

Abstract
Hyperactivation of CD4+ T cells is a hallmark of untreated HIV-1 infection. The antigenic specificities of activated CD4+ T cells and the underlying mechanisms leading to their activation remain thus far elusive. We report here that during HIV rebound the dynamics of HIV-specific CD4+ T cells is highly correlated with the dynamics of CD4+ T cells specific for persistent antigens derived from various members of the herpes virus family, whereas CD4 responses towards non-persistent antigens were unaffected by HIV replication. Notably, the dynamics of HIV and herpes viral antigen-specific CD4+ T cells responses correlated with the expression level of activation markers on dendritic cells (DCs) and activated DCs were more potent in restimulating memory T cells. These data strongly suggest that HIV replication costimulates activation of CD4+ T cells specific for persistent herpes viral antigens via activation of DCs. We propose that a large proportion of activated T cells during untreated HIV infection may be specific for herpes viral antigens and identify a novel mechanism contributing to chronic immune activation in untreated HIV-1 infection.
Keywords: CD4+ T cells, chronic immune activation, dendritic cells, herpes viruses, HIV pathogenesis
INTRODUCTION
Loss of CD4+ T cells alongside with aberrant immune activation are hallmarks of HIV-1 pathogenesis. Hyperactivation of T cells is in fact one of the best predictors of progression towards AIDS (Giorgi et al, 1999; Sousa et al, 2002). The current paradigm is based on the belief that the decline of CD4+ T cells relies largely on activation-induced cell death rather than productive infection and subsequent killing of activated CD4+ T cells (Bangs et al, 2006; Douek et al, 2003; Grossman et al, 2006; Silvestri & Feinberg, 2003). The majority of activated CD4+ T cells is neither HIV-infected nor HIV-specific (Douek et al, 2002; Kaiser et al, 2007). The mechanisms underlying this bystander activation of non-HIV-specific CD4+ T cells are still poorly defined (Bangs et al, 2006). Systemically translocating lipopolysaccharide (LPS) due to increased permeability of the gut epithelium is suggested to contribute to the general immune activation (Brenchley et al, 2007). However, the exact mechanisms how this translates into activation of CD4+ T cells remain undefined.
So far no insights about the specificity—and perhaps a specificity bias—of the activated CD4+ T cells are known. Bystander activation of CD4+ T cells during HIV-1 infection has largely been described being independent of antigen specificity and is often loosely attributed to the presence of pro-inflammatory cytokines during untreated HIV-1 infection (Bangs et al, 2006, 2009). However, a dependence on antigen specificity for CD4+ T cell activation during HIV infection—for example, through gut-derived antigens (Brenchley et al, 2007), self-antigens (Rawson et al, 2007) or (reactivation of) persistent viruses (Bangs et al, 2006)—could so far not be excluded as the antigen specificities of hyperactivated CD4+ T cells in chronic HIV-1 infection are unknown.
Here we show in a large longitudinal study set-up that HIV replication, upon cessation of antiretroviral therapy (ART), specifically leads to activation of CD4+ T cells with specificities for low level persistent herpes virus antigens in the absence of detectable reactivation of these members of the herpes virus family. Thus, we propose that HIV-1 directly or indirectly provides costimulation for CD4+ T cells which is dependent on antigen specificity and requires the presence of low levels of persistent antigens. We provide evidence that ongoing HIV replication induces activation of dendritic cells (DCs) in vivo which correlates with the activation of CD4+ T cell responses specific for persistent antigens of the herpes virus family. These findings strongly suggest that DCs foster the activation of persistent viral antigen-specific CD4+ T cells through an improved efficiency in antigen presentation upon their activation during HIV rebound.
RESULTS
HIV-1 rebound drives expansion of CD4+ T cells with specificities for herpes viral antigens
CD4+ T cell depletion and chronic immune activation are major characteristics of HIV-1 infection; however, their causal relation is poorly defined. To investigate the impact of HIV replication on immune activation, with particular interest towards the specificities of CD4+ T cells which become activated during HIV recrudescence, we analysed the in vivo dynamics of HIV-specific and non-HIV-specific CD4+ T cells in a cohort of 14 patients undergoing interruption of ART. We observed that increases in plasma viral load boosted HIV-specific CD4+ T cell responses in the majority of patients. Remarkably though, cytomegalovirus (CMV) specific CD4+ T cells followed comparable dynamics despite the absence of CMV viraemia. In striking contrast, CD4+ T cells specific for tetanus toxoid (TT) or streptokinase-streptodornase (SKSD) were not influenced by HIV replication, suggesting that HIV may explicitly expand CD4+ T cells specific for persistent antigens (Supporting Fig 1 and Table 1). To corroborate and extend these findings in a larger patient cohort with more T cell specificities, we performed a detailed longitudinal analysis of 32 patients interrupting ART and thus experiencing viral rebound (patient details are summarized in Table 1). We measured the frequencies of HIV-specific CD4+ T cells, of CD4+ T cell specific for a variety of persistent antigens of the herpes virus family (CMV, Epstein–Barr virus (EBV), herpes simplex virus (HSV) 1/2, varicella-zoster virus (VZV)) and of CD4+ T cells specific for the non-persisting, bacterial antigens TT or SKSD. Due to limited availability of cells and/or the absence of detectable responses, other non-persistent antigens such as Influenza A and measles could not be included in this study.
Table 1.
Characteristics of patient cohort B
| HIV positive | Days | Max HIV (copies × 103/ml) | ||||||
|---|---|---|---|---|---|---|---|---|
| Study group | Age (years) | Gender | Before ART | On-ART | Included in study | Before ART | Off-ART | CD4 counts (cells/ml) |
| n | Median (range) | Male (%) | Month | Median (range) | Median (range) | Median (range) | Median (range) | Median (range) |
| Stop-ART | Before ART-Stop | Before ART-Slop | ||||||
| 32 | 39 (27–59) | 84 | <6 | 537 (152–1491) | 409 (238–1179) | 173 (4–31,300) | 22 (0.25–770) | 706 (347–1419) |
| Control | Before study | First time-point studied | ||||||
| 12 | 37 (31–60) | 83 | <6 | 267 (24–2395) | 858 (270–1319) | 191 (17–655) | – | 589 (265–1223) |
For each patient multiple samples before and after treatment interruption were analysed. In line with our previous results and other reports, HIV-1 recrudescence induced a dynamic response of the HIV-specific CD4+ T cell population. However, confirming our previous finding, ensuing HIV replication also affected the dynamics of CD4+ T cell responses towards non-HIV antigens, namely CD4+ T cell responses specific for the herpes family viruses CMV, EBV, HSV1/2, and VZV, all of which form latent, persistent infections. Importantly, the dynamics of the HIV-specific and the CMV-, EBV-, HSV1/2- and VZV-specific CD4+ T cells were highly correlated (Fig 1A and B). In contrast, no correlation was found between HIV-specific CD4+ T cell responses and CD4+ T cell responses specific for the two non-persistent bacterial antigens SKSD and TT. The significance of this correlation was verified by two different correlation analyses; Spearman's correlation (RS) (Fig 1B and C) and dynamical correlation analysis (Table 2) (Dubin & Mueller, 2005). The more strict dynamical correlation analysis (RD) accounts for the fact that multiple data points originate from the same individual and are thus inter-correlated (Dubin & Mueller, 2005). Furthermore, the observed correlation between the dynamics of HIV-specific CD4+ T cell responses and CD4+ T cell responses specific for persistent antigens is dependent on HIV rebound as no correlation was found in a matching control cohort which remained continuously on ART (Fig 1C). Interestingly, in the absence of HIV-1 replication SKSD- and TT-specific CD4+ T cell responses tend to correlate with the HIV-specific CD4+ T cell response. During successful, highly active ART HIV replication may be suppressed to a level that its state may be described as non-persistent and it is conceivable that the dynamics of non-persistent antigen-specific CD4+ T cells correlate.
