

Abstract
Apoptosis is a fundamental host defence mechanism against invading microbes. Inactivation of NF-κB attenuates encephalomyocarditis virus (EMCV) virulence by triggering rapid apoptosis of infected cells, thereby pre-emptively limiting viral replication. Recent evidence has shown that hypoxia-inducible factor (HIF) increases NF-κB-mediated anti-apoptotic response in clear-cell renal cell carcinoma (CCRCC) that commonly exhibit hyperactivation of HIF due to the loss of its principal negative regulator, von Hippel–Lindau (VHL) tumour suppressor protein. Here, we show that EMCV challenge induces a strong NF-κB-dependent gene expression profile concomitant with a lack of interferon-mediated anti-viral response in VHL-null CCRCC, and that multiple established CCRCC cell lines, as well as early-passage primary CCRCC cultured cells, are acutely susceptible to EMCV replication and virulence. Functional restoration of VHL or molecular suppression of HIF or NF-κB dramatically reverses CCRCC cellular susceptibility to EMCV-induced killing. Notably, intratumoural EMCV treatment of CCRCC in a murine xenograft model rapidly regresses tumour growth. These findings provide compelling pre-clinical evidence for the usage of EMCV in the treatment of CCRCC and potentially other tumours with elevated HIF/NF-κB-survival signature.
Keywords: EMCV, HIF, NF-κB, RCC, VHL
INTRODUCTION
Apoptosis is an effective host defence mechanism against invading microbes (Teodoro & Branton, 1997). While many viruses have evolved to trigger apoptosis via several mechanisms, including viral disruption of cellular metabolism and cell cycle and recognition by cytotoxic T cells (Shen & Shenk, 1995), some viruses have instead evolved to evade host programmed cell death to promote viral replication and spread. For example, viruses encode proteins such as adenovirus E1B, baculovirus p35, cowpox virus CrmA and viral FLICE-inhibitory proteins (v-FLIPs) that directly inhibit caspases (Bertin et al, 1997; Boulakia et al, 1996; Bump et al, 1995; Ray et al, 1992; Thome et al, 1997). Mutant viruses lacking these anti-apoptotic genes trigger premature apoptotic death of host cells and consequently produce lower yields of progeny virus (Brooks et al, 1995; Hershberger et al, 1992; Pilder et al, 1984). These lines of evidence support a causal relationship between programmed cell death and viral virulence.
NF-κB is a transcription factor that regulates host immune and inflammatory responses, and influences viral replication (Baeuerle & Henkel, 1994; Wang et al, 1996). Viral infection or proinflammatory cytokines such as TNF-α promote rapid E3 ubiquitin ligase-mediated degradation of inhibitory IκBα allowing the release of NF-κB to the nucleus. NF-κB transactivates numerous genes including those encoding interleukins, cytokines, acute phase proteins, and anti-apoptotic proteins (Hayden & Ghosh, 2004). In addition to the activation of the NF-κB-mediated survival response, TNF-α also triggers caspase-mediated apoptotic response whereby the eventual fate of a cell depends on the overall balance between these opposing pathways (Qi & Ohh, 2003). Cells deficient in NF-κB p65 subunit or expressing constitutively stable IκBα are therefore unable to mount NF-κB-dependent survival response and hence are highly susceptible to TNF-α-induced caspase-dependent apoptosis (Beg & Baltimore, 1996; Van Antwerp et al, 1996). These observations support the general notion that NF-κB activation is a protective response by the host to an invading pathogen. Paradoxically, however, some pathogens have strategies to potentiate NF-κB to exploit its anti-apoptotic functions to enhance their own replication and spread. For example, NF-κB p50 subunit knockout mice exhibit accelerated apoptosis of encephalomyocarditis virus (EMCV)-infected cells, which dramatically limits viral replication and consequently survive EMCV infection that otherwise readily kills wild-type (WT) mice (Schwarz et al, 1998; Sha et al, 1995). These results suggest that NF-κB-mediated anti-apoptotic response is required for unrestricted EMCV replication, spread and virulence.
Evasion of apoptosis is also a cardinal feature of cancer. For example, clear-cell renal cell carcinoma (CCRCC) is not only the most common form of kidney cancer, but also one of the most resistant tumours to conventional cancer treatments that exhibits increased NF-κB activity and expression of anti-apoptotic NF-κB-target proteins such as survivin, c-IAP1/2 and c-FLIP (Qi & Ohh, 2003; Yang et al, 2007). CCRCC cells overexpress hypoxia-inducible factor (HIF)-α subunit due to the functional inactivation of von Hippel–Lindau (VHL) tumour suppressor protein, which serves as a substrate-specifying component of an E3 ubiquitin ligase ECV (Elongins BC/Cullin 2/VHL) that catalyses oxygen-dependent polyubiquitin-mediated destruction of prolyl-hydroxylated HIF-α. Under hypoxia, the unmodified HIF-α escapes destructive recognition by ECV and associates with its constitutively stable partner HIF-β to form an active heterodimeric HIF transcription factor. HIF binds to hypoxia-responsive elements (HREs) located in the promoter/enhancer regions of numerous hypoxia-inducible genes to initiate various adaptive responses to hypoxia, such as anaerobic metabolism, erythropoiesis and angiogenesis (Kaelin, 2002; Roberts & Ohh, 2008). VHL has been implicated in facilitating TNF-α-induced apoptosis by suppressing the expression of NF-κB-dependent anti-apoptotic genes (Qi & Ohh, 2003). Therefore, functional loss of VHL observed in the vast majority of CCRCC increases anti-apoptotic protein expression, rendering these tumour cells more resistant to normal cell death processes. Recently, An and Rettig demonstrated that VHL-mediated suppression of NF-κB activity is dependent on HIF (An & Rettig, 2005), while Yang et al showed that VHL serves as an adaptor molecule that binds and promotes the inhibitory phosphorylation of the NF-κB agonist Card9 by casein kinase 2. Downregulation of Card9 in VHL-null CCRCC normalized NF-κB activity and sensitivity to cytokine-induced cell death (Yang et al, 2007). These reports establish an association between HIF and VHL to the NF-κB survival pathway.
EMCV is a single positive-stranded RNA picornavirus with a large host range, infecting numerous mammals and birds. However, only a small number of animal species appear to be adversely affected, including swine, non-human primates and mice. Importantly, a causal relationship between EMCV infection of humans and illness has never been established (Brewer et al, 2001; Moran et al, 2005). Here, we asked whether the hallmark of cancer cells to evade apoptosis could be exploited for tumour-specific oncolytic killing via EMCV that requires elevated anti-apoptotic properties of host cells for viral replication, spread and virulence. Using multiple genetically engineered established CCRCC cell lines, early-passage primary CCRCC cultured cells and CCRCC xenografts in a murine model, we provide pre-clinical evidence that NF-κB-dependent replication and virulence of EMCV targets tumour cells that exhibit elevated HIF status for selective and effective killing.
