing phosphorylation of key TGF-β effector Smad3 (Fig S2D). These observations indicate that the myofibroblast properties of junD−/− cells do not result from activation of the TGF-β pathway and strongly suggest that JunD regulates another process, which contributes to the phenotype.
wt fibroblasts incubated with conditioned medium from junD−/− cells exhibited accumulation of SM-α-actin containing stress fibres (Fig 4A), further arguing for the role of a secreted factor. The expression of the chemokine CXCL12 was increased in junD−/− fibroblasts (Fig S2E). Since we observed that junD-dependent CXCL12 accumulation in the stroma was critical for tumour growth and spread, we next investigated if the myofibroblastic phenotype detected in junD-deficient cells may be dependent upon CXCL12. Addition of exogenous CXCL12 into the culture medium of wt cells was sufficient to increase the proportion of SM-α-actin containing stress fibres (Fig 4B). Conversely, silencing of CXCL12 decreased the level of SM-α-actin, the formation of stress fibres, the number of AJ and the proportion of mature FA in junD−/− cells, while the control siRNA had no effect (Fig 4C; Fig S2F). To further validate the CXCL12-dependent autocrine loop in fibroblasts, we confirmed that the CXCR4 receptor was expressed in fibroblasts and detected at the cellular surface (Fig S2G). Moreover, as expected from elevated CXCR4 activity, junD-deficient fibroblasts accumulated phosphorylated forms of ERK1/2, a typical response elicited by this G-protein coupled receptor (Fig 4D). Because small Guanosine triphosphate (GTP)-binding proteins of the Rho family play a central role in regulation of the actin-based cytoskeleton and cell movement, we looked at their activation status by pull-down assays (Fig 4E). Although RhoA and Rac protein levels were comparable between wt and junD−/− fibroblasts (input, Fig 4E and data not shown), junD-deficient cells exhibited higher levels of the GTP-bound form of RhoA as compared to wt fibroblasts (Fig 4E, left part). However, in parallel experiments, we did not detect accumulation of GTP-Rac in junD−/− cells (Fig 4E, right part). Furthermore, treatment of junD−/− fibroblasts with exoenzyme C3 transferase (CT03), a drug that inhibits RhoA Guanosine diphosphate (GDP)/GTP exchange activity, severely affected SM-α-actin polymerization, the formation of F-actin containing stress fibres, the number of AJ and the assembly of FA (Fig 4F). Taken as a whole, these data strongly suggest that the CXCL12/CXCR4 pathway activates the RhoA-GTPase and in turn promotes myofibroblastic properties.
Figure 4. CXCL12 is necessary and sufficient for promoting myofibroblast properties.
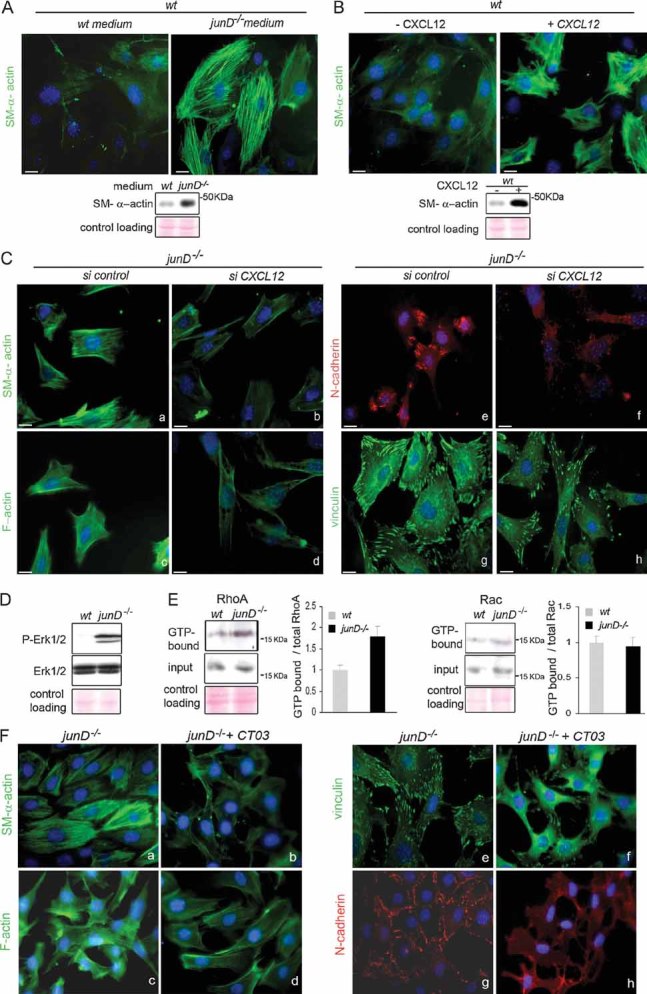
- SM-α-actin staining in wt fibroblasts after incubation in wt or junD−/−-conditioned medium. At the bottom is the corresponding Western blot.
- SM-α-actin staining in wt fibroblasts after addition of exogenous CXCL12 protein. At the bottom is the corresponding Western blot.
- Representative immunofluorescence of myofibroblast markers in junD−/− fibroblasts after transfection with a scramble siRNA (si control) (a,c,e,g) or with a CXCL12-directed siRNA (si CXCL12) (b,d,f,h).
- Western blots from wt and junD−/− whole cell extracts showing p44 and p42 MAPK (Erk1/2) and their phosphorylated forms.
- Representative GST pull-down assays on wt and junD−/− fibroblasts for RhoA (left panel) and Rac (right panel). GTP-bound form and total amount (input) of each protein are shown. Histograms show relative Rho- or Rac-GTP levels normalized to their respective total protein amounts.
- Representative immunofluorescence of myofibroblast markers in junD−/− fibroblasts either untreated (a,c,e,g) or incubated with exoenzyme C3 transferase (b,d,f,h). Scale bars = 10 µm.
HIF-1 is necessary and sufficient for converting fibroblasts into myofibroblasts
It has been shown that hypoxic gradients regulate CXCL12 through HIF induction (Ceradini et al, 2004). Since we demonstrated that HIF-1α protein accumulates in junD−/− fibroblasts and mice (Gerald et al, 2004; Laurent et al, 2008), the up-regulation of CXCL12 in junD−/− fibroblasts could be mediated, at least partly, through HIF. Specific inhibition of HIF-1α by siRNA strongly reduced HIF-1α mRNA levels (Fig S3A) and decreased the expression of its target gene, CXCL12 (Fig S2E). Moreover, HIF-1α inhibition reduced the proportion of SM-α actin- and F-actin-containing stress fibres in junD−/− fibroblasts, as well as the number and size of AJ and mature FA (Fig 5A; Fig S3B). These observations indicate that HIF-1α is a key regulator of the contractile features of junD−/− fibroblasts. To further establish whether HIF was sufficient to establish myofibroblast properties, we treated wt fibroblasts with desferrioxamine (DFO), an iron chelator that mimicked hypoxia and promoted accumulation of HIF-1α (Fig S3C). Accumulation of HIF-1α in wt cells stimulated polymerization of SM-α-actin and F-actin-containing stress fibres, as well as formation of AJ and mature FA (Fig 5B; Fig S3D). Moreover, DFO treatment also increased RhoA activity in wt cells (Fig 5C, left part), whilst the same treatment had no effect on Rac activity (Fig 5C, right part). Therefore, treatment of fibroblasts with hypoxia-mimetic DFO was sufficient for activation of RhoA and differentiation into myofibroblasts.
Figure 5. Oxidative stress-mediated HIF-1α accumulation promotes myofibroblast properties.
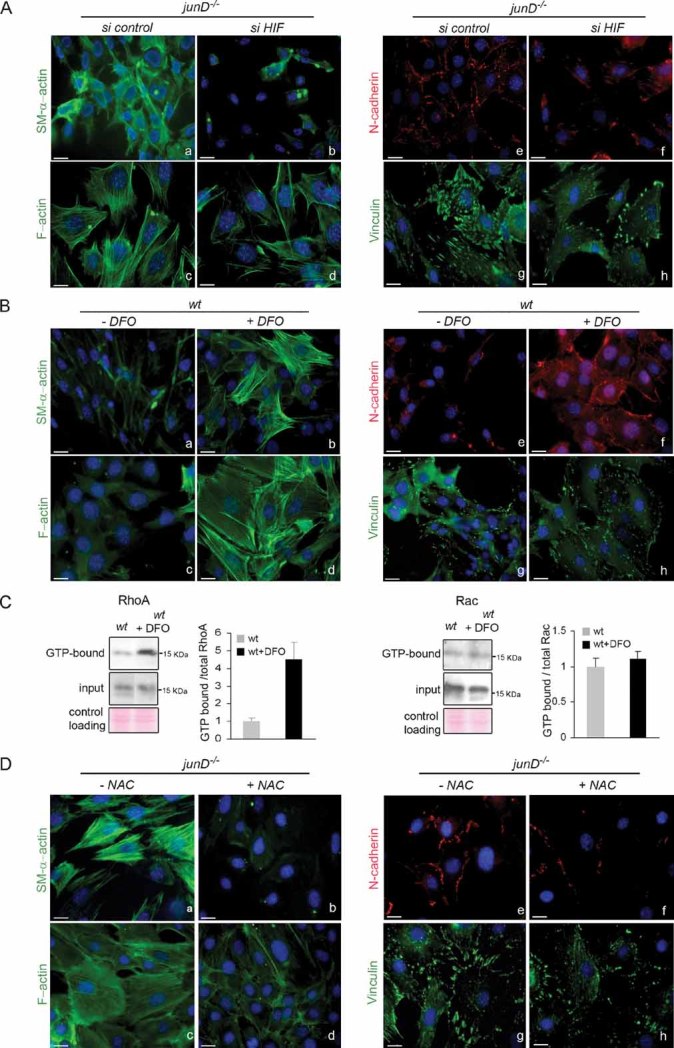
- Representative immunofluorescence of myofibroblast markers in junD−/− fibroblasts after transfection with a scramble siRNA (si control) (a,c,e,g) or with an HIF-1α-directed siRNA (si HIF) (b,d,f,h).