Figure 1. Study cohort B: Dynamics of HIV-specific CD4+ T cells and CD4+ T cells specific for persistent viral antigens correlated after HIV rebound.

- Dynamics of HIV (triangles)- and CMV (circles)-specific CD4+ T cell responses in two representative patients with (#2) or without (#C12) ART-Stop. The shaded area depicts pVL.
- Correlation analysis of HIV-specific CD4+ T cell responses and other antigen-specific CD4+ T cell responses in the ART-stop cohort.
- Correlation analysis of HIV-specific CD4+ T cell responses and other antigen-specific CD4+ T cell responses in the control cohort.
- Each individual point represents one measurement. N indicates the number of measurements. Spearman's nonparametric correlation was performed with a 2-tailed significance; p indicates the p-value and RS the Spearman's correlation coefficient. Frequencies of antigen-specific CD4+ T cells are indicated as spot forming cells (SFC) per 1x106 purified CD4+ T cells.
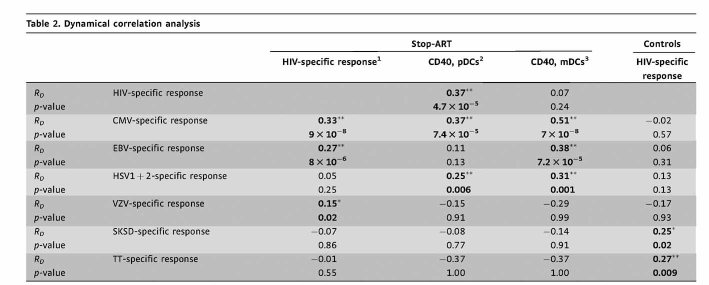 |
1“specific response” indicates “specific CD4+T cell response”.
2CD40 expression level on mDCs.
3CD40 expression level on pDCs.
RD, dynamic correlation coefficient.
4p < 0.05.
5p < 0.01.
Bold indicates statistically significant correlations.
Thus, an increase in plasma virus load (pVL) boosted HIV-specific CD4+ T cell responses, indicating that HIV reactivation, and hence an increase of antigen load promotes HIV-specific CD4+ T cell expansion. The similar dynamics of the HIV-, CMV-, EBV-, HSV1/2- and VZV-specific CD4+ T cell responses demonstrates that rebounding HIV replication does not exclusively affect the frequencies of HIV-specific CD4+ T cells but also of CD4+ T cells with specificities for other persistent antigens.
It is conceivable that upon HIV rebound, persistent latent infections such as CMV, EBV, HSV and VZV might also reactivate in vivo and hence induce stimulation of the respective CD4+ T cells. To investigate this, plasma samples from all patients were analysed for CMV and EBV viraemia by quantitative polymerase chain reaction (PCR) at baseline (‘on-ART’) and at a time-point at which CMV-specific CD4+ T cell frequencies started to increase (‘off-ART’). We chose to quantify CMV and EBV DNA in the plasma as this should more accurately reflect virus reactivation compared to DNA analysis within whole peripheral blood mononuclear cells (PBMCs), where CMV and EBV DNA content will largely reflect latent viral genomes (Compston et al, 2008; Kaur et al, 2003; Torre-Cisneros et al, 2005). CMV or EBV DNA copies were only detected in 3 of 44 analysed plasma samples after cessation of ART, all other samples were below detection limit, indicating that no systemically measurable CMV and EBV reactivation occurred (Supporting Table 2). In addition, significant CMV reactivation was unlikely to occur in our patient cohort as they were not immunosuppressed owing to early initiation of therapy during the acute phase of infection and owing to the early stage of HIV disease progression. Moreover, CD4 counts were in all cases above 350 cells/µl when ART was stopped. In support of this notion, a study conducted in the Rhesus macaque model showed that CMV reactivation is only detected in the late stages of disease during pronounced immunosuppression which was associated with high loss of CMV-specific immunity (Kaur et al, 2003). While it formally cannot be excluded that potentially low levels of CMV and EBV replication may occur locally and thus contribute to the stimulation of the respective CD4+ T cell responses, our correlated kinetic data suggest a direct link between HIV viraemia and CD4+ T cell responses specific for HIV and the investigated persistent herpes viruses.
Taken together, our results strongly suggest that HIV rebound exerts a ‘costimulatory’ function for the activation of CD4+ T cells with specificities for persistent viral antigens. ‘Costimulation’ implies that the activation of the CD4+ T cells is also dependent on the presence of (low levels of) cognate antigen. It is known for CMV, EBV and HSV that low levels of viral DNA are constantly detectable in the saliva (Griffin et al, 2008), supporting the notion that virus antigens, albeit at minute concentrations, are constantly produced. In the absence of HIV replication, these low levels of locally produced antigens might not suffice to induce full activation of specific CD4+ T cells. However, in the presence of HIV replication, the stimulation threshold of these persistent antigen-specific CD4+ T cells might be lowered through overall changes of the milieu allowing for their full activation and expansion.
HIV-driven activation of CMV-specific CD4+ T cells can be directly linked to markers of chronic immune activation
As we had observed that HIV recrudescence leads to the expansion of CD4+ T cells which are specific for persistent antigens, we hypothesized that HIV rebound also induced the up-regulation of activation markers which are indicative for ‘chronic immune activation’ in HIV-1-infected individuals. In five representative patients CD38 and HLA-DR expression was analysed on- and off-ART on total CD4+ and CMV-specific CD4+ T cells (Fig 2A and B).
Figure 2. CMV-specific CD4+ and CD8+ T cells up-regulate activation markers upon HIV rebound.
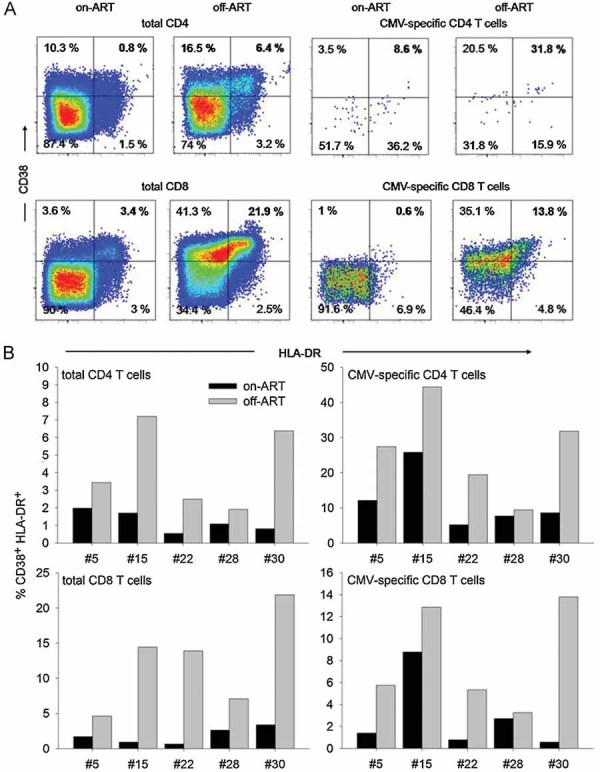
CD38 versus HLA-DR expression was analysed on total and CMV-specific CD4+ and CD8+ T cells in the same patients on- and off-ART.