RESULTS
Suppression of NF-κB activity in MEFs and CCRCC cells impairs EMCV virulence
Virulence of EMCV is positively associated with the NF-κB-mediated survival pathway. In particular EMCV is highly pathogenic to mice, causing myocarditis and dilated cardiomyopathy, invariably killing normal healthy mice whereas NF-κB p50−/− mice survive EMCV infections (Sha et al, 1995). Schwarz et al subsequently showed that mouse embryonic fibroblasts (MEFs) derived from p50−/− or p65−/− mice are resistant to EMCV-induced cytotoxicity, and revealed that NF-κB-mediated upregulation of anti-apoptotic signalling is responsible for increased EMCV viral replication and the resulting lytic death of WT MEFs. In the absence of NF-κB activity, p50−/− and p65−/− cells were able to undergo rapid apoptosis upon EMCV infection prior to viral replication, thus pre-emptively preventing progeny viral overload and spread (Schwarz et al, 1998). Together, these studies indicate that the inactivation of NF-κB transcription factor via loss of components, p50 or p65, results in severely attenuated EMCV virulence.
Indirect activation of NF-κB pathway due to aberrant oncogenic signalling is a common phenomenon in many types of cancer, which increases the capacity for tumour cells to evade apoptosis and gain a survival advantage over normal, untransformed cells. We asked whether indirect perturbation of NF-κB is sufficient to influence the susceptibility of cells to EMCV-induced cytotoxicity. NEMO (also known as IKK-γ) is a critical component of a tripartite IκBα-Kinase (IKK) complex required for phosphorylation and subsequent degradation of IκBα, thereby activating NF-κB. Therefore, loss of NEMO results in decreased NF-κB-mediated signalling due to the constitutive stabilization of IκBα (Kim et al, 2003). NEMO−/− MEFs showed markedly reduced levels of cytotoxicity than WT MEFs upon EMCV infection as measured by propidium iodide staining at regular intervals (Fig 1A), which, consistent with the observation in p50−/− and p65−/− MEFs, suggests that NF-κB inactivation ultimately protects cells from EMCV-induced death. Similar observations were noted in parallel experiments using Annexin V and Trypan Blue exclusion cell viability assays (data not shown). These results support the notion that the functionality of NF-κB can profoundly affect cellular susceptibility to EMCV-induced killing, which may therefore provide therapeutic rationale for an oncolytic EMCV-based approach in the treatment of cancers with elevated NF-κB signalling.
Figure 1. NF-κB pathway influences cellular susceptibility to EMCV-induced cytotoxicity.

- NEMO promotes susceptibility of MEFs to EMCV-induced death. Equal amounts of total cell lysates from WT and NEMO−/− MEFs were immunoblotted with anti-NEMO and anti-Vinculin antibodies (upper panel). WT and NEMO−/− MEFs were infected with EMCV (MOI = 0.1) and viable cells were counted at 2 h intervals post-infection by Annexin V-FITC/propidium iodide staining. Experiments were conducted in triplicate and error bars represent standard errors (lower graph). Two-way ANOVA was applied for statistical analysis between treatments and time points. * and *** denote p < 0.05 and p < 0.001, respectively.
- NEMO promotes susceptibility of 786-O CCRCC cells to EMCV-induced death. 786-O cells transfected with NEMO-specific siRNA (siNEMO) or scrambled non-targeting siRNA (siCON) (left panel) were infected with or without EMCV (MOI = 0.1), and viable cells counted 18 h post-infection by Trypan Blue exclusion assay (right graph).
- Activated NF-κB pathway promotes susceptibility of 786-O CCRCC cells to EMCV-induced death. 786-O cells were treated with or without JSH-23 (10 µM) in the presence or absence of LPS (10 µg/ml) and the level of nuclear NF-κB visualized by immunoblotting (left panel). 786-O cells treated with or without JSH-23 were challenged with EMCV (MOI = 0.1) and viable cells were counted 18 h post-infection by Trypan Blue exclusion assay (right graph). C1 + C2 denote two splice isoforms of nuclear restricted pre-mRNA binding protein hnRNP. Experiments were performed in triplicate and error bars represent standard errors, independent Student's t-test was used to analyse difference between groups.
We next asked whether NF-κB influenced EMCV virulence in VHL-null 786-O CCRCC cells, which are known to exhibit elevated NF-κB activity (An & Rettig, 2005), via molecular manipulation of NEMO. 786-O cells with transient siRNA-mediated knockdown of endogenous NEMO (786-O + siNEMO) were resistant to EMCV virulence relative to 786-O cells transfected with scrambled siRNA (786-O + siCON) (Fig 1B). In parallel, treatment of 786-O cells with a small molecule JSH-23, an aromatic diamine that inhibits nuclear translocation of NF-κB (Shin et al, 2004) significantly reduced both NF-κB nuclear localization upon LPS treatment and susceptibility to EMCV virulence (Fig 1C). These results suggest that the susceptibility of CCRCC cells to EMCV virulence is significantly influenced by NF-κB activity. Notably, comparable levels of endogenous NEMO expression were observed between patient-derived CCRCC tumour and matched normal kidney tissue samples, as well as between 786-O (VHL−/−; HIF1α−/−) CCRCC cells or isogenically matched HA-tagged WT VHL-reconstituted cells (786-VHL) and normal renal proximal tubule epithelial cells (RPTEC) (Supporting Information Fig 1).
Loss of VHL enhances NF-κB-mediated survival signalling upon EMCV challenge
We asked whether the loss of VHL influences susceptibility of CCRCC cells, which frequently harbour loss-of-function mutation or loss of VHL, to EMCV. 786-O or 786-VHL cells were challenged with EMCV and analysed for NF-κB-dependent gene expression profile by Affymetrix array and real-time semi-quantitative RT-PCR analyses. EMCV challenge induced NF-κB-regulated genes such as IL-8, IL-6 and TNF-α to a significantly greater extent in 786-O cells as compared to 786-VHL cells (Table 1 and Fig 2). The markedly enhanced NF-κB-mediated gene expression observed upon EMCV challenge in the absence of VHL is likely due to the pre-existing activation, and therefore readiness, of NF-κB-mediated signalling pathway resulting from VHL loss (An & Rettig, 2005; Qi & Ohh, 2003).
Table 1.
NF-κB-dependent genes are preferentially induced by EMCV in 786-O cells compared to 786-VHL cells
| Affymetrix probe | Gene name | Gene title | 7B6-O VSV | 786-VHL VSV | 786-0 EMCV | 786-VHL EMCV |
|---|---|---|---|---|---|---|
| 211506_s_at | IL8 | Interleukin-8 | 1.23 | 0.54 | 17.15 | 6.50 |
| 205207_at | IL6 | Interleukin-6 (interferon, beta 2) | 3.48 | 1.87 | 6.50 | 2.64 |
| 231779_at | IRAK2 | Interleukin-1 receptor-associated kinase 2 | 1.87 | 1.07 | 4.92 | 1.41 |
| 201502_s_at | NFKBIA | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha | 2.30 | 2.14 | 2.83 | 1.41 |
| 227345_at | TNFRSF10D | Tumor necrosis factor receptor superfamily, member 10d | 1.07 | 0.76 | 2.83 | 1.52 |
| 209294_x_at | TNFRSF10B | Tumor necrosis factor receptor superfamily, member 10b | 1.32 | 1.15 | 2.30 | 1.74 |
| 203927_at | NFKBIE | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, epsilon | 1.41 | 1.00 | 2.14 | 1.00 |
| 231775_at | TNFRSF10A | Tumor necrosis factor receptor superfamily, member 10a | 1.07 | 0.93 | 2.00 | 1.23 |
Affymetrix microarray expression analysis of NF-κB-regulated genes induced upon EMCV or VSV challenge in 786-O cells and 786-VHL cells. The complete gene expression data have been deposited on the ArrayExpress database maintained by the European Bioinformatics Institute (accession number: E-MTAB-294).