- Representative immunofluorescence of myofibroblast markers in wt fibroblasts either untreated (a,c,e,g) or incubated with DFO (b,d,f,h).
- Representative GST pull-down assays for RhoA (left panel) and Rac (right panel) on wt fibroblasts with or without DFO.
- Representative immunofluorescence of myofibroblast markers in junD−/− fibroblasts either untreated (a,c,e,g) or incubated with NAC (b,d,f,h). Scale bars = 10 µm.
Finally, to investigate if myofibroblast properties detected in junD−/− cells were dependent upon chronic oxidative stress, we subjected them to long-term antioxidant treatment. Culturing junD−/− fibroblasts with N-acetylcysteine (NAC) has been shown to decrease ROS content and HIF protein levels (Gerald et al, 2004). This treatment collectively decreased the contractile features of junD-deficient cells (Fig 5D; Fig S3E), further demonstrating the role of ROS in myofibroblastic differentiation. These results suggest redox-dependent accumulation of HIF stimulates the CXCL12/CXCR4 signalling pathway, triggers activation of RhoA and thereby elicits myofibroblast features.
Human HER2-amplified tumours accumulate CXCL12 and myofibroblasts in their stroma
Having established that myofibroblast content correlated with an increased risk of tumour cell dissemination and that stress-induced CXCL12-dependent signalling played a key role in acquisition of myofibroblast properties, we next investigated the potential relevance of these findings in humans. In that purpose, we analysed the pattern of expression of CXCL12/CXCR4, the myofibroblast content and the possible link with oxidative stress in three classes of human breast cancers, chosen according to their distinct invasive properties and clinical outcomes. We compared the stromal properties of (1) Lum-A breast carcinomas, a subtype associated with a good prognosis, (2) HER2-amplified adenocarcinomas, a subset of aggressive tumours characterized by amplification of the HER2/ERBB2 oncogene and high rate of nodal metastases and (3) basal-like cancers (BLC), another type of aggressive and highly proliferative tumours, albeit less prone to lymph node metastases than HER2 ones. We first investigated the expression pattern of CXCL12/CXCR4 in both stromal and tumour compartments by performing immunohistochemical staining using tissue microarrays (TMA) from HER2, BLC and Lum-A primary tumours (Fig 6; Table S2). Expression of CXCL12 was significantly increased in HER2-neoplasic cells (Fig 6Aa–c) compared to BLC (Fig 6Ad–f) or Lum-A (Fig 6Ag–i) tumour cells. CXCR4 was also strongly expressed in HER2 (Fig 6Ba–c) and BLC (Fig 6Bd–f) epithelial compartment but to a lesser extent in Lum-A (Fig 6Bg–i), as it has been previously reported (Li et al, 2004; Muller et al, 2001). Interestingly, HER2-amplified tumours exhibited the highest expression levels of both CXCL12 and CXCR4 in the fibroblastic compartment (Fig 6Aa–c and Ba–c), compared to BLC (Fig 6Ad–f and Bd–f) or Lum-A (Fig 6Ag–i and Bg–i; Table S2).
Figure 6. CXCL12 and CXCR4 levels are increased in the stroma of HER2-driven breast cancer.
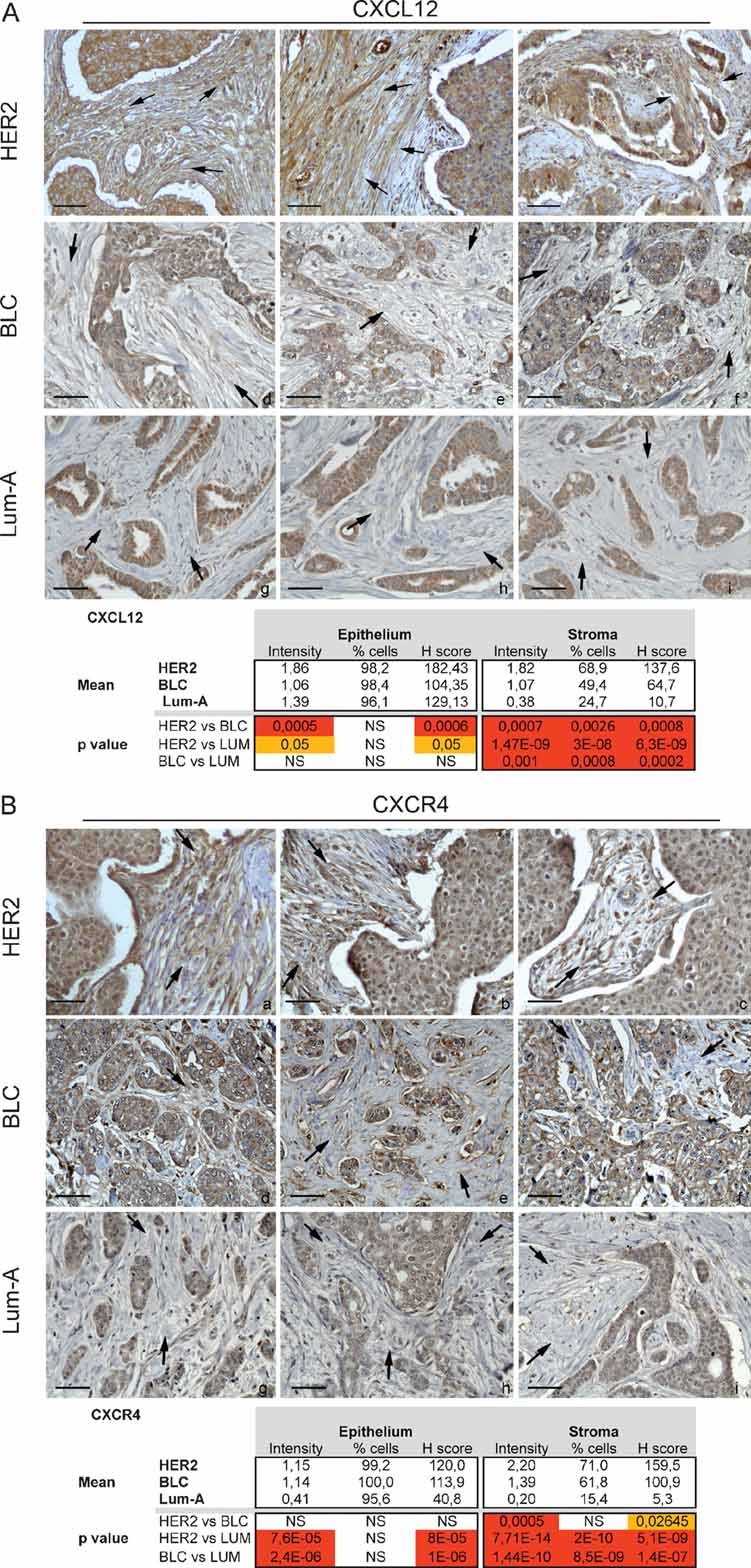
- Sections and histological analysis of HER2 (a–c), BLC (d.f) and Lum-A (g–i) human breast tumours using CXCL12-specific antibody.
- Sections and histological analysis of HER2 (a–c), BLC (d.f) and Lum-A (g–i) human breast tumours using CXCL4-specific antibody. Scale bars = 40 µm.