- Shows stainings of one representative patient (#30). Numbers indicate % of CD38+ HLA-DR+ cells gated on total or CMV-specific CD4+ and CD8+ T cells.
- Percentage of CD38+ HLA-DR+ cells among total or CMV-specific CD4+ and CD8+ T cells in five patients on- compared to off-ART. CMV-specific T cells were identified as IFN-γ+ cells upon restimulation with CMV peptides.
Indeed, the percentage of CD38+ HLA-DR+ cells among total CD4+ T cells was significantly (p < 0.05, paired t-test; data pairs analysed N = 5) increased off-ART. Simultaneously, these activation markers were up-regulated significantly (p < 0.05, paired t-test; data pairs analysed N = 5) to even higher degrees on CMV-specific CD4+ T cells (Fig 2B). Thus, HIV-1 rebound mediated costimulation is therefore reflected both by the expansion of CMV-specific CD4+ T cells as well as by up-regulation of the classical activation markers HLA-DR and CD38 which characterize chronic immune activation in HIV-1 infection. Although our longitudinal quantification of antigen-specific T cell responses was limited to CD4+ T cells, we also analysed whether viral rebound led to increased immune activation within total and CMV-specific CD8+ T cell populations, in particular since the immune activation status of CD8+ T cells is the best predictor of disease progression (Giorgi et al, 1999). Up-regulation of activation markers on total CD8+ and CMV-specific CD8+ T cells occurred in all tested patients to similar degrees as was observed for CD4+ T cells (Fig 2A and B). In addition, preliminary observations in a small number of patients from study cohort A suggest that also HIV- and CMV-specific CD8+ T cell responses exhibit similar dynamics upon treatment interruption (Supporting Fig 2). These observations lend support to the hypothesis that HIV-1 rebound might not only costimulate activation of CD4+ T cells with specificities for persistent herpes viruses, but also the respective CD8+ T cells. We are currently enrolling patients for a new study to address this important question.
HIV rebound is not associated with elevated levels of plasma LPS, sCD14 or pro-inflammatory cytokines
We next investigated several potential mechanisms by which HIV-1 rebound might costimulate the activation of CD4+ T cells specific for persistent herpes viral antigens. We hypothesized that potentially increased levels of LPS or pro-inflammatory cytokines off-ART might drive the costimulation of CD4+ T cell responses. LPS and pro-inflammatory cytokines could mediate direct costimulatory effects on CD4+ T cells or act indirectly via activation of antigen-presenting cells. To test this hypothesis, we longitudinally measured plasma LPS levels and pro-inflammatory cytokine levels on- and off-ART. Five patients with relative high levels of HIV rebound (>1.5 × 105 RNA copies/ml) were chosen as it seemed most likely that altered LPS or cytokine levels would be detectable in these individuals. LPS translocation from the gut is suggested to contribute to chronic immune activation in HIV-1 infection and was shown to be elevated in the chronic phase of the disease (Brenchley et al, 2007). In our study, plasma LPS levels were not affected by HIV-1 rebound (Fig 3). The absence of increased levels of LPS (>100 pg/ml) was verified by four different tests designed to detect endotoxin. It should be noted that our patient cohort initiated ART during the acute phase of HIV infection and might thus exhibit reduced gastrointestinal damage and LPS translocation. Further, with our measurements in the plasma we cannot exclude locally elevated levels of LPS, e.g. in the gut-associated lymphoid tissue. In addition, we also measured soluble CD14 (sCD14) as a surrogate biomarker for increased levels of LPS (Brenchley et al, 2007). In line with our LPS measurements, sCD14 levels were not affected by HIV-1 rebound (Fig 3) and did not correlate with the magnitude of the HIV-specific CD4+ T cell response or the levels of LPS in the few samples where we could detect LPS (Supporting Fig 3). Comparable to the LPS analyses, pro-inflammatory cytokines (IL-2, -6, -10, IFNγ, TNFα, IFNα) remained below detection limit in the studied plasma samples, even during HIV rebound (Fig 3). Again, this does not exclude locally elevated levels at sites of inflammation and/or virus replication. The failure to detect elevated cytokine levels in the plasma is not unexpected as it was recently published that even during primary infection elevated levels of plasma cytokines were not detectable in all patients and increased rather transiently, e.g. for IFNα also with very rapid kinetics (Stacey et al, 2009). The timing of our samples might thus have missed transient increases. Of note, pro-inflammatory cytokines could be detected during the acute phase of primary HIV-1 infection in samples from the same patients (data not shown).
Figure 3. Plasma LPS levels, sCD14 and pro-inflammatory cytokines are not elevated during HIV rebound.

Plasma LPS levels (open diamonds), sCD14 (closed triangles) and plasma cytokine concentrations (closed circles) were measured longitudinally before and during HIV rebound in five representative patients. Shaded areas depict pVL. The detection limits for the plasma cytokines were: IL-2, 8 pg/ml; IL-6, 10 pg/ml; IL-10, 10 pg/ml; IFNγ, 40 pg/ml; TNFα, 10 pg/ml; IFNα, 25 pg/ml.
HIV-1 rebound drives activation of dendritic cells
Since we detected a preferential activation of CD4+ T cells specific for persistent but not of those specific for non-persistent antigens, we reasoned that HIV rebound might be associated with the activation and maturation of DCs which renders these cells more efficient in antigen presentation and might thus allow CD4+ T cell stimulation despite the low levels of the persistent antigens. Our analyses did not support a dominant effect of LPS and pro-inflammatory cytokines, however, several in vitro (Fonteneau et al, 2004; Harman et al, 2006; Smed-Sorensen et al, 2005) and ex vivo (Barron et al, 2003; Dillon et al, 2008; Mandl et al, 2008; Tilton et al, 2008) studies indicate that HIV itself directly affects the maturation status of DCs. In particular CD40 was suggested to be expressed at increased levels on blood DCs from HIV-infected individuals, as assessed in cross-sectional studies (Barron et al, 2003; Dillon et al, 2008). We decided to measure directly the impact of HIV recrudescence on ex vivo CD40 and CD86 expression levels on plasmacytoid (pDCs) and myeloid (mDCs) blood dendritic cells within individual patients. To this end we longitudinally assessed these activation markers on pDCs and mDCs in a subset of 15 rebound patients and 5 control patients (Figs 4 and 5). Longitudinal samples were analysed from the same time-points as were studied for CD4+ T cell responses, LPS and cytokine levels. While changes in CD86 expression were too small to draw firm conclusions (data not shown), CD40 expression was significantly up-regulated on mDCs as well as pDCs upon HIV rebound (Figs 4B and 5). Most importantly, the levels of CD40 expression were positively correlated with the magnitude of CD4+ T cell responses specific for HIV, CMV, EBV and HSV (Fig 5A and Table 2). No such correlation was found between CD40 expression levels on DCs and the size of the CD4+ T cell response against the two non-persistent antigens (Fig 5B). In the control cohort (patients under continuous ART) the expression levels of CD40 on mDCs and pDCs were about 1 log lower than after HIV rebound in the ART-Stop cohort and were not significantly modulated over the time course of analysis. Furthermore, in these patients, the CD40 expression levels did not correlate with HIV-specific CD4+ T cell frequencies (Fig 5C).
Figure 4. CD40 expression on peripheral blood dendritic cells increases during viral rebound and follows similar dynamics as HIV- and CMV-specific CD4+ T cell responses.

- Flow cytometric gating strategy for peripheral blood myeloid DCs (mDCs) (lin−, HLA-DR+, CD123−+, CD11c+), and plasmacytoid DC (pDCs) (lin−, HLA-DR+, CD123+, CD11c−).