Figure 2. Loss of VHL in CCRCC cells accentuates NF-κB-mediated survival signalling upon EMCV challenge.
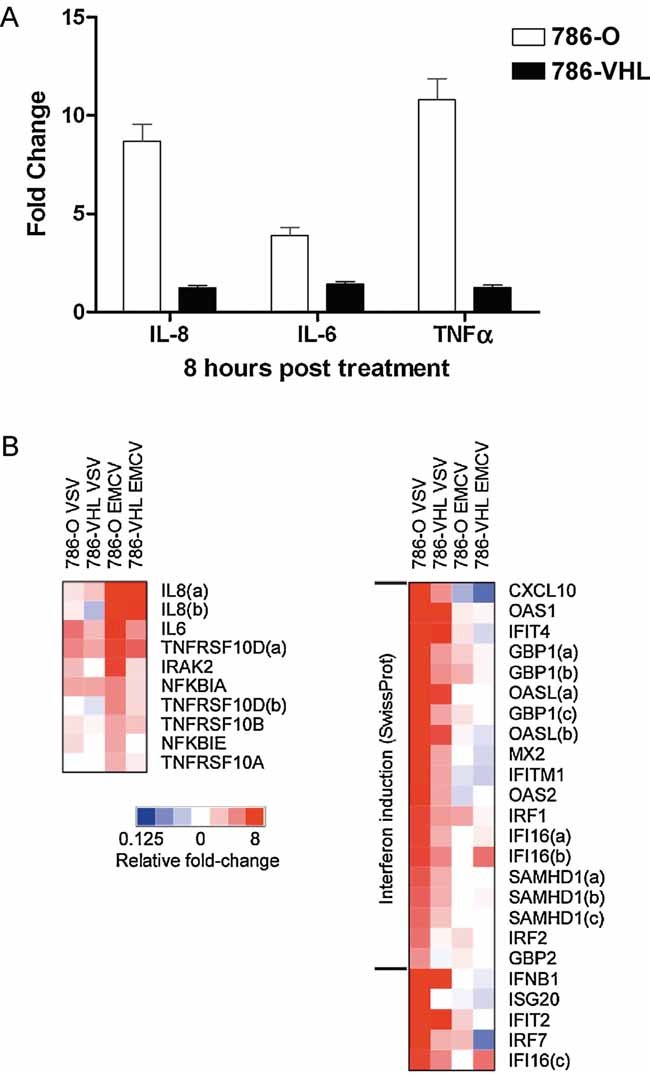
- Loss of VHL in 786-O cells accentuates NF-κB-mediated transcription upon EMCV challenge. Steady-state 1L8, 1L6 and TNF-α mRNA levels were measured by real-time PCR in 786-O (open bars) and 786-VIIL (solid bars) cells infected with EMCV (MOI = 0.1) for 8 h. Experiments were performed in triplicate and error bars represent standard errors.
- Heatmap representation NF-κB-dependent and IFN/IFN-stimulated gene expression in 786-O and 786-VHL cells challenged with EMCV or VSV. Relative gene expression levels of NF-κB-dependent (left panel) and IFN/IFN-stimulated (right panel) genes in 786-O and 786-VHL cells challenged with EMCV or VSV. Actual expression levels as determined by Affymetrix microarray expression analysis arc shown in Tables 1 and 2.
Previously, we demonstrated that 786-O cells mounted a stronger anti-viral interferon-mediated response than 786-VHL cells upon vesicular stomatitus virus (VSV) challenge, and thereby protecting 786-O cells against VSV-induced virulence (Hwang et al, 2006). In contrast, EMCV infection induced a modest to negligible interferon response (Table 2). EASE analysis of transcripts preferentially induced by VSV in 786-O cells compared to 786-VHL cells yielded ‘interferon induction’ as it is most enriched SwissProt category (EASE score = 1.10E−19; Fisher's Exact Test score = 1.06E−21). However, EMCV-infected 786-O cells showed no significant enrichment of ‘interferon induction’ category (EASE score = 1). In addition, while VSV infection of 786-O cells for 8 h induced a 74-fold increase in IFN-β transcript levels, EMCV infection had a negligible influence on IFN-β expression (0.93-fold change) (Table 2). Moreover, unlike EMCV, VSV did not generate further NF-κB signalling response irrespective of VHL status (Table 1). These results indicate that EMCV and VSV cause strikingly different VHL-dependent responses in CCRCC cells. The lack of interferon-mediated anti-viral response in combination with elevated NF-κB-mediated anti-apoptotic response in 786-O cells upon EMCV challenge suggest that the loss of VHL will likely boost EMCV replication and render CCRCC cells susceptible to EMCV-induced cytotoxicity.
Table 2.
EMCV induces markedly weaker IFN/IFN-stimulated gene expression than VSV
| Affymetrix gene | Gene title | 786-O VSV | 786-VHL VSV | 786-O EMCV | 786-VHL EMCV |
|---|---|---|---|---|---|
| Interferon induction (SwissProt) | |||||
| 204533_A⌉CXCL10 | Chemokine (C-X-C motif) ligand 10 | 17.15 | 2.64 | 0.50 | 0.27 |
| 205552_S_OAS1 | 2′,5′-Oligoadenylate synthetase 1, 40/46 kDa | 17.15 | 6.96 | 1.23 | 1.15 |
| 204747_A⌉IFIT4 | Interferon-induced protein with tetratricopeptide repeats 4 | 14.93 | 5.66 | 1.32 | 0.71 |
| 231577_S_GBP1 | Guanylate binding protein 1, interferon-inducible, 67 kDa | 12.13 | 2.30 | 1.32 | 1.00 |
| 205660_A⌉OASL | 2′-5′-Oligoadenylate synthetase-like | 11.31 | 4.59 | 1.15 | 0.76 |
| 204994_A⌉MX2 | Myxovirus (influenza virus) resistance 2 (mouse) | 9.85 | 2.14 | 1.00 | 0.71 |
| 214022_S_IFITM1 | Interferon induced transmembrane protein 1 (9–27) | 6.96 | 2.30 | 0.76 | 0.66 |
| 204972_A⌉0AS2 | 2′-5′-Oligoadenylate synthetase 2, 69/71 kDa | 6.50 | 2.14 | 0.71 | 1.00 |
| 202531_A⌉IRF1 | Interferon regulatory factor 1 | 5.28 | 2.46 | 2.14 | 1.15 |
| 208966_X_IFI16 | Interferon, gamma-inducible protein 16 | 4.92 | 2.00 | 1.00 | 1.23 |
| 234987_A⌉SAMHD1 | SAM domain and HD domain 1 | 4.29 | 2.00 | 0.93 | 0.93 |
| 203275_A⌉IRF2 | Interferon regulatory factor 2 | 3.25 | 1.15 | 1.41 | 1.00 |
| 202748_A⌉GBP2 | Guanylate binding protein 2, interferon-inducible | 2.64 | 0.87 | 1.23 | 1.07 |
Affymetrix microarray expression analysis of IFN and genes identified as being involved in ‘Interferon Induction’ by EASE induced upon EMCV or VSV challenge in 786-O and 786-VHL cells. The complete gene expression data have been deposited on the ArrayExpress database maintained by the European Bioinformatics Institute (accession number: E-MTAB-294).