We next evaluated the proliferation rate and myofibroblastic content in these three classes of human breast cancers. In agreement with the previously known characteristics of these tumours, Ki67 nuclear staining showed that the proliferation rate—as detected in both tumour and stromal compartments—was higher in BLC (Fig S4Ad–f), than HER2 (Fig S4Aa–c), itself significantly higher than in Lum-A (Fig S4Ag–i). These observations suggest that the high rate of lymph node metastasis in HER2-derived tumours compared to BLC is not strictly correlated to the proliferation rate. We also evaluated the recruitment of macrophages in each type of breast cancer using CD68-specific marker (Fig S4B). Both forms of aggressive breast cancers (HER2 and BLC) exhibited a clear enhanced immune response, when compared to the non-aggressive one (Lum-A) (Fig S4B). In contrast, there was no significant difference in macrophages recruitment between HER2 and BLC, suggesting that immune response does not drive increased metastatic potential into lymph nodes in HER2 tumours. To further evaluate the influence of the fibroblastic component of the stroma in each tumour type, we analysed the proportion of myofibroblasts using an SM-α-actin marker (Fig 7A). Among the total population of CAFs, the percentage of SM-α-actin-positive cells and the intensity of SM-α-actin staining were significantly higher in HER2- and BLC-derived stroma (Fig 7Aa–c; Fig 7Ad–f) than in Lum-A (Fig 7Ag–i). Moreover, we detected a clear difference in the general fibroblasts content in these three subtypes of tumours (Fig 7B). For each tumour, analysis was based on percentage of fibroblasts compared to total cells forming the tumour. The maximum proportion of stromal fibroblasts in BLC was evaluated at 40–45%, while it reached 60–65% in HER2 or Lum-A tumours. Thus, there were qualitative and quantitative differences in CAFs properties in HER2, BLC and Lum-A tumours. Lum-A tumours exhibited high fibroblasts content but low expression of SM-α-actin; BLC tumours showed high expression of SM-α-actin but low fibroblasts content; HER2-amplified tumours exhibited both high fibroblasts content and high expression of SM-α-actin, indicating that this subtype of breast cancers accumulate the highest proportion of myofibroblasts.
Figure 7. HER2 tumours exhibit high content of myofibroblasts and display a stress response signature.
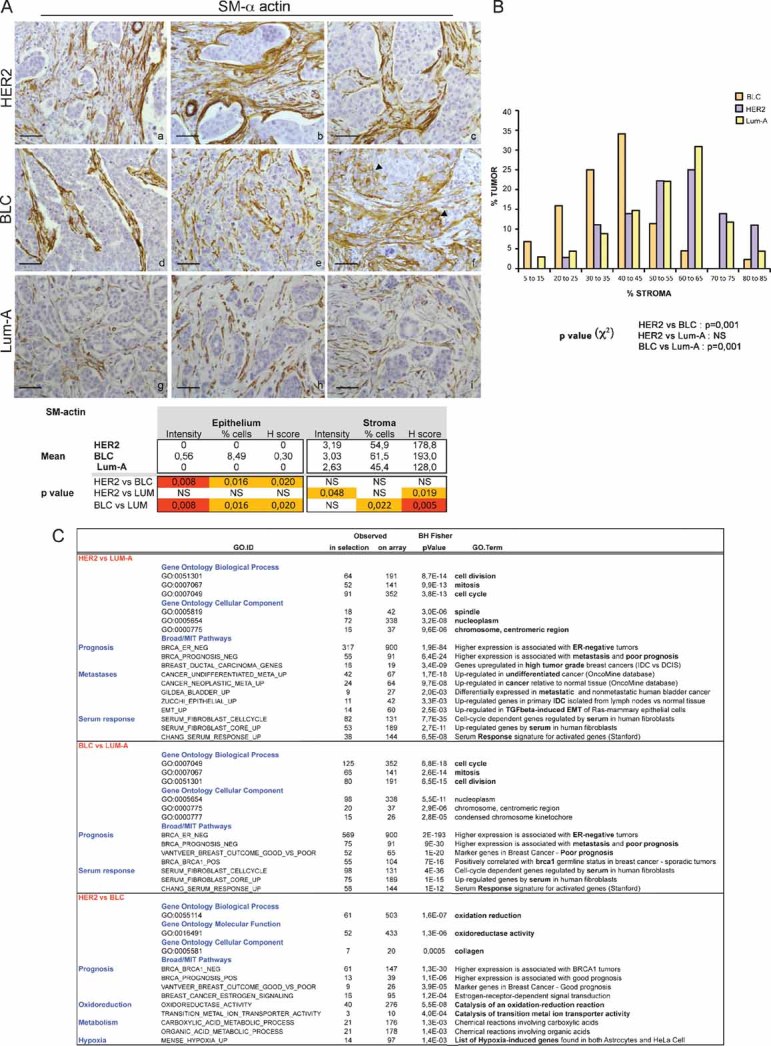
- Representative staining of SM-α-actin in HER2 (a–c), BLC (d–f) and Lum-A (g–i) human breast tumours. Arrowheads indicate SM-α-actin staining in epithelial cells in BLC (f). p-values are as in Fig 6.
- Representative graph of the percentage of fibroblasts compared to total cells forming the tumour in each breast cancer subtype. p-values by χ2 test are highly significant between BLC and HER2 or Lum-A (≤0.001) and non-significant between HER2 and Lum-A.
- Gene Ontology pathways significantly at least 2-fold up-regulated in HER2 versus Lum-A, BLC versus Lum-A and HER2 versus BLC, as indicated. p-values by Fisher Exact test adjusted using the Benjamini–Hochberg correction are indicated.
Interestingly, in HER2-driven tumours, the myofibroblastic content was significantly correlated with the stromal expression levels of CXCL12 (Pearson correlation coefficient: r = 0.56; p = 5 × 10−4). Even more importantly, the myofibroblastic content of HER2-amplified tumours was also significantly correlated with the metastatic rate in lymph nodes (r = 0.76; p = 0.01), further indicating that myofibroblasts promote migration of tumour cells in lymph nodes. Taken together, these data indicate that accumulation of CXCL12 in the stroma of HER2-amplified tumours is associated with high myofibroblasts content, which impacts tumour spreading in lymph nodes.
HER2-amplified tumours display a gene expression profile involved in oxido-reduction
Since HER2-tumours demonstrated correlated metastatic rate, myofibroblasts content and CXCL12 staining, we next wondered whether genes regulating oxidative stress could be abnormally expressed in this set of tumours. We first performed unsupervised analysis and pathway enrichment studies using all Gene Ontology (GO) terms and the global test methods to examine the predominant signatures in HER2, BLC and Lum-A tumours. The GO terms oxidation–reduction (GO: 0055114) (Fig S5A) and oxido-reductase activity (GO: 0016491) (Fig S6A) appeared among the 100 most significantly deregulated pathways (at the 36th and 83th positions, respectively) with P-value smaller than 2 × 10−16. We confirmed this difference using hierarchical clustering and principal component analysis (Fig S5B–D; Fig S6B–D), further highlighting that these pathways were able to discriminate HER2, BLC and Lum-A tumours. In order to better characterize HER2-specific expression pattern, we next performed supervised clustering according to the tumour subtypes. We defined significantly up-regulated genes in HER2 versus Lum-A, BLC versus Lum-A and HER2 versus BLC and submitted the identified set of genes to GO analysis (Fig 7C). As expected according to the clinical outcomes of the tumours, the up-regulated genes in BLC versus Lum-A or HER2 versus Lum-A were involved in cell cycle regulation or associated with specific signatures known to denote estrogen-negative tumours or poor prognosis. Interestingly, when comparing up-regulated genes in HER2 versus BLC, the first identified statistically relevant GO signature was involved in oxido-reduction (Fig 7C). This confirmed the global test analysis and indicated that one of the major differences identified between these two aggressive breast cancer subtypes—HER2 and BLC—was dependent upon oxido-reduction. Genes that are up-regulated in HER2 tumours when compared to BLC have been directly linked to H2O2 generation (NADPH oxidase, Nox4), production of fatty acid hydroxyperoxides (lipoxygenases), oxidative deamination of collagens and elastin (lysyl oxidases), metabolism of xenobiotics (cytochromes P450) (Table S3). Moreover, genes known to be induced upon oxidative stress (such as NQO1) or hypoxia (LOX) were up-regulated in HER2 versus BLC tumours. Thus, these data indicate that HER2-amplifying tumours exhibit an expression profile characteristic of a stress response, when compared to BLC. Finally, in order to reconcile parts of this study based on mouse and human analyses, we have tested JunD expression pattern in breast cancers. We did not observe any difference in junD mRNA levels in Lum-A, BLC and HER2 tumours. In contrast, we detected significant variations in the subcellular localization of the protein (Fig 8A). Although high levels of JunD were detected in the nucleus of Lum-A tumours, this nuclear localization was reduced in BLC and almost undetectable in HER2-amplified tumours (Fig 8A). Although the involved mechanism remains unknown, exclusion of JunD from the nucleus can efficiently inactivate it and suggests that JunD is far less active in HER2 than in BLC or Lum-A, further correlating with the oxidative stress signature of this breast cancer subtype.
Figure 8. JunD nuclear exclusion in HER2-tumours and model describing ROS, as key players in the reciprocal cross-talk between tumour cells and surrounding fibroblasts.