- CD40 expression levels on pDCs (open diamonds, red line) and on mDCs (open circles, blue line) are plotted together with CMV (closed circles)- and HIV (closed triangles)-specific CD4+ T cell responses in two representative patients with (#22) or without (#C9) ART Stop. Shaded areas indicate pVL.
Figure 5. CD40 expression on peripheral blood dendritic cells correlates with the dynamics of the HIV-, CMV-, EBV- and HSV1+2-specific CD4+ T cell responses during viral rebound.
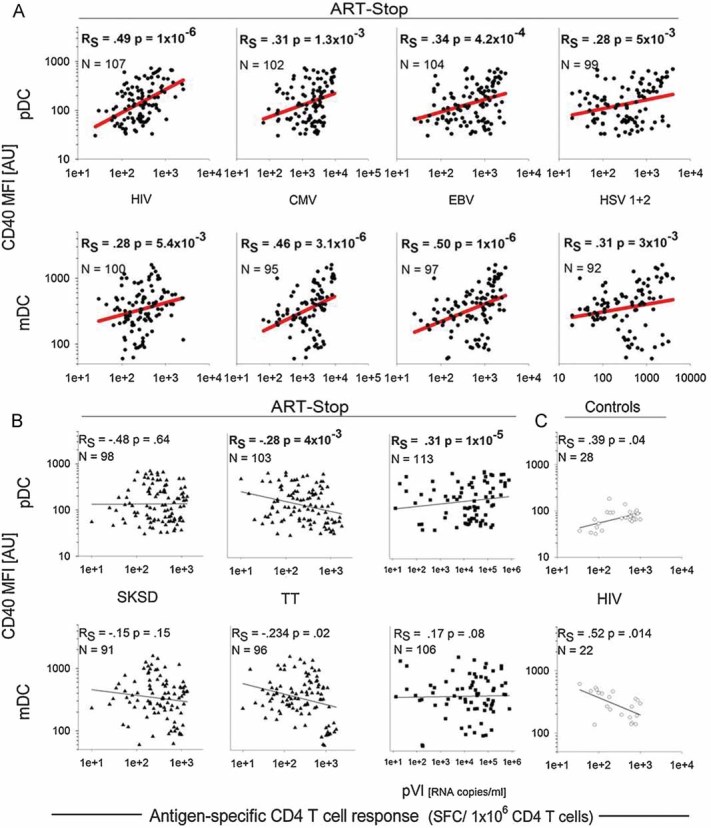
- Correlation analysis between CD40 expression levels and the frequencies of HIV- and Herpes virus-specific CD4+ T cell responses and HIV plasma viral load.
- Correlation analysis between CD40 expression levels and the frequencies of non-persistent antigen-specific CD4+ T cell responses and HIV plasma viral load.
- Low expression levels of CD40 on mDC and pDC in the control cohort.
- Each individual point represents one measurement. N indicates the number of measurements. Spearman's nonparametric correlation was performed with a 2-tailed test of significance; p indicates the p-value and RS the Spearman's correlation coefficient.
Although we failed to detect elevated levels of LPS upon treatment interruption and viral rebound (which would be a potent activator of DCs), nor did we detect marked variations of sCD14 levels (as surrogate marker for increased LPS levels), we nonetheless tested whether sCD14 levels correlated with the activation status of DCs. We could not detect a significant positive correlation between sCD14 and CD40 expression levels on pDCs or mDCs (Supporting Fig 3).
Activated DCs are more potent in reactivating CMV-specific CD4+ T cell responses
Our hypothesis that CD4+ T cells specific for herpes viral persistent antigens are more effectively restimulated by activated DCs, especially in the setting of limiting amounts of antigen, requires experimental validation. We therefore generated immature monocyte-derived DCs in vitro, activated those either with LPS or with AT-2-inactivated HIV particles (iHIV) and pulsed them with limiting concentrations of overlapping CMV-derived peptides. These DCs were then used to restimulate autologous purified bulk CD4+ T cells from CMV-positive donors. Activated DCs (either by LPS or by iHIV) consistently induced markedly enhanced proliferation of CMV-specific CD4+ T cells compared to non-activated DCs (Fig 6A–C) (p < 0.05, paired t-test; Number of individuals tested N = 3), indicating that also antigen-experienced CD4+ T cells profit from the activation status of DCs, in particular under limiting antigen concentrations. So far it has only been shown that iHIV-activated DCs enhance allogenic T cell responses—which are mostly naïve T cells (Harman et al, 2006).
Figure 6. Activated DCs are more potent in restimulating CMV-specific CD4+ T cells.

- Representative histograms showing proliferation of CD4+ T cells as measured by CFSE dilution. Purified bulk CD4+ T cells from CMV seropositive donors were stimulated with autologous monocyte-derived DCs (MDDCs) which had been activated by LPS, AT-2-inactivated HIV particles (iHIV) or were left unactivated. MDDCs were pulsed with or w/o limiting amounts of overlapping CMV-derived peptides. The dashed line indicates culture with unpulsed MDDCs, the solid line indicates culture with CMV peptide-loaded MDDCs. The indicated percentage of CFSE low cells refers to stimulation with CMV peptide-pulsed MDDCs.
- Proliferative responses of CD4+ T cells stimulated with CMV-loaded MDDCs in 3 healthy CMV-positive donors. MDDCs were activated with LPS.
- Proliferative responses of CD4+ T cells stimulated with CMV-loaded MDDCs in 3 healthy CMV-positive donors. MDDCs were activated with iHIV particles.
- Error bars indicate standard deviation between 3 samples in the same experiment.
DISCUSSION
The mechanisms underlying hyperactivation of non-HIV-specific CD4+ T cells during chronic HIV-1 infection are still poorly understood. Here, we show that HIV replication preferentially costimulates activation of CD4+ T cells with specificities for persistent herpes virus family members. We provide strong circumstantial evidence that this activation (in the absence of reactivation of the herpes viruses) is mediated through HIV replication induced activation of DCs, which could in turn provide more efficient presentation of low level persistent antigens to the CD4+ T cells. As this mechanism requires the presence of cognate antigen, it excludes CD4+ T cells with specificities for non-persistent antigens. Indeed, in our in vivo study we have not observed that HIV rebound would affect CD4+ T cell responses specific for non-persistent antigens similarly to those with specificities for persistent antigens.
There is other evidence in the literature suggesting specific effects of HIV infection on CMV and other persistent antigen specific CD4+ T cells. For instance, in a cross-sectional study, higher frequencies of CMV versus mumps-specific CD4+ T cells were reported in HIV positive compared to uninfected individuals (Waldrop et al, 1997). Elevated frequencies of blood CMV-specific IFNγ-secreting CD4+ T cells were also reported in another study comparing HIV progressors to LTNP or HIV-uninfected individuals (Harari et al, 2004a). In addition, elevated frequencies of CMV-specific CD4+ T cells were found to correlate with stronger HIV-specific CD4+ T cell responses (Pitcher et al, 1999). Furthermore, in the case of CD8+ T cell responses, CMV- and EBV-specific CD8+ T cells were described to exhibit a more activated phenotype during primary HIV infection compared to flu-specific CD8+ T cells (Doisne et al, 2004).