Loss of VHL enhances EMCV replication and increases CCRCC cell cytotoxicity
We next asked whether VHL influences EMCV viral replication. Lysates from EMCV-challenged 786-O and 786-VHL cells were added to L929 mouse fibroblast monolayers, which were then monitored for cytotoxicity in which an increase in L929 cytotoxicity would be indicative of an increase in EMCV titre. As predicted, 786-O cells with strong hypoxic signature, as indicated by elevated expression of hypoxia-inducible GLUT-1 (Fig 3A, left panel), and correspondingly enhanced capacity for NF-κB activation produced orders of magnitude higher EMCV titres than in 786-VHL cells with low NF-κB activity, demonstrating enhanced EMCV replication in CCRCC cells devoid of VHL (Fig 3A, right panel). EMCV challenge produced similar orders of magnitude higher EMCV titres in another distinct VHL−/− CCRCC cell line ectopically expressing empty plasmid (RCC4-MOCK) than in WT VHL reconstituted isogenic counterpart (RCC4-VHL) (Fig 3B).
Figure 3. Loss of VHL in CCRCC cells enhances EMCV replication.
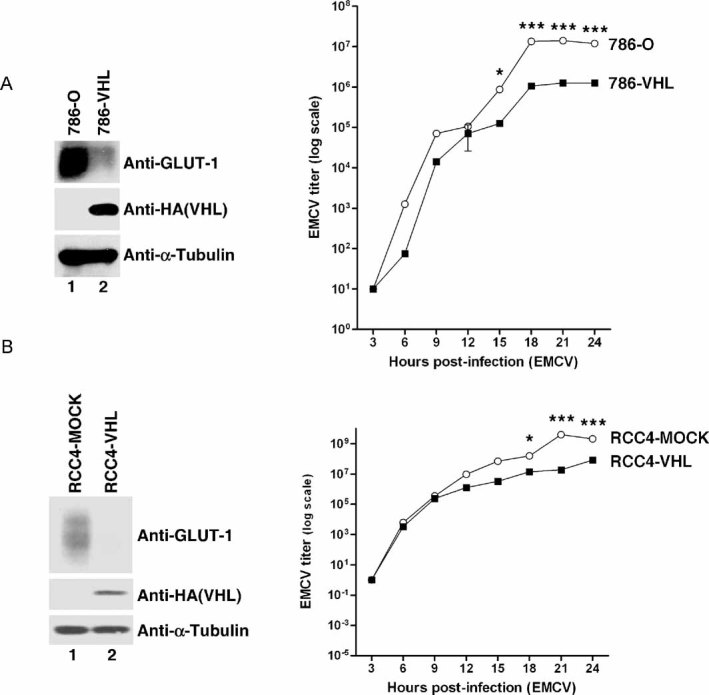
- Loss of VHL in 786-O cells enhances EMCV replication. 786-O and 786-VHL cells were lysed and immunoblotted with anti-HA, anti-GLUT-1 and anti-α-Tubulin antibodies (left panel). Cells were challenged with EMCV (MOI = 0.01) and cumulative virus titre was evaluated at 3 h intervals post-infection (right graph). Two-way ANOVA was applied for statistical analysis between treatments and time points. * and *** denote p < 0.05 and p < 0.001, respectively.
- Loss of VHL in RCC4 cells enhances EMCV replication. RCC4-MOCK and RCC4-VHL cells were lysed and immunoblotted with anti-HA, anti-GLUT-1 and anti-α-Tubulin antibodies (left panel). Cells were challenged with EMCV (MOI = 0.01) and cumulative virus titre was evaluated at 3 h intervals post-infection (right graph). Two-way ANOVA was applied for statistical analysis between treatments and time points. * and *** denote p < 0.05 and p < 0.001, respectively.
Next, we asked whether the marked increase in EMCV replication resulting from the loss of VHL would be accompanied by enhanced sensitivity of CCRCC cells to EMCV-induced death. While 786-VHL cells remained viable, the vast majority of 786-O cells were killed following a 14 h challenge with EMCV (Fig 4A). Consistent with this notion, another VHL-null CCRCC UMRC2 cells were effectively killed post-EMCV challenge while VHL-reconstituted UMRC2-VHL or CAK1-1 CCRCC cells, which express endogenous WT VHL, remained viable after 24 h post-EMCV challenge (Supporting Information Fig 2). Moreover, 786-O cells yielded approximately 500-fold lower tissue culture infecting dose (TCID50) than 786-VHL cells (Fig 4B), and ultrastructural electron microscopic examination confirmed viral cytopathologic effect as indicated by increased appearance of peri-cytoplasmic aggregation of organelles in 786-O cells infected with EMCV (Fig 4C, asterisk and open arrows). As predicted, 786-VHL cells infected with EMCV exhibited signs of early nuclear condensation indicative of apoptosis (Supporting Information Fig 3). These results suggest that the loss of VHL leads to a significant increase in susceptibility to EMCV due to a dramatic increase in viral replication and subsequent lytic death. In contrast, cells possessing functional VHL remain relatively unharmed due to premature apoptosis upon initial EMCV infection, thereby severely attenuating viral replication and spread. Notably, EMCV challenge of early-passage (less than 4 months) primary CCRCC cultured cells derived from freshly excised human CCRCC tissue samples similarly resulted in marked cell death 48 h post-infection (Fig 4D).
Figure 4. Loss of VHL enhances susceptibility of CCRCC cells to EMCV-induced cytotoxicity.

- Loss of VHL enhances susceptibility of 786-O cells to EMCV-induced cytotoxicity. 786-O and 786-VHL cells infected with EMCV (MOI = 0.1) were visualized by phase contrast microscopy at various time points (right panel) and viable cells counted at 2 h intervals post-infection by Trypan Blue exclusion assay (left graph). Experiments were conducted in triplicate and error bars represent standard errors. Two-way ANOVA was applied for statistical analysis between treatments and time points. * and *** denote p < 0.05 and p < 0.001, respectively.
- VHL-dependent killing by EMCV occurs in a dose-dependent manner. 786-O and 786-VHL cells were infected with twofold serial dilutions of EMCV (starting from MOI = 1) for 24 h. Live cells were stained with 0.05% crystal violet and quantified by integrated density analysis. Nonlinear regression was performed and TCID50 was calculated in GraphPad Prism. Experiments were performed in quadruplicate and error bars represent standard errors.