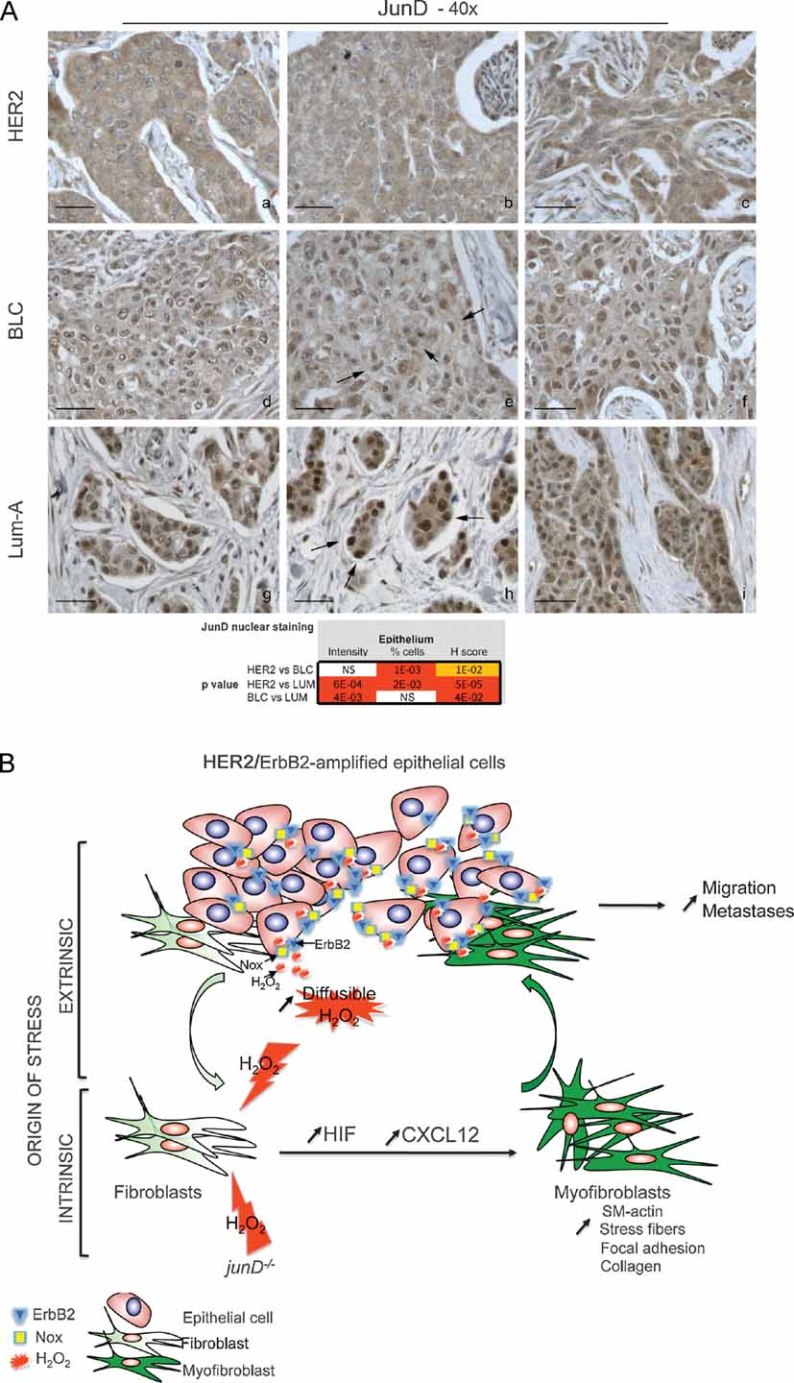
- Representative staining of JunD in HER2 (a–c), BLC (d–f) and Lum-A (g–i) human breast tumours. p-values by χ2 test are highly significant between BLC and HER2 or Lum-A.
- Model: In carcinoma, chronic oxidative stress promotes the conversion of fibroblasts into myofibroblasts, contractile cells with high migration properties capacity. Pro-invasive myofibroblast properties result from ROS-mediated accumulation of the pro-angiogenic HIF-1α factor and the CXCL12 chemokine. The origin of oxidative stress can be either intrinsic or extrinsic to fibroblasts. Indeed, fibroblasts may acquire genetic alteration, such as junD inactivation, which will intrinsically increase ROS contents. Since genetic alteration in stromal fibroblasts may be rare events, stress may also more often originate from tumour cells themselves. Accordingly, we identified a signature characteristic of stress–response in HER2-amplified tumours. In this set of tumours, stress can originate from non-exclusive mechanisms, such as JunD nuclear exclusion, up-regulation of Nox4 or HER2/ERBB2 amplification per se, since it has been previously demonstrated that growth factor-stimulated RTKs enhance H2O2 levels through activation of Nox. H2O2 is highly diffusible and can easily cross-cellular membranes to act on surrounding fibroblasts. Acute stress in fibroblasts increases levels of HIF and CXCL12 that, in turn, convert fibroblasts into myofibroblasts. These highly contractile SM-α-actin-expressing cells would subsequently promote migration and dissemination of neoplastic cells.
In conclusion, HER2-amplified tumours were characterized by high expression of CXCL12 and accumulation of myofibroblasts and revealed an associated stress response signature, all features that may correlate with their high tendency to metastasize in lymph nodes. Taking both mouse and human studies, our data underline the role of persistent oxidative stress on metastatic spread through the conversion of fibroblasts into myofibroblasts. We have used our data to establish a proposed model, as described in Fig 8B.
DISCUSSION
An initial step in understanding the mechanisms of stromal reaction in tumour progression is to fully define the reactive stroma phenotype and its formation. In the present study, by combining mouse models and studies on human materials, we uncover a new ROS-dependent mechanism that impacts on tumourigenesis. Collectively, our data indicate that the oxidative stress-mediated accumulation of HIF-1α stimulates the CXCL12/CXCR4 signalling axis that converts fibroblasts into myofibroblasts and is associated with a high rate of metastases in both mouse and human adenocarcinomas.
Role of myofibroblasts in metastatic spread
The current evidence considers the surrounding tumour microenvironment as an important determinant in the final outcome of cancer. It has been clearly established that CAFs, one of the most abundant stromal components, promote tumour cell proliferation and angiogenesis (Allinen et al, 2004; Bhowmick et al, 2004; Kalluri & Zeisberg, 2006; Littlepage et al, 2005; Olumi et al, 1999; Orimo et al, 2005; Tlsty & Coussens, 2006). Moreover, CAFs have been recently shown to orchestrate tumour inflammation (Erez et al, 2010), further highlighting a new interaction between fibroblasts and immune cells. The link between inflammation and cancer has also been well demonstrated. Indeed, inflammatory immune cells are highly recruited in carcinomas and promote tumour growth and metastases (Condeelis & Pollard, 2006; Coussens & Werb, 2002; de Visser et al, 2006; Erez et al, 2010; Grivennikov & Karin, 2010; Joyce & Pollard, 2009; Karin & Greten, 2005; Mantovani et al, 2008; Radisky & Radisky, 2007; van Kempen et al, 2006). Our work complements these views and assigns to CAFs an essential role in increasing the risk of metastatic dissemination by modulating SM-α-actin expression in a CXCL12-dependent manner. Indeed, the myofibroblast-enriched junD-deficient stroma accelerates tumour onset and increases the number and the size of metastases. Moreover, the HER2 class of human breast cancers, which displays a high rate of lymph node metastases, exhibits a significantly correlated high proportion of SM-α-actin-expressing fibroblasts. In contrast, increased rate of metastasis was not correlated with enhanced immune response, suggesting that other mechanism exists. Similar to our findings, experimental and clinical data support the notion that stromal myofibroblasts participate in tumour development. Moreover, recent data show that genetic inactivation of Pten in stromal fibroblasts of mouse mammary glands accelerates tumour initiation and progression by massive remodelling of extracellular matrix (ECM) and increased angiogenesis (Trimboli et al, 2009). In xenograft mouse models, myofibroblasts were shown to stimulate growth of human breast cancer cells (Olumi et al, 1999; Orimo et al, 2005). Moreover, in vitro co-cultures of myofibroblasts and tumour epithelial cells demonstrated that myofibroblasts promote invasion of breast, colon, pancreas and squamous carcinoma cells (Casey et al, 2008; De Wever et al, 2004; Hwang et al, 2008; Lewis et al, 2004). Similarly, progression of in situ to invasive breast carcinoma is promoted by fibroblasts and inhibited by normal myoepithelial cells (Hu et al, 2008). Furthermore, mesenchymal stem cells and derivatives within tumours promote breast cancer metastasis (Karnoub et al, 2007). In clinical studies, the abundance of stromal myofibroblasts predicts human disease recurrence. Tumours with abundant myofibroblasts are associated with significantly shorter event-free survival rates for stages II and III human colorectal cancers (Tsujino et al, 2007). In addition, in lung and breast adenocarcinomas, myofibroblast content is significantly correlated with lymph node metastasis and shortened patient survival (Tokunou et al, 2001; Yazhou et al, 2004). Accordingly, we show here that HER2-amplifying invasive adenocarcinomas, human breast cancers that display high rate of lymph node metastasis, are associated with the highest proportion of myofibroblasts, when compared to BLC or Lum-A. Taken together, these data emphasize that tumour spreading could be facilitated by the myofibroblastic component of the stroma.