Such a mechanism of HIV mediated costimulation of CD4+ T cells with specificities for persistent viruses requires the presence of low levels of cognate antigen, but would be independent of overt reactivation of heterologous viruses. In case of persistent, latent herpes virus infections, low levels of persistent viral antigens are very likely to be continuously present in the host. Although it is impossible to quantify the amount of this low level antigen expression (even in the mouse, Reddehase et al, 2008), several indirect measures strongly support the presence of low level antigen expression; compared to TT-specific CD4+ T cells (non-persistent antigen), most CMV-, EBV-, HSV- and VZV-specific CD4+ T cells exhibit a more effector–memory phenotype which is likely to be indicative for repetitive antigen encounter and persistent, low antigen load (Harari et al, 2004b; Jones et al, 2007). Furthermore, in the murine and human CMV infection, a slow accumulation of CMV-specific T cells with effector–memory phenotypes is observed and has been ascribed to constant low level exposure to cognate antigen (Karrer et al, 2003; Komatsu et al, 2003; Snyder et al, 2008). In humans, DNA of CMV, EBV and HSV is regularly shed into the saliva suggesting that low antigen load should be present systemically (Griffin et al, 2008). In case of HIV-infected individuals, the quantity of shedding was reported to be independent of therapy status (to our knowledge ART has no direct antiviral effects on viruses of the herpes family), CD4 counts or pVL (except for EBV and pVL) (Griffin et al, 2008), which is in line with our data showing that CMV or EBV are not systemically reactivated in our patient cohort upon ART interruption. Although we cannot completely exclude simultaneous reactivation of persistent viruses, perhaps restricted to specific anatomical sites, as a major driver for the activation of the respective CD4+ T cells, this seems rather unlikely as we observed a highly synchronous, concerted activation of herpes virus family-specific and HIV-specific CD4+ T cells following ART interruption. This rather points to a common source for activation. In addition, the viruses studied here persist in different cell types and are likely to depend on different stimuli for their reactivation from latency, which makes a simultaneous reactivation upon ART interruption to appear even less plausible. Apart from the ‘common’ members of the herpes virus family as studied here, KSHV (the causative oncovirus of Kaposi's Sarcoma) has increased prevalence in HIV-infected individuals and is associated with a negative disease outcome. It also establishes persistent infection, which is in immunocompetent individuals usually well suppressed and free of symptoms. KSHV is very likely relevant for our study as well as it could serve as an additional source of persistent low levels of antigen (Edelman, 2005).
Our data suggest that more efficient presentation of very low levels of persistent antigens by activated DCs is responsible for the activation of persistent-antigen specific CD4+ T cells during HIV replication. This defines a new role for DCs in HIV pathogenesis. As shown here and previously by others in vitro (Fonteneau et al, 2004; Harman et al, 2006; Smed-Sorensen et al, 2005; Tilton et al, 2008) and in vivo (Barron et al, 2003; Dillon et al, 2008), HIV has direct activatory effects on DCs, likely through viral-derived TLR 7/8 ligands such as ssRNA. Our data highlight further that TLR-ligand activated DCs or DCs activated by iHIV particles are superior in their ability to restimulate antigen-experienced T cells compared to their non-activated counterparts—in particular at limiting antigen concentrations. In line with our results it has been reported that TLR-ligand activated DCs induce enhanced CMV-specific CD4+ T cell responses (Lore et al, 2003).
Our data complement a recent study—which, however, has not been confirmed by others (Lederer et al, 2009)—showing that activation of DCs upon simian immunodeficiency virus (SIV) infection is at least partially impaired in a non-pathogenic model of SIV infection, in contrast to a pathogenic model of SIV infection or the HIV-driven activation of human DCs (Mandl et al, 2008). The differential activation of DCs from natural hosts compared to non-natural hosts upon SIV/HIV exposure was ascribed to inherent differences in TLR-mediated signalling and resulted in strong type 1 interferon production in DCs from non-natural monkey hosts or from humans (Mandl et al, 2008). Type 1 interferons have been shown to provide a potent ‘signal 3’ for the activation of T cells (Haring et al, 2006; Havenar-Daughton et al, 2006; Pape et al, 1997).
A current paradigm postulates that systemic microbial translocation from the gut leads to increased systemic LPS levels in HIV-infected individuals due to increased permeability of the gut epithelium (Brenchley et al, 2007). A recent study, however, failed to detect changes in levels of LPS and sCD14 during disease progression (Redd et al, 2009).
How elevated LPS levels would specifically translate into an increased activation status of T cells remains to be defined. We therefore analysed in our patient cohort whether increased systemic LPS levels were evident after ART interruption and whether they would correlate with the observed DC activation and the dynamics of T cell responses. We could not detect elevated LPS levels in the plasma, rendering it unlikely that LPS is the major driving force for the in vivo activation of DCs. However, it cannot be excluded that elevated LPS levels are present at specific anatomical sites such as in intestinal tissue and impact locally on the activation of DCs. In line with the absence of systemic detectable LPS upon ART interruption, we were not able to detect inflammatory cytokines in plasma upon HIV rebound. The absence of systemically elevated cytokine levels is not surprising as it was recently published that even during primary infection elevated levels of plasma cytokines were not detectable in all patients and were present only very transiently (Stacey et al, 2009).
Innate stimuli especially TLR ligands as well as pro-inflammatory cytokines can directly lead to the activation of DCs but are also thought to be able to directly impact on T cell activation—a mechanism which is so far not well defined. A direct role for TLR ligands in the activation of T cells has been shown for costimulation of CD4+ T cells, e.g. by TLR7/8 ligands in the presence of low levels of simultaneous T-cell antigen receptor (TCR) stimulation (Caron et al, 2005; Kabelitz, 2007). Such a costimulatory effect of TLR ligands on activation of T cells in the presence of limiting antigen levels would well fit with our in vivo data and cannot be excluded, perhaps in addition to the TLR-mediated activation of DCs. Additionally, a recent in vitro model of bystander activation showed that soluble factors secreted upon TCR stimulation by CD4+ T cells can induce a partially activated phenotype in bystander CD4+ T cells, but fail to induce their proliferation (in the absence of TCR stimulation) (Bangs et al, 2009). Furthermore, as mentioned earlier, inflammatory cytokines such as type 1 interferons or IL-12 have been shown to provide a potent ‘signal 3’ for the activation of murine T cells in close proximity with signals 1 and 2 (Haring et al, 2006; Havenar-Daughton et al, 2006; Way et al, 2007).
In conclusion, innate stimuli and cytokines which are associated with HIV replication may provide costimulatory signals to CD4+ T cells either directly or as we show here, more likely indirectly through activation of DCs (Fig 7). As discussed above, this costimulation by HIV requires the presence of at least minimal levels of cognate antigen to induce full activation of CD4+ T cells, since only persistent-antigen specific CD4+ T cells were activated in our study.
Figure 7. Proposed mechanisms of how HIV replication induces activation of CD4+ T cells with specificities for persistent viral antigens.

In the absence of HIV replication—and thus in the absence of inflammation and/or HIV-derived pathogen-associated molecular patterns (PAMPs)—dendritic cells do not show an activated phenotype and are not very efficient in restimulating CD4+ T cells specific for low levels of latent persistent viral antigens (left side). However, in the presence of ongoing HIV replication, DCs exhibit an activated phenotype and are more effective in restimulating CD4+ T cells specific for latent persistent viral antigens in particular in the setting of limiting antigen levels, as is likely the case in vivo for these latent viral infections (right side). Activation of DCs might be triggered by PAMPs (either HIV derived such as ssRNA or perhaps derived from bacteria which translocate systemically from the gut lumen) or by pro-inflammatory cytokines such has TNFα or type I interferons which are locally produced in tissues with active HIV replication. In addition pro-inflammatory cytokines may exert direct costimulatory effects on CD4+ T cells.