- Visualization of EMCV infection of 786-O cells by light microscopy. 786-O cells uninfected or infected with EMCV (MOI = 0.1) for 7 and 15 h were visualized by electron microscopy. Solid arrow represents virus particle formation in infected cells. Asterisk and open arrows represent lytic cell death. Bars represent 1 µm.
- Patient-derived primary CCRCC cells are effectively killed by EMCV. Early passage primary CCRCC cells generated from surgically excised human CCRCC tumours were infected with EMCV (MOI = 0.1 and 1.0) and viable cells were counted at 24 and 48 h post-infection by Trypan Blue exclusion assay. Con denotes uninfected cells. Statistical differences between indicated groups were analysed using the independent Student's t-test.
CCRCC cell susceptibility to EMCV virulence is HIF-dependent
We showed previously that loss of VHL leads to enhanced NF-κB-regulated survival response, consequently protecting VHL-null CCRCC cells from TNF-α-induced apoptosis (An & Rettig, 2005; Qi & Ohh, 2003) (see also Supporting Information Fig 4), and An and Rettig subsequently demonstrated that HIF2α upon VHL loss activates EGFR/PI3K/Akt/IKK-α signalling cascade leading to NF-κB activation (An & Rettig, 2005; Qi & Ohh, 2003). The present results suggest that the loss of VHL in HIF2α-overexpressing 786-O (HIF1α−/−) CCRCC cells increases NF-κB-mediated anti-apoptotic response and sensitivity to EMCV-induced cytolysis. Therefore, we asked whether the observed VHL-dependent susceptibility to EMCV-induced death is HIF2α-dependent. We measured cytotoxicity of 786-O cells stably expressing HA-VHL mutants with varying abilities to degrade HIF2α upon EMCV challenge. Similar to 786-O cells, 786-VHL (Y112H, VHL disease-associated mutant with reduced capacity to bind and degrade HIF-α; Fig 5A) cells were highly sensitive to EMCV-induced killing, whereas 786-VHL (L188V, VHL disease-associated mutant capable of binding and promoting degradation of HIF-α; Fig 5A) cells, like 786-VHL cells, were considerably (approximately 50-fold) more resistant to EMCV (Fig 5B). These results are consistent with the notion that VHL-mediated EMCV susceptibility is dependent on HIF2α regulation.
Figure 5. Susceptibility of CCRCC cells to EMCV virulence is HIF2α-dependent.

- VHL disease-causing mutants Y112H and L188V have varying abilities to regulate the HIF pathway. [35S]methionine-labelled in vitro translated empty plasmid (MOCK), HA-tagged WT or the indicated mutant VHL proteins were mixed with equal amounts of biotinylated HIF1α[ODD-OH] or hydroxylation-defective HIF1α[ODD(PA)] peptides for 30 min at 37°C. Reaction mixtures were then subjected to biotin affinity pull-down (PD) via streptavidin-coated beads and bound radiolabelled HA-VHL proteins were resolved on SDS–PAGE and visualized by autoradiography (left panel). Unmixed radiolabelled in vitro translated HA-tagged WT or the indicated mutant VHL proteins were also immunoprecipitated (IP) with excess anti-HA antibody, resolved on SDS–PAGE and visualized by autoradiography as an indication of equal loading (left graph, lanes 1–4). 786-O, 786-VHL(WT), 786-VHL(Y112H) and 786-VHL(L188V) cells were lysed and immunoblotted with anti-HA, anti-GLUT-1 and anti-α-Tubulin antibodies (right panel).
- HIF regulation by VHL influences susceptibility of CCRCC cells to EMCV-induced killing. 786-VHL(Y112H) and 786-VHL(L188V) cells were infected with two-fold serial dilutions of EMCV (starting from MOI = 1) for 24 h. Live cells were stained with 0.05% crystal violet and quantified by integrated density analysis. Nonlinear regression was performed and TCID50 was calculated in GraphPad Prism. Experiments were performed in quadruplicate and error bars represent standard errors.
- siRNA-mediated inhibition of HIF2α decreases susceptibility to EMCV-induced death. 786-O cells were transiently transfected with a control scrambled siRNA (siCON) or HIF2α-specific siRNA (siHIF2α). After 24 h, steady-state mRNA levels of HIF2α were measured by real-time qPCR (left graph) and cells were challenged with EMCV (MOI = 0.1). Live cells were quantified by Trypan Blue exclusion assay at 18 h post-infection (right panel). Experiments were performed in triplicate and error bars represent standard errors.
- Small molecule inhibitor chetomin-mediated inhibition of HIF2α activity decreases susceptibility to EMCV-induced death. 786-O and 786-VHL cells were treated with or without 50 nM chetomin for 16 h and then challenged with EMCV at the indicated MOI for 18 h. Live cells were stained with 0.05% crystal violet and quantified by integrated density analysis. Experiments were conducted in triplicate and error bars represent standard errors.
To directly address this notion, 786-O cells were transiently transfected with HIF2α-targeting siRNA or non-targeting control siRNA and then challenged with EMCV. Attenuation of endogenous HIF2α expression (Fig 5C, left graph) dramatically decreased the susceptibility of 786-O cells to EMCV-induced cytotoxicity (Fig 5C, right panel). Similar results were observed using another distinct HIF2α-specific siRNA (data not shown) and as expected, siRNA-mediated knockdown of HIF2α lowered the expression of HIF-target genes such as endothelin 1 (Edn1) and glucose transporter 1 (GLUT1) (data not shown). In parallel, HIF2α activity was inhibited in 786-O cells using chetomin, a small molecule inhibitor of HIF-mediated transcription (Hwang et al, 2006; Kung et al, 2004), prior to EMCV challenge. In the presence of chetomin, 786-O cells displayed reduced sensitivity to EMCV-induced killing (Fig 5D) and as expected, diminished HIF-mediated transactivation (data not shown). Moreover, glioblastomas, like many other solid tumours, have significant hypoxic signature and HIF1 has been shown to play a critical role in gliomagenesis (Kaur et al, 2005). Treatment of T98G glioblastoma cells with hypoxia mimetic cobalt chloride, which promotes HIF-α stabilization (Maxwell et al, 1999), sensitized T98G cells to EMCV virulence (Supporting Information Fig 5). These results suggest that the susceptibility of tumour cells to EMCV virulence is, at least in part, mediated via HIF.
CCRCC xenografts are susceptible to EMCV-induced killing in vivo
We next asked whether EMCV could kill CCRCC cells in vivo. Engineered CCRCC VHL-null 786-O cells stabling expressing HRE-driven luciferase (786-HRE-Luc) were implanted into dorsal skin-fold window-chamber on SCID mice (Sufan et al, 2009) and treated with either live EMCV or irradiated (dead) dEMCV 2 days post-implantation. 786-HRE-Luc cells express constant detectable luminescence due to the constitutively stable expression of HIF2α in the absence of VHL, which binds and transactivates HRE-driven luciferase expression. Intratumoural EMCV injections of microtumour xenografts were necessary to minimize viral dosage/load and potential systemic viral spread as EMCV is lethal in mice. Notably, while dEMCV-treated group displayed negligible changes in tumour bioluminescence (BLI) 4 days post-treatment, there was a striking diminution (over 100-fold reduction) of bioluminescent signal in EMCV-treated group (Fig 6A). Notably, EMCV infection had negligible effect on HIF2α protein stability, HRE-driven BLI and HIF-target GLUT-1 expression (Supporting Information Fig 6), suggesting that the reduction in bioluminescent signal reflects EMCV-induced tumour cell death rather than the possibility of EMCV-mediated attenuation of HIF levels without tumour killing. Consistent with this notion, immunohistochemical analysis revealed substantially larger and prominent regions of tumour necrosis in EMCV-treated xenografts in comparison to dEMCV-treated counterparts (Fig 6B). These results demonstrate that EMCV treatment effectively regresses CCRCC xenograft growth in vivo.