Myofibroblasts express genes that encode invasion associated-secreted factors, ECM proteins and ECM remodelling proteases, which may facilitate tumour cell dissemination. The transcriptome of CAFs, as well as junD-deficient fibroblasts, shows abundant expression of collagens, cytoskeleton components, cell adhesion molecules and MMPs. Moreover, we observe that HER2-breast tumours reveal a high content of various collagens, when compared to BLC. Interestingly, imaging of invading co-cultures of squamous cell carcinoma with stromal fibroblasts revealed that the leading cell is always a fibroblast (Gaggioli et al, 2007). In this study, tumour cells migrate within tracks of ECM molecules, secreted by the leading fibroblasts, whose migration is dependent on Rho-GTPase. Thus, myofibroblasts trigger both deposition and proteolysis of ECM molecules that promote migration of cancer cells. Accordingly, collagen density in mammary tissue significantly increases tumour formation and results in an invasive phenotype with high numbers of lung metastases (Provenzano et al, 2008). In accordance with the prevalent role of tumour microenvironment in cancer cell dissemination, a stromal gene expression signature, similar to that observed during wound healing, predicts human breast and prostate cancer progression and patient survival (Bacac et al, 2006; Chang et al, 2004, 2005; West et al, 2005). All these data strongly suggest that stromal myofibroblasts impact tumour aggressiveness.
Oxidative stress-dependent origin of myofibroblasts
Although the importance of CAFs in tumour development is becoming clear, their origin is still controversial and the basis of their myofibroblast characteristics debated (Haviv et al, 2009; Hinz et al, 2007; Ostman & Augsten, 2009). In adenocarcinomas, it has been suggested that myofibroblasts derive from epithelial cells throughout EMT (Kalluri & Weinberg, 2009; Neilson, 2006; Radisky et al, 2007; Zavadil et al, 2008). Although some data indicate that the stromal compartment, when microdissected from human breast cancers, exhibits genetic alterations (Eng et al, 2009), the proportion of karyotypic alterations in fibroblasts remain less frequent than in cancer cells (Qiu et al, 2008). In addition to EMT, recent data from human breast cancer and animal models established that tumour-associated myofibroblasts can also derive from haematopoietic or mesenchymal stem cells from the bone marrow (Direkze et al, 2004; Ishii et al, 2003; LaRue et al, 2006; Mishra et al, 2008; Mori et al, 2005; Studeny et al, 2004). Local resident fibroblasts or fibroblasts stimulated by members of the TGF-β family have also been considered as a major source of CAFs (Kalluri & Zeisberg, 2006; Mueller et al, 2007; Ostman & Augsten, 2009). Consistently, we demonstrate here that fibroblasts can differentiate into myofibroblasts upon stress in vitro, effect which is reversed by long-term antioxidant treatment. Moreover, our study shows different patho-physiological conditions (junD-deficient animals, HER2-amplified breast adenocarcinomas), in which stress response is associated with myofibroblast accumulation. Thus, our data are consistent with previous published data and further argue that myofibroblasts can originate from stress-exposed fibroblasts.
Many distinct biological circumstances can stimulate oxidative stress in tumours. The origin of oxidative stress in tumours can be either intrinsic or extrinsic to fibroblasts. Acquisition of genetic alterations (e.g. p53 loss) in the stroma, either induced by cancer cells or by chemotherapy, may allow a local production of ROS that would further promote the appearance of myofibroblasts. In addition, MMPs, such as MMP3, a remodelling enzyme that is up-regulated in the earliest stage of human breast cancer, have been shown to modulate activity of the mitochondrial respiratory chain, subsequently enhancing ROS content and stimulating tumour progression (Radisky et al, 2005). MMP3 was also defined as a major factor secreted from senescent fibroblasts (Parrinello et al, 2005), which constitute an inflammatory environment that compromises the structure and the functions of the surrounding tissue (Coppe et al, 2008). Interestingly, genotoxic stress and persistent DNA damage signalling promote the development of senescent fibroblasts that resemble reactive stroma and stimulate tumour growth (Rodier et al, 2009). Thus, stress-signalling originating from tumour epithelial cells may modulate local environment. In that respect, the stress–response signature that we detect in HER2-amplified tumours, expressing high levels of ERBB2 receptor, is interesting. It has been shown that stimulation of receptor tyrosine kinase (RTK) by growth factors, such as epidermal growth factor (EGF), is associated with ROS generation (mainly H2O2) through activation of non-phagocytic Nox (Aslan & Ozben, 2003). ROS production is crucial for RTK-downstream signalling pathways and stimulation of proliferation. Application of an antioxidant treatment to HER2-transformed cells demonstrates that ROS are important for their proliferation and survival (Preston et al, 2001; Wang et al, 2005). In agreement with these data, the present study shows that genes that are directly linked to H2O2 synthesis, such as Nox4, are significantly up-regulated in HER2-amplifying tumours. Interestingly, HER2-amplified tumours display significant exclusion of JunD from the nuclear compartment, when compared to BLC and Lum-A. Nox4 has been already shown to be up-regulated in junD-deficient cells (Gerald et al, 2004). Taken together, these data suggest that amplification and constitutive activation of ERBB2 is associated with reduced JunD activity and massive ROS production, potentially through Nox activation. Since H2O2, despite short half-life, can easily diffuse among cellular membranes, H2O2 production by HER2-neoplasic cells may have an impact on surrounding fibroblasts and promote their conversion into myofibroblasts. Accordingly, HER2-amplified tumours exhibit high content in myofibroblasts, which is significantly correlated with lymph node metastases. Thus, oncogene-mediated hyperplasia may trigger a stress-mediated stromal reaction that initiates a vicious cycle promoting tumour invasion.
Role of HIF and CXCR4/CXCL12 signalling pathways in myofibroblast differentiation and metastases
We uncover a novel cell autonomous HIF-mediated mechanism that regulates differentiation of fibroblasts into myofibroblasts and may enhance tumour spreading. Previously, a stroma-derived predictor of poor prognosis, consisting of angiogenic and hypoxic gene expression, was discovered (Finak et al, 2008). Similarly, it has been suggested that a hypoxia-induced HIF-mediated response reflects metastatic potential in soft tissue sarcomas (Francis et al, 2007). Finally, HIF-1α expression was correlated to aggressiveness in breast cancers, especially in node positive HER2-driven carcinomas (Giatromanolaki et al, 2004; Gruber et al, 2004; Vleugel et al, 2005; Yamamoto et al, 2008). Consistently, we identified a hypoxia-related signature among the genes that are significantly up-regulated in HER2 tumours, when compared to BLC. Thus, HIF-dependent signature has been closely linked to aggressive phenotype in human cancers, further arguing for the deleterious effect of chronic stress.
In addition to HIF, we identified the chemokine CXCL12 as a new mediator for the formation of contractile features in myofibroblasts. CXCL12 has already been involved in tumour growth and metastatic spread, mostly through its role in chemoattraction (Muller et al, 2001; Ostman & Augsten, 2009). Indeed, CXCL12 stimulates carcinoma cell proliferation and recruitment of endothelial precursor cells (Littlepage et al, 2005). Moreover, CXCR4-positive tumour cells are attracted to CXCL12-expressing metastatic organs, through a chemotactic gradient (Zlotnik, 2008). Furthermore, CXCL12 has been defined as a master regulator of trafficking of haematopoietic- and cancer-stem cells (Gelmini et al, 2008). Finally, CXCL12-dependent tumour cell migration has also been associated with macrophages and their cross-talk with tumour cells (Joyce & Pollard, 2009). In agreement with this previously identified chemoattractive function, we detected a significant increase in recruitment of inflammatory cells in our mouse model of mammary adenocarcinomas. In contrast, this chemoattractive effect was not detected in the transplanted tumour model, despite clear accumulation of CXCL12. Similarly, HER2-human breast cancers did not show enhanced rate of CD68-positive cells when compared to BLC, while CXCL12 accumulated. In contrast, SM-α-actin-expressing fibroblasts accumulated in the stroma of the two mouse models used in this study. Thus, our study complements previous published data about CXCL12 by deciphering a new function for this chemokine in stromal fibroblasts and acquisition of contractile properties. Indeed, we show here that CXCL12 is necessary and sufficient to convert normal fibroblasts into myofibroblasts. Accordingly, CXCL12 has been previously shown to induce intracellular actin polymerization in lymphocytes (Bleul et al, 1996). We show here also that HER2 human breast cancers, which are associated with increased risk of nodal metastasis, exhibit a statistically significant increase in CXCL12 and CXCR4 expression in the stroma. Similarly, CXCR4 expression in breast cancers has been correlated with survival and metastatic development (Kang et al, 2005; Li et al, 2004; Muller et al, 2001). This suggests that there may be some value in developing CXCR4-blocking antibodies for lymph node-positive HER2 patients, in addition to the already existing treatments (Baselga & Swain, 2009). Many reports have associated stromal CXCL12-positive staining with increased aggressive metastatic foci in human cancers (Kryczek et al, 2007). Fibroblast-derived CXCL12 stimulates the invasion of oral squamous cell carcinoma and CXCL12 blocking antibodies reduce the level of invasion (Daly et al, 2008). In our model of fibroblasts, CXCL12 triggers activation of the Rho family of GTPases and increases migratory properties, as it has been shown in melanoma cells and inflammatory cells (Bartolome et al, 2004; Tan et al, 2006). Moreover, previous studies on the pro-invasive capacity of myofibroblasts revealed an essential role of the RhoA/Rho-kinase axis in cancer cell invasion (De Wever et al, 2004; Nguyen et al, 2005). Altogether, these data suggest that activation of RhoA triggered by CXCL12/CXCR4 signalling in stressed fibroblasts could contribute to dissemination of tumour cells.