As a consequence a large proportion of hyperactivated CD4+ T cells which appear upon HIV infection may be specific for persistent, non-HIV antigens. Activation of DCs by HIV likely plays a critical role in this process and defines a new antigen-dependent mechanism of bystander activation of CD4+ T cells, unravelling a further constituent of the chronic immune activation and CD4+ T cell loss in HIV infection.
In addition, our findings indicate a new role for persistent herpes virus infections in HIV-1 pathogenesis. Herpes virus infections are well described to facilitate HIV-1 acquisition (HSV-2) and are major drivers of AIDS mortality (CMV reactivation). We describe here that they might also contribute significantly to chronic immune activation. This underpins the beneficial role of early initiation of ART as a means to limit immune activation in order to preserve T cell immunity during HIV-1 infection.
MATERIALS AND METHODS
Study population
We recruited two study cohorts (A and B). In study cohort A, eight HIV-positive individuals who initiated ART during the chronic phase of infection and six patients who initiated ART during acute infection were included (Supporting Table 1) (Trkola et al, 2005). In the study cohort B, 44 HIV-positive individuals were recruited who initiated ART during the acute phase of infection (Table 1 and Supporting Table 3). These patients participated in the Zurich primary HIV-1 infection study (ZPHI), a prospective long-term observational single centre study. In this protocol patients are offered early ART during the acute phase of infection and after 1 year of suppressed viraemia (<40 HIV-1 RNA copies/ml plasma) patients can choose to interrupt ART. The following criteria were used to define acute infection: (a) acute retroviral syndrome (ARS) and negative or indeterminate Western blot in the presence of a positive p24 Ag and/or detectable plasma HIV-1 RNA or (b) documented seroconversion with or without symptoms within 90 days. Study details are listed under www.clinicaltrials.gov; ID NCT00537966. All individuals of study cohort B were screened positive for CMV, EBV, HSV1 + 2 and VZV by serology prior to enrolment. In study cohort B, 32 patients interrupted ART (rebound cohort) and 12 patients remained continuously on ART (control cohort). Approval of the ethical committee and written informed consent from all subjects were obtained according to the guidelines of the University Hospital Zurich.
Quantification of CMV and EBV DNA
CMV and EBV DNA were quantified in plasma samples as previously described (Yun et al, 2000; Zingg et al, 1999). The detection limit was 200 copies/ml as values below 200 copies cannot be reproduced with statistical confidence.
Generation of MDDCs
Monocyte-derived dendritic cells (MDDCs) were generated from freshly isolated PBMCs according to standard procedures as described elsewhere (Harman et al, 2006).
IFN-γ ELISpot assay
CD4+ T cells were isolated by magnetic microbeads (Miltenyi) from cryopreserved PBMCs and co-cultured with 10% fresh autologous MDDCs. Cells were stimulated in an IFN-γ ELISpot as described elsewhere (Oxenius et al, 2000). Briefly, cells were stimulated over-night with an HIV gag pool (NIH AIDS Reagent), CMV, HSV1 + 2, VZV lysates (Virion), EBV lysate (Virusys), an SKSD preparation (Cyanamid Iberica, SA) or TT (gift from Novartis Vaccines). The number of spot forming cells (SFC) was calculated by subtracting an unstimulated control as background. Spots were quantified using an automated spot counter (AID). If not stated differently, antigen-specific CD4+ T cell responses are shown as SFC/1 × 106 CD4+ T cells.
The paper explained
PROBLEM
Continuous depletion of CD4+ T helper cells, key players of the adaptive immune system, is a hallmark of chronic HIV-1 infection and progression to AIDS. Chronic hyperactivation of CD4+ T cells is thought to be key to their depletion. Hyperactivated CD4+ T cells are not necessarily HIV-infected or HIV-specific; little is known about their antigenic specificities and the mechanisms by which HIV causes this bystander activation of CD4+ T cells.
RESULT
We show that active HIV-1 replication induces in vivo bystander activation of CD4+ T cells which are specific for persistent herpes virus antigens, whereas no activation was observed for CD4+ T cells with specificity for non-persistent antigens. This suggests that bystander activation depends on the presence of (low levels of) cognate antigen. Bystander T cell activation is likely to depend on HIV-induced maturation of DCs since their in vivo activation status closely correlates with the magnitude of bystander T cell activation.
IMPACT
We propose that HIV-1 driven activation of DCs results in more efficient presentation of herpes viral antigens, resulting in enhanced activation of herpes-virus specific CD4+ T cells, thereby providing a scenario which would account for HIV-1-associated activation of non-HIV-specific CD4+ T cells. These findings provide new insights into HIV pathogenesis and how HIV-1 manipulates the immune system potentially to its own favour.
Flow cytometry
For CD4+ T cell immunophenotyping cryopreserved PBMCs were thawed and rested over-night before stimulation for 6 h with or without CMV pp65 and IE-1 overlapping peptide pools in the presence of Monensin- and Brefeldin-A (Sigma). Cells were surface stained with anti-CD4 Am-Cyan, anti-CD3 Pacific-blue, anti-CD38 APC, anti-HLA-DR APC-H7 (all BD) followed by intracellular staining with anti-IFN-γ PE-Cy7 (BD) and anti-IL-2 FITC (Biolegend). CMV-specific CD4+ T cells were identified as IFN-γ+ in the stimulated sample.
For DC analysis cryopreserved PBMCs were thawed and immediately surface stained with anti-lineage-1 (lin-1: CD3, CD14, CD16, CD19, CD20, CD56) FITC, anti-CD34 FITC, anti-HLA-DR APC-H7, anti-CD123 PerCp-Cy5.5 (all BD), anti-CD11c Biotin (Biolegend) and anti-CD40 PE (Ancell), followed by staining with Streptavidin PE-Cy7 (BD).
Data were collected using an LSRII flow cytometer (BD). Data files were analysed using FlowJo software (Tree Star, Inc.).
LPS levels
EDTA (ethylenediaminetetraacetic acid) plasma samples were diluted 1 to 10 with endotoxin-free water and incubated for 10 min at 70°C. Plasma LPS was quantified with the LAL Endochrome Kit (CharlesRiver) according to the manufacturer's protocol for low range detection. Additionally the same samples were analysed with the LAL QCL-1000 Kit (Lonza) and two kinetic kits (Endosafe Endochrome-K, CharlesRiver and LAL Kinetic QCL, Lonza). Only data from the LAL Endochrome Kit are shown as it proved to be the most sensitive assay.
Soluble CD14 (sCD14) and cytokines
Plasma samples were tested for IL-2, -6, -10, IFN-γ and TNF-α by Cytometric Bead Array (BD) according to the manufacturer's protocol. IFN-α (Interferonsource) and sCD14 (R&D Systems) were measured by commercially available ELISA (enzyme-linked immunosorbent assay) kits according to the manufacturer's protocols.
In vitro proliferation assays
CD4+ T cells from healthy CMV-positive donors were purified from freshly isolated PBMCs by magnetic microbeads (Miltenyi) and labelled with 5 µM CFSE (carboxyfluorescein diacetate, succinimidyl ester, Invitrogen). For LPS activation, MDDCs were loaded over-night with 0.1 or 0.01 µg/ml CMV pp65 and IE-1 peptide pools and then stimulated for 24 h with 0.5 µg/ml LPS (Sigma). For iHIV activation, MDDCs were stimulated for 24 h with AT-2 iHIV particles (50 µg/ml p24) or matching concentrations of control microvesicles (gift from J. Lifson) and then pulsed for 1 h with CMV peptide pools. Purified CD4+ T cells were stimulated with 1% autologous MDDCs for 6 days and analysed by flow cytometry as described above. This assay was performed in RPMI-medium (Invitrogen) supplemented with 10% human serum.