Figure 6. EMCV regresses human CCRCC xenograft growth in murine dorsal skin-fold window chamber model.

- EMCV kills HIF-overexpression tumour cells in vivo. 786-HRE-Luc cells were implanted in dorsal dermis in 6 SCID mice and tumour xenograft visualized 1 day before virus treatment (left panels). Mice were segregated into two groups of 3 where one group received intratumoural injection of live EMCV (bottom panels) while the other group received intratumoural injection of irradiated dead (d) EMCV (top panels), and tumour BLI signals visualized and measured 4 days post-treatment. Average BLI signals from each group measured in photons/seconds/area/steradian are shown with error bars representing standard errors (graph). Representative longitudinal images from the same mouse from each group are also shown (left panels).
- EMCV kills tumour cells by necrosis. Haematoxylin and eosin (H&E) staining was performed on the resected xenografts post-termination of treatment. Representative images from each group are shown at 100× magnification. Dashed line, viable/necrotic interface; V, viable cells; N, necrotic cells; a, adipocytes; bv, blood vessels.
DISCUSSION
Neoplastic transformation invariably includes acquired mechanisms to evade programmed cell death. While this cardinal feature generally makes tumour cells more resistant to apoptosis than normal cells, it concomitantly presents tumour-specific attributes that can be exploited in the design of novel therapeutics. One such attribute is that tumour cells often have impaired antiviral responses and are therefore more susceptible to viral infection than normal cells. Multiple mechanisms underlie these antiviral defects, which have been targeted in the development of oncolytic virus strategies to combat specific cancers, and several different species of oncolytic viruses have since been taken to phase I and II clinical trials (Aghi & Martuza, 2005). For example, some cancers with impaired IFN responses appear to be particularly sensitive to infection and killing by a number of VSV strains, while cancers harbouring defects in p53 responses or exhibiting abnormal cell cycle properties have been targeted by herpes virus and adenovirus-based constructs (O'Shea, 2005; Stojdl et al, 2003). In addition, cancer cells with active Ras signalling pathway have been targeted by reoviruses (Norman & Lee, 2005). An advantage of oncolytic viruses to target tumour mass is that after killing the initially infected cells, new viral progeny is released, which can amplify the oncolytic effect until the whole tumour is eradicated while, importantly, leaving normal healthy tissue relatively unaffected (Chiocca, 2002; Demers et al, 2003). For example, a phase III trial comprising more than 50 patients with head and neck tumours showed a 70% response to a combination of chemotherapy and intratumoural adenovirus application compared to a 30% response for chemotherapy alone (Wakimoto et al, 2004).
Functional inactivation of VHL is the principal cause of both sporadic CCRCC and hereditary VHL disease, which is characterized by the development of multiple hypervascular tumours including CCRCC, establishing VHL as the ‘gatekeeper’ of the renal epithelium (Roberts & Ohh, 2008). CCRCC is one of the most resistant tumours to radiation and chemotherapy, and patients with metastatic CCRCC have a median overall survival of 13 months. While radical or partial nephrectomy remains to be the prevailing treatment option for localized CCRCC, tumours can recur post-operatively in one-third of patients as distant metastases, and only 4–6% of these tumours respond to chemotherapy (Cohen & McGovern, 2005). For localized CCRCC, EMCV could be delivered intratumourally via percutaneous approach, as it is often performed for percutaneous needle biopsy (Laguna et al, 2009) or ablative therapies (Miao et al, 2001). A systemic application of EMCV could be an option for metastatic CCRCC patients with infaust prognosis who cannot be treated surgically and do not respond to other conventional treatment options.
How a patient might respond to EMCV treatment is a concern that would require attention for any future clinical application. The pathogenicity of EMCV in mice certainly renders it challenging for further pre-clinical testing in a traditional mouse models. This is, however, an issue that is not restricted to this particular oncolytic virus candidate. For example, the avian virus Newcastle Disease Virus (NDV) and Vaccinia virus (wyeth strain) replicate poorly in mouse tissues, consequently complicating the determination of and often exaggerating their therapeutic window in xenograft mouse tumour models (Williamson et al, 1990). Measles virus cannot replicate in mouse tissues since its receptor is restricted to human cells. Complex transgenic mouse models (e.g. addition of human CD46 gene and deletion of interferon receptor) are therefore required to carry out pre-clinical testing of this oncolytic virus (Sellin & Horvat, 2009). Despite these limitations, all of these viruses have progressed into clinical testing. Moreover, mouse model system could potentially be developed in NF-κB p50−/− or p65−/− background, which are resistant to EMCV-induced cytotoxicity (Schwarz et al, 1998; Sha et al, 1995), to circumvent the pathogenic limitation of EMCV in WT mice. There is also limited number of rat tumour models and since EMCV is not pathogenic for rats, this rodent species could be adapted for human CCRCC xenograft for pre-clinical analysis. The exact toxicity profile of the laboratory attenuated strain of EMCV has yet to be determined in non-human primates but these future studies could further aid in the clinical development of this oncolytic virus candidate.
The extent of host immune response triggered after administration of high doses of EMCV poses a potential caveat for the efficacy of EMCV as an oncolytic agent. However, another picornavirus, Seneca Valley Virus, has been successfully and safely administered intravenously to patients at doses of 1e11 pfu/patient while NDV PV701 has also been administered at multiple doses exceeding 1e11 pfu/patient without deleterious side-effects (Laurie et al, 2006). Despite these examples of promising administration of high dose virus titres, pre-clinical and eventual clinical studies are necessary to determine the extent of host immune response at various EMCV doses to better define the therapeutic window. As with other anti-cancer therapies, the possibility of EMCV treatment leading to the selection of tumour cells possessing or developing resistance to EMCV infection is another concern to consider for future studies, including combining EMCV with conventional and other anti-cancer treatment protocols, including Tyrosine Kinase Inhibitors.