In conclusion, our data support the conclusion that production of CXCL12 by stromal fibroblasts is tightly regulated by oxidative stress in an HIF-dependent manner. Most probably, this enhances metastatic spread by increasing the migratory potential of both tumour cells and their associated stroma. These data provides new insights into the contribution of oxidative stress to the tumour-associated microenvironment and may contribute to a better knowledge of mechanisms stimulating cell dissemination.
MATERIALS AND METHODS
Cell culture and siRNA
Independent immortalized cell lines derived from wt or junD−/− embryos were generated using a conventional 3T3 protocol as previously described in Gerald et al (2004). Experiments were performed at least in triplicate on three independent cell lines of each genotype. Treatments of cells were performed for 16 h by addition of exogenous CXCL12 (100 nM), a kind gift of Arenzana-Seistedos, for 4 h with DFO (100 µM) (D9533-Sigma) and 3 h with exoenzyme C3 transferase (1 µg/ml) (CT03A-cytoskeleton). Long-term antioxidant treatment, using NAC (500 µM) (A9165-Sigma), was applied for 20 days, with addition of the product every 2 days. For siRNA experiments, cells were transfected with 25–50 nM siRNA using Dharmafect1 reagent (Dharmacon). siRNA sequences targeting mouse HIF-1α and CXCL12 were, respectively, 5′-CCCUAUAUCCCAAUGGAUG-3′ and 5′-CAACGUCAAGCAUCUGAAA-3′.
Mouse strains and graft experiments
Due to male sterility, the junD-deficient mice were maintained through the breeding of heterozygous animals. Ras junD+/+ and Ras junD−/− mice have been obtained by crossing junD+/− mice with MMTV-Ha-Ras mouse mammary tumour model (Sinn et al, 1987). Mice were checked weekly for tumour growth. On average, tumours appeared in 15-month-old animals. Tumour volume was determined by the use of a calliper and the following equation: 0.5 × [length × (width)2]. For immunohistochemistry, tumours were fixed in 4% paraformaldehyde (PFA) for 1 h 30 min at room temperature and then embedded in gelatin 15%/sucrose 7.5%. Ten micrometre sections were incubated with antibody against SM-α-actin (A2547, clone1A4, Sigma), CXCL12 (MAB350, R&D system), CD31 (7388-50, Abcam), E-cadherin (4065, Cell Signaling), Vimentin (RV202, Abcam), Podoplanin (Abcam, ab11936), F4/80 (ab6640, Abcam) or coloured with Masson's trichrome or haematoxyline-eosin-saffranin (HES). Fibroblasts, myofibroblasts, epithelial cells, macrophages and haematopoietic cells were specifically stained using vimentin, SM-actin, E-cadherin, F4/80 and CD45-specific antibodies, respectively. For quantification, three different tumours of each genotype were analysed and three sections from distinct areas of each tumour were evaluated. Staining intensity and percentage of labelled cells were scored for SM-actin, vimentin and E-cadherin positive cells. Numbers of CD45- or F4/80-positive cells per tumour surface were also quantified. Statistical analysis were done using student test. Graft experiments were performed using 10-month-old mice. Single cell suspensions containing 2 × 106 B16F10 cells in 200 µl were injected subcutaneously. The mice were checked daily for tumour growth and tumours were measured using callipers. Tumours and lungs were collected when tumours reach appreciatively 2 cm3. Removals were fixed in 10% formol, sectioned in paraffin (5 µm) and coloured. When required, mice have been treated daily with 3 µg of control- or CXCL12-specific siRNA (sequence above), previously validated on cells. The Institut Curie ethical committee approved all experiments.
Gene expression analysis
Whole genome expression profiling of wt and junD−/− fibroblasts were performed using mouse expression beadchip (Sentrix Mouse-6 v1.1) from Illumina (see Supporting Information). Three RNA samples were pooled according to their genotype. Pooled RNA were used to synthesize cRNA and hybridized to Illumina mouse-6 expression arrays (version 1). Detected probe sets were selected (n = 46,673) and further analysed using beadstudio software. The background was subtracted and intensities normalized using cubic spline algorithm. Only at least 2-fold up-regulated genes with a significant P-value (p < 0.05) (n = 1934) in junD−/− versus wt cells were taken into account. The comparison between lists of genes obtained in independent gene expression analyses was carried out using the hypergeometric law via Fisher's Exact test in R (The R development Core Team, R: A Language and Environment for Statistical Computing, Version 2.8.1, 2008).
Gene expression profiling and pathway enrichment analysis in human breast cancers
Only human tumours with a high content in epithelial tissue (at least 65%) have been used. Total RNA were extracted from frozen tumours with TRIzol reagent (Life Technology, Inc.) and purified using the RNeasy MinElute Cleanup kit (Qiagen). RNA quality was checked on an Agilent 2100 bioanalyser. Samples were analysed on Human Genome U133 Plus 2.0 array (Affymetrix), according to manufacturer's procedures. Log-intensity values were normalized using the GC-RMA algorithm. Probes, with log-intensity value smaller than 3.5, were discarded. A linear model was then fitted to detect and correct any batch and hybridization effects. All GO pathways were retrieved using the R software and Bioconductor. We first applied the global test method proposed by Goeman et al (2004), investigating whether the expression pattern of a group of genes is significantly related to a clinical outcome of interest. We also performed supervised comparative analysis using Welch test and adjusted p-values using Benjamini–Hochberg procedure (R-Multitest package).
Immunofluorescence and immunoblotting
Fluorescence microscopy was performed as previously described with few modifications (Mechta et al, 1997). In brief, cells were fixed in 4% PFA for 30 min, permeabilized in 0.01% sodium dodecyl sulphate (SDS) for 10 min, rinsed twice in phosphate buffered saline (PBS) solutions and blocked for 30 min in 10% foetal calf serum (FCS). Cells were stained with 4,6-diamidino-2-phenylindole (DAPI) (50 µg/ml, Roche) for DNA detection, together with specific antibody recognizing SM-α-actin (A2547 clone 1A4, Sigma 1/400), vinculin (V9131 clone hVIN-1, Sigma 1/1000), N-cadherin (SC-7939, Santa Cruz 1/250), tensin (610064, BD Biosciences 1/500) or FAK (F2918, Sigma 1/1000), followed by either fluorescein isothiocyanate (FITC)-coupled or texas red-coupled secondary antibody (Amersham). F-actin was visualized with FITC-phalloidine (P5282, Sigma 1/1000). Slides were examined using a Zeiss Axioplan 2 and images were acquired with identical exposure times and settings using a digital camera (Photometrix Quantix). Fluorescence image analysis was performed using the ImageJ software (Rasband, WS., ImageJ, U.S National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997–2008). After background subtraction, the mean fluorescence intensity of SM-α-actin and F-actin was measured considering all cells per field per condition (n ≥ 20 cells), from at least three independent experiments. In order to count the FA and the adherent junctions and measure their size, we used the ‘analysis particle’ tool of ImageJ (n ≥ 50 per condition). The FA size was measured as the length of the FA in the direction perpendicular to the cell boundary. For immunoblotting analysis, whole cell extracts and Western blotting was performed as in Gerald et al (2004) using antibodies described above. Blots were incubated with horseradish peroxidase-conjugated secondary antibody (Amersham) followed by detection with enhanced chemoluminescence and exposed to autoradiography.
The Paper Explained
PROBLEM
While the tumour microenvironment is known to contribute to tumour progression, the role of carcinoma-associated fibroblasts (CAFs) remains controversial and their origin unclear. This study addresses the hypothesis that chronic oxidative stress can modulate tumour growth and spread by modulating surrounding tumour fibroblasts.
RESULTS
We took advantage of the chronic oxidative stress resulting from junD deletion to examine the role of reactive oxygen species (ROS) in tumour development. In this model, CAFs derive from stress-exposed fibroblasts and promote metastatic dissemination of neoplastic cells. Pro-invasive myofibroblast properties resulted from ROS-mediated accumulation of the pro-angiogenic HIF-1α and the pro-inflammatory chemokine CXCL12 that activated the RhoA-GTPase. Invasive HER2-human breast adenocarcinomas, characterized by high rate of lymph node metastases, exhibit a correlated stromal accumulation of both CXCL12 and myofibroblasts and display an associated oxido-reduction signature, indicating the relevance of our findings in human cancers.
IMPACT
HER2-amplifying human breast adenocarcinomas, a breast cancer molecular subtype associated with a very poor prognosis and lymph node metastases, express high levels of CXCL12 in the stroma. Our study raises the intriguing possibility that, in addition to current treatments, CXCR4-blocking antibodies may be effective in combating tumour metastasis in lymph node-positive HER2 patients.