Statistical analysis
Spearman's correlation analysis with a two-tailed test of significance was performed using SPSS software (SPSS). Dynamical correlation analysis was performed as described elsewhere (Dubin & Mueller, 2005). This method quantifies the covariation of components of multivariate longitudinal data. It estimates a correlation for the trend rather than the magnitude of the measured parameters. Standard R-Software was used for analysis. p-Values <0.01 were considered significant.
Author contributions
A.H., M.R., P.R., W.B. and H.K. performed experiments. A.H., M.R. and F.G. analysed results. A.H. and A.O. made the figures and wrote the first draft of the paper. A.T., H.G. and A.O. designed the research and wrote the paper. H.G. enrolled patients.
For more information
The Swiss HIV Cohort Study: http://www.shcs.ch/
Acknowledgments
We are grateful to our patients for their commitment and thank C. Grube, B. Hasse, U. Karrer, R. Oberholzer, L. Aceto, R. Laffer, U. von Both, K. Thierfelder and D. Braun for excellent patient care; M. Smith, A. Manrique, F. Burgener, D. Klimpel and C. Leemann for technical help; U. Karrer and K. Wanke for supportive data; I. Nievergelt and C. Vögtli for administrative assistance; R. Weber for continuous support of the ZPHI study; J. Lifson for providing us with AT-2-iHIV and control vesicles and Novartis for the kind gift of TT. This work was supported by the ETH Zurich, the Swiss National Science Foundation (Grant Nos. 310000-120739 to AT, 324730-116035 to HFG, 310030-113947 to AO) and the Horten Foundation. A.T. is an Elizabeth Glaser Scientist supported by the Elizabeth Glaser Pediatric AIDS Foundation.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
References
- Bangs SC, McMichael AJ, Xu X-N. Bystander T cell activation—implications for HIV infection and other diseases. Trends Immunol. 2006;27:518–524. doi: 10.1016/j.it.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Bangs SC, Baban D, Cattan HJ, Li CK-F, McMichael AJ, Xu X-N. Human CD4+ memory T cells are preferential targets for bystander activation and apoptosis. J Immunol. 2009;182:1962–1971. doi: 10.4049/jimmunol.0802596. [DOI] [PubMed] [Google Scholar]
- Barron M, Blyveis N, Palmer B, MaWhinney S, Wilson C. Influence of plasma viremia on defects in number and immunophenotype of blood dendritic cell subsets in human immunodeficiency virus 1-infected individuals. J Infect Dis. 2003;187:26–37. doi: 10.1086/345957. [DOI] [PubMed] [Google Scholar]
- Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2007;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H, Delneste Y. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-ψ production by memory CD4+ T cells. J Immunol. 2005;175:1551–1557. doi: 10.4049/jimmunol.175.3.1551. [DOI] [PubMed] [Google Scholar]
- Compston LI, Sarkobie F, Li C, Candotti D, Opare-Sem O, Allain J-P. Multiplex real-time PCR for the detection and quantification of latent and persistent viral genomes in cellular or plasma blood fractions. J Virol Methods. 2008;151:47–54. doi: 10.1016/j.jviromet.2008.03.023. [DOI] [PubMed] [Google Scholar]
- Dillon SM, Robertson KB, Pan SC, Mawhinney S, Meditz AL, Folkvord JM, Connick E, McCarter MD, Wilson CC. Plasmacytoid and myeloid dendritic cells with a partial activation phenotype accumulate in lymphoid tissue during asymptomatic chronic HIV-1 infection. J Acquir Immune Defic Syndr. 2008;48:1–12. doi: 10.1097/QAI.0b013e3181664b60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doisne J-M, Urrutia A, Lacabaratz-Porret C, Goujard C, Meyer L, Chaix M-L, Sinet M, Venet A. CD8+ T cells specific for EBV, cytomegalovirus, and influenza virus are activated during primary HIV infection. J Immunol. 2004;173:2410–2418. doi: 10.4049/jimmunol.173.4.2410. [DOI] [PubMed] [Google Scholar]
- Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S, et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature. 2002;417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- Douek DC, Picker LJ, Koup RA. T cell dynamics in HIV-1 infection. Annu Rev Immunol. 2003;21:265–304. doi: 10.1146/annurev.immunol.21.120601.141053. [DOI] [PubMed] [Google Scholar]
- Dubin JA, Mueller H-G. Dynamical correlation for multivariate longitudinal data. J Am Stat Assoc. 2005;100:872–881. [Google Scholar]
- Edelman DC. Human herpesvirus 8—a novel human pathogen. Virol J. 2005;2:78. doi: 10.1186/1743-422X-2-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonteneau J-F, Larsson M, Beignon A-S, McKenna K, Dasilva I, Amara A, Liu Y-J, Lifson JD, Littman DR, Bhardwaj N. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J Virol. 2004;78:5223–5232. doi: 10.1128/JVI.78.10.5223-5232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi JV, Hultin LE, McKeating JA, Johnson TD, Owens B, Jacobson LP, Shih R, Lewis J, Wiley DJ, Phair JP, et al. Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J Infect Dis. 1999;179:859–870. doi: 10.1086/314660. [DOI] [PubMed] [Google Scholar]
- Griffin E, Krantz E, Selke S, Huang M-L, Wald A. Oral mucosal reactivation rates of herpesviruses among HIV-1 seropositive persons. J Med Virol. 2008;80:1153–1159. doi: 10.1002/jmv.21214. [DOI] [PubMed] [Google Scholar]
- Grossman Z, Meier-Schellersheim M, Paul WE, Picker LJ. Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nat Med. 2006;12:289–295. doi: 10.1038/nm1380. [DOI] [PubMed] [Google Scholar]
- Harari A, Petitpierre S, Vallelian F, Pantaleo G. Skewed representation of functionally distinct populations of virus-specific CD4 T cells in HIV-1-infected subjects with progressive disease: changes after antiretroviral therapy. Blood. 2004a;103:966–972. doi: 10.1182/blood-2003-04-1203. [DOI] [PubMed] [Google Scholar]
- Harari A, Vallelian F, Pantaleo G. Phenotypic heterogeneity of antigen-specific CD4 T cells under different conditions of antigen persistence and antigen load. Eur J Immunol. 2004b;34:3525–3533. doi: 10.1002/eji.200425324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring JS, Badovinac VP, Harty JT. Inflaming the CD8+ T cell response. Immunity. 2006;25:19–29. doi: 10.1016/j.immuni.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Harman AN, Wilkinson J, Bye CR, Bosnjak L, Stern JL, Nicholle M, Lai J, Cunningham AL. HIV induces maturation of monocyte-derived dendritic cells and Langerhans cells. J Immunol. 2006;177:7103–7113. doi: 10.4049/jimmunol.177.10.7103. [DOI] [PubMed] [Google Scholar]
- Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting edge: the direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol. 2006;176:3315–3319. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- Jones L, Black Antony P, Malavige Gathsaurie N, Ogg Graham S. Phenotypic analysis of human CD4 T cells specific for immediate early 63 protein of varicella-zoster virus. Eur J Immunol. 2007;37:3393–3403. doi: 10.1002/eji.200737648. [DOI] [PubMed] [Google Scholar]
- Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19:39–45. doi: 10.1016/j.coi.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Kaiser P, Joos B, Niederost B, Weber R, Gunthard HF, Fischer M the Swiss HIV Cohort Study. Productive human immunodeficiency virus type 1 infection in peripheral blood predominantly takes place in CD4/CD8 double-negative T lymphocytes. J Virol. 2007;81:9693–9706. doi: 10.1128/JVI.00492-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karrer U, Sierro S, Wagner M, Oxenius A, Hengel H, Koszinowski UH, Phillips RE, Klenerman P. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J Immunol. 2003;170:2022–2029. doi: 10.4049/jimmunol.170.4.2022. [DOI] [PubMed] [Google Scholar]
- Kaur A, Kassis N, Hale CL, Simon M, Elliott M, Gomez-Yafal A, Lifson JD, Desrosiers RC, Wang F, Barry P, et al. Direct relationship between suppression of virus-specific immunity and emergence of cytomegalovirus disease in simian AIDS. J Virol. 2003;77:5749–5758. doi: 10.1128/JVI.77.10.5749-5758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu H, Sierro S, Cuero AV, Klenerman P. Population analysis of antiviral T cell responses using MHC class I-peptide tetramers. Clin Exp Immunol. 2003;134:9–12. doi: 10.1046/j.1365-2249.2003.02266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer S, Favre D, Walters K-A, Proll S, Kanwar B, Kasakow Z, Baskin CR, Palermo R, McCune JM, Katze MG. Transcriptional profiling in pathogenic and non-pathogenic SIV infections reveals significant distinctions in kinetics and tissue compartmentalization. PLoS Pathog. 2009;5:e1000296. doi: 10.1371/journal.ppat.1000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lore K, Betts MR, Brenchley JM, Kuruppu J, Khojasteh S, Perfetto S, Roederer M, Seder RA, Koup RA. Toll-like receptor ligands modulate dendritic cells to augment cytomegalovirus- and HIV-1-specific T cell responses. J Immunol. 2003;171:4320–4328. doi: 10.4049/jimmunol.171.8.4320. [DOI] [PubMed] [Google Scholar]
- Mandl JN, Barry AP, Vanderford TH, Kozyr N, Chavan R, Klucking S, Barrat FJ, Coffman RL, Staprans SI, Feinberg MB. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med. 2008;14:1077–1087. doi: 10.1038/nm.1871. [DOI] [PubMed] [Google Scholar]
- Oxenius A, Price DA, Easterbrook PJ, O'Callaghan CA, Kelleher AD, Whelan JA, Sontag G, Sewell AK, Phillips RE. Early highly active antiretroviral therapy for acute HIV-1 infection preserves immune function of CD8+ and CD4+ T lymphocytes. Proc Natl Acad Sci USA. 2000;97:3382–3387. doi: 10.1073/pnas.97.7.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape K, Khoruts A, Mondino A, Jenkins M. Inflammatory cytokines enhance the in vivo clonal expansion and differentiation of antigen-activated CD4+ T cells. J Immunol. 1997;159:591–598. [PubMed] [Google Scholar]
- Pitcher CJ, Quittner C, Peterson DM, Connors M, Koup RA, Maino VC, Picker LJ. HIV-1-specific CD4+ T cells are detectable in most individuals with active HIV-1 infection, but decline with prolonged viral suppression. Nat Med. 1999;5:518–525. doi: 10.1038/8400. [DOI] [PubMed] [Google Scholar]
- Rawson PM, Molette C, Videtta M, Altieri L, Franceschini D, Donato T, Finocchi L, Propato A, Paroli M, Meloni F, et al. Cross-presentation of caspase-cleaved apoptotic self antigens in HIV infection. Nat Med. 2007;13:1431–1439. doi: 10.1038/nm1679. [DOI] [PubMed] [Google Scholar]
- Redd AD, Dabitao D, Bream JH, Charvat B, Laeyendecker O, Kiwanuka N, Lutalo T, Kigozi G, Tobian AAR, Gamiel J, et al. Microbial translocation, the innate cytokine response, and HIV-1 disease progression in Africa. Proc Natl Acad Sci USA. 2009;106:6718–6723. doi: 10.1073/pnas.0901983106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddehase MJ, Simon CO, Seckert CK, Lemmermann N, Grzimek NK. Murine model of cytomegalovirus latency and reactivation. Curr Top Microbiol Immunol. 2008;325:315–331. doi: 10.1007/978-3-540-77349-8_18. [DOI] [PubMed] [Google Scholar]
- Silvestri G, Feinberg MB. Turnover of lymphocytes and conceptual paradigms in HIV infection. J Clin Invest. 2003;112:821–824. doi: 10.1172/JCI19799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smed-Sorensen A, Lore K, Vasudevan J, Louder MK, Andersson J, Mascola JR, Spetz A-L, Koup RA. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J Virol. 2005;79:8861–8869. doi: 10.1128/JVI.79.14.8861-8869.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity. 2008;29:650–659. doi: 10.1016/j.immuni.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa AE, Carneiro J, Meier-Schellersheim M, Grossman Z, Victorino RMM. CD4 T cell depletion is linked directly to immune activation in the pathogenesis of HIV-1 and HIV-2 but only indirectly to the viral load. J Immunol. 2002;169:3400–3406. doi: 10.4049/jimmunol.169.6.3400. [DOI] [PubMed] [Google Scholar]
- Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, Lebedeva M, DeCamp A, Li D, Grove D, et al. Induction of a striking systemic cytokine cascade prior to peak viraemia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83:3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilton JC, Manion MM, Luskin MR, Johnson AJ, Patamawenu AA, Hallahan CW, Cogliano-Shutta NA, Mican JM, Davey RT, Jr, Kottilil S, et al. Human immunodeficiency virus viremia induces plasmacytoid dendritic cell activation in vivo and diminished alpha interferon production in vitro. J Virol. 2008;82:3997–4006. doi: 10.1128/JVI.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre-Cisneros J, del Castillo M, Caston JJ, Castro MC, Perez V, Collantes E. Infliximab does not activate replication of lymphotropic herpesviruses in patients with refractory rheumatoid arthritis. Rheumatology. 2005;44:1132–1135. doi: 10.1093/rheumatology/keh696. [DOI] [PubMed] [Google Scholar]
- Trkola A, Kuster H, Rusert P, Joos B, Fischer M, Leemann C, Manrique A, Huber M, Rehr M, Oxenius A, et al. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat Med. 2005;11:615–622. doi: 10.1038/nm1244. [DOI] [PubMed] [Google Scholar]
- Waldrop SL, Pitcher CJ, Peterson DM, Maino VC, Picker LJ. Determination of antigen-specific memory/effector CD4+ T cell frequencies by flow cytometry: evidence for a novel, antigen-specific homeostatic mechanism in HIV-associated immunodeficiency. J Clin Invest. 1997;99:1739–1750. doi: 10.1172/JCI119338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way SS, Havenar-Daughton C, Kolumam GA, Orgun NN, Murali-Krishna K. IL-12 and type-I IFN synergize for IFN-γ production by CD4 T cells, whereas neither are required for IFN-γ production by CD8 T cells after Listeria monocytogenes infection. J Immunol. 2007;178:4498–4505. doi: 10.4049/jimmunol.178.7.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun Z, Lewensohn-Fuchs I, Ljungman P, Vahlne A. Real-time monitoring of cytomegalovirus infections after stem cell transplantation using the TaqMan polymerase chain reaction. Transplantation. 2000;69:1733–1736. doi: 10.1097/00007890-200004270-00037. [DOI] [PubMed] [Google Scholar]
- Zingg W, Bossart W, Berli E, Nadal D. Detection and quantification of cell-free Epstein–Barr virus by polymerase chain reaction and subsequent DNA enzyme immunoassay. J Virol Methods. 1999;79:141–148. doi: 10.1016/s0166-0934(99)00013-0. [DOI] [PubMed] [Google Scholar]