Upregulation of NF-κB-response pathway is another feature common to many cancers (Karin et al, 2002), and the elevated NF-κB survival response via HIF2α upon the loss of its negative regulator VHL is thought to contribute to the refractory nature of CCRCC to radiation and chemotherapy (An & Rettig, 2005). Intriguingly, NF-κB activity has been shown to potentiate replication and virulence of a particular picornavirus EMCV with a large host range, but non-pathogenic to humans. For example, inhibition of NF-κB via p50 subunit knockout dramatically limits EMCV replication due to premature apoptosis of virally infected cells and consequently protects p50−/− mice from EMCV-induced death, which otherwise readily kills WT mice (Sha et al, 1995). We therefore reasoned that human CCRCC with loss of VHL might be particularly susceptible to EMCV virulence due to HIF-mediated elevation of NF-κB survival pathway. Consistent with this notion, we show here that CCRCC cells devoid of functional VHL with high HIF1/2α levels are highly susceptible to EMCV virulence, lengthening the time of viral replication prior to viral burst and subsequent spread. In contrast, restoration of VHL or attenuation of HIF2α or NF-κB provides cellular protection against EMCV due to rapid apoptosis of infected cells prior to viral replication. Expression profiling of mRNA derived from EMCV-infected CCRCC cells supports the observation that HIF potentiates the induction of NF-κB-dependent genes. Interestingly, this subset of genes appears to be particularly induced by EMCV much more strongly than by another virus VSV. Since it has been demonstrated that NF-κB-mediated suppression of apoptosis is required for EMCV virulence, the potentiation of NF-κB activity by HIF-α likely contributes to the enhanced EMCV replication and cell death in tumour cells with hyperactive HIF. These pre-clinical findings suggest for the first time a potential oncolytic use of EMCV in the management of CCRCC.
Additional mechanism for the preferential killing of HIF-α-positive cells by EMCV may involve protein expression from the virus’ internal ribosome entry site (IRES). It is well established that hypoxia inhibits protein synthesis due to a marked reduction of translation from most cellular mRNA (Kim & Jang, 2002; Schepens et al, 2005). An exception is HIF-α protein, which is continuously produced as a result of enhanced polypyrimidine tract-binding protein (PTB) interaction with its IRES sequence in 5′ untranslated region. EMCV also has an IRES site that is regulated by PTB and is one of the most frequently used IRES in gene therapy today (Ngoi et al, 2004; Schepens et al, 2005). Interestingly, Nevins et al have shown that the translation mediated by EMCV IRES is more efficient than IRES from other cellular genes examined including XIAP, Apaf-1, DAP5, BiP, and c-Myc and that translation from the EMCV IRES is enhanced under anoxia (Nevins et al, 2003). Thus, unlike many viruses that must, but may be unable to, overcome hypoxia-mediated inhibition of protein synthesis for viral replication, it is possible that hypoxia or increased HIF activity common in solid tumours due to the general oxygen-sensing pathway or various oncogenic mutations on a growing list of genes such as PTEN, TSC and VHL may inadvertently exacerbate EMCV replication and virulence. Thus, the potential range of tumour targets for EMCV as an oncolytic agent could very well extend beyond kidney cancer.
MATERIALS AND METHODS
Cells
786-O (VHL-/-; HIF1α-/-) CCRCC cells, renal proximal tubule epithelial cells (RPTEC) and L929 mouse fibroblast cells were obtained from the American Type Culture Collection (Rockville, MD). 786-O subclones ectopically expressing WT hemagglutinin (HA)-VHL or mutant HA-VHL (Y112H) or HA-VHL (L188V) were previously described (Hwang et al, 2006; Kung et al, 2004). RCC4 (VHL-/-) CCRCC subclones stably expressing HA-VHL (RCC4-VHL) or empty plasmid (RCC4-MOCK) were generously provided by Dr. Wafik S. El-Deiry. WT and NEMO-/- MEF cells were generously provided by Dr. Tak Wah Mak (Ontario Cancer Institute, Princess Margaret Hospital). 786-HRE-Luc cells were generated as described previously (Sufan et al, 2009). All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated foetal bovine serum (Sigma–Aldrich, Oakville, ON) at 37°C in a humidified 5% CO2 atmosphere. Patient-derived early-passage primary CCRCC cultured cells were collected from freshly excised human CCRCC tissue samples donated under UHN Research Ethics Board approval and cultured according to published methods (Park et al, 2004).
Antibodies
Anti-α-tubulin antibody was obtained from Sigma–Aldrich. Anti-vinculin antibody was obtained from Upstate (Lake Placid, NY). Anti-GLUT-1 and anti-hnRNP C1 + C2 antibodies were obtained from Abcam (Cambridge, MA). Anti-HA antibody (12CA5) was obtained from Roche Molecular Biochemicals (Laval, PQ). Anti-NEMO (IKK-γ) and NF-κB antibody (p65) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Chemicals
Chetomin, TNF-α and JSH-23 were obtained from Alexis Biochemicals (San Diego, CA), Upstate and Calbiochem (San Diego CA), respectively.
Immunoblotting
Western blotting was performed as described previously. In brief, cells were lysed in EBC buffer (50 mM Tris [pH 8.0], 120 mM NaCl, 0.5% NP-40) supplemented with a cocktail of protease and phosphatase inhibitors (Roche), boiled in sodium dodecyl sulphate (SDS)-containing sample buffer, and then size-fractionated by SDS–PAGE. Resolved proteins were then electrotransferred onto polyvinylidene difluoride membrane (Bio-Rad Laboratories, Hercules, CA), immunoblotted with the various antibodies, and visualized by chemiluminescence (Roche).
Subcellular fractionation
Biochemical fractionation of cells was performed as previously described (Watson et al, 2006).
Trypan Blue exclusion assay
Cells were seeded at a density of 100,000 cells per well in 6-well plates and incubated overnight. Cells were treated with either 100 ng/ml TNF-α or EMCV (at various MOI). At regular time intervals, live and dead cells were pooled and resuspended in serum-free DMEM. Cells were incubated with 0.4% Trypan Blue (Gibco/Invitrogen, Burlington, ON) for 5 min at room temperature. The total number of cells and the number of cells that incorporated the dye were counted in triplicate at each time point using a hemocytometer.
Flow cytometry detection of cytotoxicity
Cells were stained with Annexin V-FITC and propidium iodide using the Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences, Mississauga, ON) and analysed by flow cytometry on a FACSAria Cell-Sorting System (BD Biosciences).
RNA extraction and cDNA synthesis
RNA was isolated using RNease Mini kit (Qiagen, Mississauga, ON) and treated with DNase I (Ambion, Austin, TX) for 30 min to remove residual DNA. RNA concentration was determined by spectrophotometry, and 2.5 or 5 µg of total RNA from each sample was reverse transcribed by oligo dT priming. Reverse transcription was performed by incubating the RNA with 1 µl of 0.5 µg/µl oligo dT and DNase/RNase-free water (Sigma–Aldrich) to a total volume of 11 µl at 70°C for 10 min. Four microlitres of 5× First Strand Buffer, 2 µl of 0.1 M DTT, 1 µl of 10 mM dNTP and 1 µl of SuperScript II enzyme (all from Gibco/Invitrogen) were added to the reaction and the mixture was incubated for 1.5 h at 42°C. The SuperScript was then inactivated by incubating the samples at 70°C for 15 min. These cDNA samples were stored at −20°C until used in real-time PCR experiments.