Immunohistochemistry on human breast carcinomas
Sections of paraffin-embedded tissue (3 µm) were stained using streptavidin-peroxidase protocol, immunostainer Benchmark, Ventana, Illkirch, France with specific antibodies recognizing CXCR4 (1/50; ab2074, Abcam), CXCL12 (1/100; ab9797, Abcam), Ki67 (1/200; MIB-1, Dakocytomation), SM-α actin (1/400; A2547, Sigma), CD68 (clone KP1 M081401-2, Dako) and JunD (1/100; sc-74, Santa Cruz) (see also Fig S7). TMA from 36 HER2, 44 BLC and 23 Lum-A tumours were composed using three cores of tumour tissue per case and one core of normal tissue (1 mm of diameter each) and hybridized simultaneously. Invasive HER2-amplified carcinomas have been defined according to ERBB2 immunostaining using ASCO's guideline. Among invasive ductal carcinomas, the BLC immunophenotype was defined as follows: ER−PR− ERBB2− with the expression of at least one of the following markers: KRT5/6+, EGF-R+, kit+. Lum-A tumours were ER+. For quantification, three sections from distinct areas of each tumour were evaluated independently by at least two different investigators. A score, associated with a colour code, was given as a function of the percentage of positive cells and the staining intensity. The colour code is as the following: white = no or weak signal; yellow = moderate; orange = high; red = intense. Experiments were approved by the ethics committee of the Institut Curie and informed consent was obtained from all included patients prior to inclusion in the study.
Migration assay
Migration assays were performed by using Corning polycarbonate Transwell 24-well plates. Cells (7 × 105) were seeded to the upper chamber of each well (6.5 mm in diameter, 8 µm pore size). Medium containing 7% FCS was placed in the lower compartment of the chamber. After 24 h at 37°C, any remaining cells on the upper membrane surface were removed by careful wiping with a cotton swab, and the filters were fixed and stained with 0.2% crystal violet solution in 20% methanol for 15 min. The colouration was removed using acetic acid (10%) and absorbance at 530 nm was used to measure the proportion of migrating cells adhering to the under surface of the filter. The number of total cells was evaluated by using unwrapped chambers.
RhoA- and Rac-pull-down assay
RhoA and Rac activity were quantified by measuring the amounts of RhoA-GTP and Rac-GTP precipitated in a pull-down assay from cell lysates, using the GTPase-binding domain of Rhotekin (RBD) or p21-activated kinase, PAK, (PBD), fused to the glutathione-S-transferase (GST-RBD or GST-PBD, respectively). Briefly, 107 cells were lysed in lysis buffer (200 mM NaCl, 0.5% NP-40, 1 mM ethylenediaminetetraacetic acid (EDTA) pH 8.0, 20 mM Tris–HCl pH 8.0). Whole cell extracts (500 µg) were added to GST-RBD or GST-PBD beads. The pull-down reaction mixtures were incubated for 45 min at 4°C with gentle agitation. The supernatants were removed by brief centrifugation and the precipitated proteins were subjected to immunoblot analysis using monoclonal antibody to RhoA (2117, Cell Signaling) or to Rac1/2/3 (2465, Cell Signaling). The intensities of the bands of GTP-bound RhoA and Rac were quantified by ImageJ and normalized to the total amount of the corresponding protein in whole lysates.
Statistical analysis
All experiments were performed at least three times. Differences were considered to be statistically significant at values of p ≤ 0.05 by Student's t-test and Mann Whitney test. Graphs show mean and standard error of mean using Student's t-test. Single, double and triple asterisks indicate statistically significant differences: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.005. All survival analyses were carried out using Kaplan–Meier method and log-rank test in R (refer above).
Acknowledgments
We thank M. Yaniv, A. Aurias, D. Williamson and D. Lallemand for critical reading and interesting comments of the manuscript. We are grateful to Arenzana-Seistedos and H. Lortat-Jacob for fruitful discussions about this work. We thank G. Laurent for assistance in graft experiments, T. Gruosso for help in FACS experiments, A. Sadou for GST-pull down assays and F. Assayag for the gift of B16F10 cells. We acknowledge the Institut Curie Breast Cancer Study Group, headed by B. Sigal-Zafrani, A. Fourquet and R. Salmon for providing human breast tumours. We are grateful to all members of the animal facilities of Curie Institute for their helpful expertise. The experimental work was supported by grants from the Institut National de la Santé et de la Recherche Médicale (Inserm), the Institut Curie, the Fondation de France, the Agence Nationale de la Recherche, the French National Institut of Cancer (INCa) and the Association pour la Recherche contre le Cancer.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
FMG and AT participated in the conception and design of the experiments. AT, DG, BB, MC, MCP and DB performed the experiments on mouse models and derived cells. AT, GD, SL, MR, MHS, TD and OD participated in the studies of human samples. XSG and AVS provided all human samples used in this study. CL and GR contributed to the statistical analyses of the data. FMG directed the work and wrote the paper with suggestions and comments from all authors.
For more information
Oncomine data base:
Breast cancer information:
F. Mechta-Grigoriou's laboratory:
http://www.curie.fr/recherche/themes/detail_equipe.cfm/lang/_gb/id_equipe/318.htm
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6:17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Aslan M, Ozben T. Oxidants in receptor tyrosine kinase signal transduction pathways. Antioxid Redox Signal. 2003;5:781–788. doi: 10.1089/152308603770380089. [DOI] [PubMed] [Google Scholar]
- Bacac M, Provero P, Mayran N, Stehle JC, Fusco C, Stamenkovic I. A mouse stromal response to tumor invasion predicts prostate and breast cancer patient survival. PLoS ONE. 2006;1:e32. doi: 10.1371/journal.pone.0000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett JM, Ellis IO, Dowsett M, Mallon EA, Cameron DA, Johnston S, Hall E, A'Hern R, Peckitt C, Bliss JM, et al. Human epidermal growth factor receptor 2 status correlates with lymph node involvement in patients with estrogen receptor (ER) negative, but with grade in those with ER-positive early-stage breast cancer suitable for cytotoxic chemotherapy. J Clin Oncol. 2007;25:4423–4430. doi: 10.1200/JCO.2007.11.0973. [DOI] [PubMed] [Google Scholar]
- Bartolome RA, Galvez BG, Longo N, Baleux F, Van Muijen GN, Sanchez-Mateos P, Arroyo AG, Teixido J. Stromal cell-derived factor-1alpha promotes melanoma cell invasion across basement membranes involving stimulation of membrane-type 1 matrix metalloproteinase and Rho GTPase activities. Cancer Res. 2004;64:2534–2543. doi: 10.1158/0008-5472.can-03-3398. [DOI] [PubMed] [Google Scholar]
- Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9:463–475. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1) J Exp Med. 1996;184:1101–1109. doi: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey TM, Eneman J, Crocker A, White J, Tessitore J, Stanley M, Harlow S, Bunn JY, Weaver D, Muss H, et al. Cancer associated fibroblasts stimulated by transforming growth factor beta1 (TGF-beta 1) increase invasion rate of tumor cells: a population study. Breast Cancer Res Treat. 2008;110:39–49. doi: 10.1007/s10549-007-9684-7. [DOI] [PubMed] [Google Scholar]
- Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, Botstein D, Brown PO. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Nuyten DS, Sneddon JB, Hastie T, Tibshirani R, Sorlie T, Dai H, He YD, van't Veer LJ, Bartelink H, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA. 2005;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly AJ, McIlreavey L, Irwin CR. Regulation of HGF and SDF-1 expression by oral fibroblasts—implications for invasion of oral cancer. Oral Oncol. 2008;44:646–651. doi: 10.1016/j.oraloncology.2007.08.012. [DOI] [PubMed] [Google Scholar]
- de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, Mareel M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004;18:1016–1018. doi: 10.1096/fj.03-1110fje. [DOI] [PubMed] [Google Scholar]
- Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, Alison MR, Wright NA. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64:8492–8495. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- Eng C, Leone G, Orloff MS, Ostrowski MC. Genomic alterations in tumor stroma. Cancer Res. 2009;69:6759–6764. doi: 10.1158/0008-5472.CAN-09-0985. [DOI] [PubMed] [Google Scholar]
- Erez N, Truitt M, Olson P, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- Eyden B, Banerjee SS, Shenjere P, Fisher C. The myofibroblast and its tumours: a review. J Clin Pathol. 2009;62:236–249. doi: 10.1136/jcp.2008.061630. [DOI] [PubMed] [Google Scholar]
- Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, Andre S, Piccart M, Campone M, Brain E, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med. 2009;15:68–74. doi: 10.1038/nm.1908. [DOI] [PubMed] [Google Scholar]
- Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- Francis P, Namlos HM, Muller C, Eden P, Fernebro J, Berner JM, Bjerkehagen B, Akerman M, Bendahl PO, Isinger A, et al. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics. 2007;8:73. doi: 10.1186/1471-2164-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- Gelmini S, Mangoni M, Serio M, Romagnani P, Lazzeri E. The critical role of SDF-1/CXCR4 axis in cancer and cancer stem cells metastasis. J Endocrinol Invest. 2008;31:809–819. doi: 10.1007/BF03349262. [DOI] [PubMed] [Google Scholar]
- Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Giatromanolaki A, Koukourakis MI, Simopoulos C, Polychronidis A, Gatter KC, Harris AL, Sivridis E. c-erbB-2 related aggressiveness in breast cancer is hypoxia inducible factor-1alpha dependent. Clin Cancer Res. 2004;10:7972–7977. doi: 10.1158/1078-0432.CCR-04-1068. [DOI] [PubMed] [Google Scholar]
- Goeman JJ, van de Geer SA, de Kort F, van Houwelingen HC. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20:93–99. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Karin M. Inflammation and oncogenesis: a vicious connection. Curr Opin Genet Dev. 2010;20:65–71. doi: 10.1016/j.gde.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber G, Greiner RH, Hlushchuk R, Aebersold DM, Altermatt HJ, Berclaz G, Djonov V. Hypoxia-inducible factor 1 alpha in high-risk breast cancer: An independent prognostic parameter? Breast Cancer Res. 2004;6:R191–R198. doi: 10.1186/bcr775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haviv I, Polyak K, Qiu W, Hu M, Campbell I. Origin of carcinoma associated fibroblasts. Cell Cycle. 2009;8:383–395. doi: 10.4161/cc.8.4.7669. [DOI] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y, et al. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13:394–406. doi: 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii G, Sangai T, Oda T, Aoyagi Y, Hasebe T, Kanomata N, Endoh Y, Okumura C, Okuhara Y, Magae J, et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem Biophys Res Commun. 2003;309:232–240. doi: 10.1016/s0006-291x(03)01544-4. [DOI] [PubMed] [Google Scholar]
- Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- Kang H, Watkins G, Douglas-Jones A, Mansel RE, Jiang WG. The elevated level of CXCR4 is correlated with nodal metastasis of human breast cancer. Breast. 2005;14:360–367. doi: 10.1016/j.breast.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol. 2007;292:C987–C995. doi: 10.1152/ajpcell.00406.2006. [DOI] [PubMed] [Google Scholar]
- LaRue AC, Masuya M, Ebihara Y, Fleming PA, Visconti RP, Minamiguchi H, Ogawa M, Drake CJ. Hematopoietic origins of fibroblasts. I. In vivo studies of fibroblasts associated with solid tumors. Exp Hematol. 2006;34:208–218. doi: 10.1016/j.exphem.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Laurent G, Solari F, Mateescu B, Karaca M, Castel J, Bourachot B, Magnan C, Billaud M, Mechta-Grigoriou F. Oxidative stress contributes to aging by enhancing pancreatic angiogenesis and insulin signaling. Cell Metab. 2008;7:113–124. doi: 10.1016/j.cmet.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Lewis MP, Lygoe KA, Nystrom ML, Anderson WP, Speight PM, Marshall JF, Thomas GJ. Tumour-derived TGF-beta1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. Br J Cancer. 2004;90:822–832. doi: 10.1038/sj.bjc.6601611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell. 2004;6:459–469. doi: 10.1016/j.ccr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Littlepage LE, Egeblad M, Werb Z. Coevolution of cancer and stromal cellular responses. Cancer Cell. 2005;7:499–500. doi: 10.1016/j.ccr.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Mechta F, Lallemand D, Pfarr CM, Yaniv M. Transformation by ras modifies AP1 composition and activity. Oncogene. 1997;14:837–847. doi: 10.1038/sj.onc.1200900. [DOI] [PubMed] [Google Scholar]
- Mechta-Grigoriou F, Gerald D, Yaniv M. The mammalian Jun proteins: redundancy and specificity. Oncogene. 2001;20:2378–2389. doi: 10.1038/sj.onc.1204381. [DOI] [PubMed] [Google Scholar]
- Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW, Banerjee D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008;68:4331–4339. doi: 10.1158/0008-5472.CAN-08-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori L, Bellini A, Stacey MA, Schmidt M, Mattoli S. Fibrocytes contribute to the myofibroblast population in wounded skin and originate from the bone marrow. Exp Cell Res. 2005;304:81–90. doi: 10.1016/j.yexcr.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Mueller MM, Fusenig NE. Friends or foes—bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- Mueller L, Goumas FA, Affeldt M, Sandtner S, Gehling UM, Brilloff S, Walter J, Karnatz N, Lamszus K, Rogiers X, et al. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am J Pathol. 2007;171:1608–1618. doi: 10.2353/ajpath.2007.060661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- Neilson EG. Mechanisms of disease: fibroblasts—a new look at an old problem. Nat Clin Pract Nephrol. 2006;2:101–108. doi: 10.1038/ncpneph0093. [DOI] [PubMed] [Google Scholar]
- Nguyen QD, De Wever O, Bruyneel E, Hendrix A, Xie WZ, Lombet A, Leibl M, Mareel M, Gieseler F, Bracke M, et al. Commutators of PAR-1 signaling in cancer cell invasion reveal an essential role of the Rho-Rho kinase axis and tumor microenvironment. Oncogene. 2005;24:8240–8251. doi: 10.1038/sj.onc.1208990. [DOI] [PubMed] [Google Scholar]
- Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–5011. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth—bystanders turning into key players. Curr Opin Genet Dev. 2009;19:67–73. doi: 10.1016/j.gde.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–496. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouyssegur J, Mechta-Grigoriou F. Redox regulation of the hypoxia-inducible factor. Biol Chem. 2006;387:1337–1346. doi: 10.1515/BC.2006.167. [DOI] [PubMed] [Google Scholar]
- Preston TJ, Muller WJ, Singh G. Scavenging of extracellular H2O2 by catalase inhibits the proliferation of HER-2/Neu-transformed rat-1 fibroblasts through the induction of a stress response. J Biol Chem. 2001;276:9558–9564. doi: 10.1074/jbc.M004617200. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, Trivett M, Thompson ER, Ramakrishna M, Gorringe KL, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008;40:650–655. doi: 10.1038/ng.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky ES, Radisky DC. Stromal induction of breast cancer: inflammation and invasion. Rev Endocr Metab Disord. 2007;8:279–287. doi: 10.1007/s11154-007-9037-1. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem. 2007;101:830–839. doi: 10.1002/jcb.21186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest. 1995;95:859–873. doi: 10.1172/JCI117736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- Sinn E, Muller W, Pattengale P, Tepler I, Wallace R, Leder P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell. 1987;49:465–475. doi: 10.1016/0092-8674(87)90449-1. [DOI] [PubMed] [Google Scholar]
- Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, Champlin RE, Andreeff M. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96:1593–1603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- Tan W, Martin D, Gutkind JS. The Galpha13-Rho signaling axis is required for SDF-1-induced migration through CXCR4. J Biol Chem. 2006;281:39542–39549. doi: 10.1074/jbc.M609062200. [DOI] [PubMed] [Google Scholar]
- Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–150. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- Tokunou M, Niki T, Eguchi K, Iba S, Tsuda H, Yamada T, Matsuno Y, Kondo H, Saitoh Y, Imamura H, et al. c-MET expression in myofibroblasts: role in autocrine activation and prognostic significance in lung adenocarcinoma. Am J Pathol. 2001;158:1451–1463. doi: 10.1016/S0002-9440(10)64096-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong JL, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–1091. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujino T, Seshimo I, Yamamoto H, Ngan CY, Ezumi K, Takemasa I, Ikeda M, Sekimoto M, Matsuura N, Monden M. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin Cancer Res. 2007;13:2082–2090. doi: 10.1158/1078-0432.CCR-06-2191. [DOI] [PubMed] [Google Scholar]
- van Kempen LC, de Visser KE, Coussens LM. Inflammation, proteases and cancer. Eur J Cancer. 2006;42:728–734. doi: 10.1016/j.ejca.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Vleugel MM, Greijer AE, Shvarts A, van der Groep P, van Berkel M, Aarbodem Y, van Tinteren H, Harris AL, van Diest PJ, van der Wall E. Differential prognostic impact of hypoxia induced and diffuse HIF-1alpha expression in invasive breast cancer. J Clin Pathol. 2005;58:172–177. doi: 10.1136/jcp.2004.019885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XF, Witting PK, Salvatore BA, Neuzil J. Vitamin E analogs trigger apoptosis in HER2/erbB2-overexpressing breast cancer cells by signaling via the mitochondrial pathway. Biochem Biophys Res Commun. 2005;326:282–289. doi: 10.1016/j.bbrc.2004.11.028. [DOI] [PubMed] [Google Scholar]
- West RB, Nuyten DS, Subramanian S, Nielsen TO, Corless CL, Rubin BP, Montgomery K, Zhu S, Patel R, Hernandez-Boussard T, et al. Determination of stromal signatures in breast carcinoma. PLoS Biol. 2005;3:e187. doi: 10.1371/journal.pbio.0030187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Ibusuki M, Okumura Y, Kawasoe T, Kai K, Iyama K, Iwase H. Hypoxia-inducible factor 1alpha is closely linked to an aggressive phenotype in breast cancer. Breast Cancer Res Treat. 2008;110:465–475. doi: 10.1007/s10549-007-9742-1. [DOI] [PubMed] [Google Scholar]
- Yazhou C, Wenlv S, Weidong Z, Licun W. Clinicopathological significance of stromal myofibroblasts in invasive ductal carcinoma of the breast. Tumour Biol. 2004;25:290–295. doi: 10.1159/000081394. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Haley J, Kalluri R, Muthuswamy SK, Thompson E. Epithelial-mesenchymal transition. Cancer Res. 2008;68:9574–9577. doi: 10.1158/0008-5472.CAN-08-2316. [DOI] [PubMed] [Google Scholar]
- Zlotnik A. New insights on the role of CXCR4 in cancer metastasis. J Pathol. 2008;215:211–213. doi: 10.1002/path.2350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.