Microarray and real-time semi-quantitative PCR
Triplicate cDNA samples derived from cells following the same treatment were pooled. Microarray analysis was performed using the Human Genome U133 Plus 2.0 Array according to the manufacturer's protocol and analysed using Microarray Analysis Suite 5.0 (MAS5.0) (Affymetrix, Santa Clara, CA). The gene expression data were deposited at the ArrayExpress database maintained by the European Bioinformatics Institute (accession number: E-MTAB-294). Real-time PCR reactions were run on 386-well plates on the ABI Prism 7900HT (Applied Biosystems, Foster City, CA). Primers were designed to sequences for the desired genes using Primer Express 2.0 software (Applied Biosystems). The real-time PCR reaction mixture contains the following: 1 µl of 10× PCR buffer, 0.6 µl of 50 mM MgCl2, 0.2 µl dNTP (10 mM each of dATP, dTTP, dCTP, dGTP), 0.2 µl primer mix, 0.025 µl Platinum Taq DNA polymerase, 0.3 µl SYBR green fluorescent dye, 0.2 µl ROX internal reference dye (all from Invitrogen) and 3.4 µl blue water for a total mixture volume of 6 µl per well. Ten nanograms of cDNA was loaded per reaction well in a total volume of 4 µl yellow water. Genomic DNA derived from human placenta was used to generate standard curves for each primer tested. The housekeeping gene, TBP (TATA box binding protein) was used to normalize cDNA loading.
Antiviral assay
Various CCRCC cells were plated at a density of 20,000 cells per well in 96-well plates. Cells were incubated overnight at 37°C and then treated with various dilutions of EMCV in 0.5% FBS containing DMEM. Cells infected with EMCV were incubated for various time points and then stained with 0.05% crystal violet dye to visualize live cells.
Chetomin antiviral assay
786-O or 786-VHL cells were plated in 6-well plates at a density of 100,000 cells per well and incubated overnight at 37°C. Cells were then treated with 50 nM chetomin for 18 h prior to RNA isolation for real-time PCR or for 16 h prior to challenge with EMCV at various MOIs. Cell viability was quantified by Trypan Blue exclusion assay at 18 h post-infection. The experiment was performed in triplicate.
JSH-23 antiviral assay
786-O cells were plated in 6-well plates at a density of 100,000 cells per well and incubated overnight at 37°C. Cells were then first stimulated with 10 µg/ml LPS for 1 h, followed by 1 h treatment with 10 µM JSH-23 prior to 18 h challenge with EMCV (MOI = 0.1). Cell viability was quantified by Trypan Blue exclusion assay at 18 h post-infection. The experiment was performed in triplicate.
Cobalt chloride antiviral assay
T98G cells were plated at a density of 20,000 cells per well in a 96-well plate and incubated overnight at 37°C. Cells were either untreated or treated with 0.3 nM cobalt chloride in 10% FBS DMEM for 16 h. The cells were then challenged with different dilutions of EMCV in 0.5% FBS DMEM while maintained in the corresponding concentration of cobalt chloride. Cells infected with EMCV were incubated for 24 and 40 h respectively and then stained with 0.05% crystal violet dye to visualize live cells.
Quantification of crystal violet stained cells
Cells stained with crystal violet were scanned using a SnapScan e50 (Afga) scanner and the resulting TIFF images were imported into ImageJ. The images were converted to 8-bit grey scale and the ‘integrated density’ of each well was measured. The ‘percentage of control’ values were calculated by dividing the raw ‘integrated density’ values by the ‘integrated density’ of corresponding untreated control wells. Nonlinear regression was performed on the resulting values using a sigmoidal dose–response model with variable slope equation in GraphPad Prism version 4.0b for Mac (Mac 05 version) (GraphPad Software, San Diego, CA).
Statistical analysis
Two-way ANOVA and unpaired two-tailed Student's t-test were performed for analysing comparative data between groups using GraphPad Prism software. A significance level of 0.05 was applied for all statistical analyses.
The paper explained
PROBLEM
Clear-cell renal cell carcinoma (CCRCC), the most common form of kidney cancer, is among the most resistant of cancers to radiation and chemotherapy. The inactivation of the VHL tumour suppressor protein, the principal negative regulator of the transcription factor known as HIF, causes the vast majority of CCRCC. There is no effective treatment for advanced CCRCC.
RESULTS
Emerging evidence has shown that HIF whose overexpression is intimately linked to poor prognosis potentiates the activity of a critical cellular survival pathway mediated by NF-κB. Taking advantage of this property, this report describes the selective killing of CCRCC cells by EMCV in a manner that is dependent upon activation of HIF and the NF-κB-mediated survival pathway. EMCV acts as an oncolytic virus and suppresses human CCRCC tumour growth in a mouse dorsal skin window-chamber model system.
IMPACT
This pre-clinical study proposes the use of EMCV as an oncolytic virus aimed at eradicating kidney tumours and potentially other tumours with elevated HIF and NF-κB pathways. Cancers that are particularly resistant to conventional cancer therapies may be paradoxically more susceptible to EMCV-induced killing since this virus exploits the very nature of cancer cells to evade cell death to exacerbate its own virulence.
Virus replication assay
786-O, 786-VHL, RCC4-MOCK or RCC4-VHL cells were plated in 6-well plates at a density of 100,000 cells per well and incubated overnight in 10% FBS DMEM at 37°C. Cells were treated with EMCV for 2 h. Following two washes with PBS, 10% FBS DMEM medium was replaced. Cells were harvested by scraping and were collected together with the supernatant at indicated time-points. Cells were lysed by freeze–thaw and vortexing. Fivefold serial dilutions of the samples were added onto L929 mouse fibroblast monolayers and incubated for 48 h before being stained with 0.05% crystal violet to quantify cytopathic effects and median tissue culture infective dose (TCID50).
Electron microscopy
Examination of the specimen was conducted after the cells had been washed three times with serum-free minimum essential medium, fixed with 2% (w/v) paraformaldehyde, 2.5% (w/v) glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4, and dehydrated in a series of graded ethanol concentrations. Prepared cells were morphologically observed under a Semeddx II scanning electron microscope (Hitachi).
RNA interference
786-O cells were plated in 24-well plates at a density of 30,000 cells per well. Oligofectamine (Invitrogen) was used to transfect 60 pmol of each siRNA in Opti-MEM I (Gibco/Invitrogen) according to the manufacturer's protocol. The following siRNAs were used: (1) siHIF2α-1: 5′-GAGACGCCAUCAUCUCUCUGGAUUU-3′ (sense) and 5′-AAAUCCAGAGAGAUGAUGGCGUCUC-3′ (antisense); (2) siHIF2α-2: 5′-CAGCAUCUUUGAUAGCAGUTT-3′ (sense) and 5′-ACUGCUAUCAAAGAUGCUGTT-3′ (antisense); and (3) siRNA Negative Control Duplex, Medium GC Duplex (Invitrogen). For NEMO knockdown, 786-O cells were plated in 6-well plates at a density of 100,000 cells per well. XtremeGene (Roche) was used to transfect 160 pmol of Accell SMARTpool siRNA NEMO (Dharmacon) in Opti-MEM I (Gibco/Invitrogen) according to the manufacturer's protocol.
Biotinylated HIF-α(ODD) peptide in vitro binding assay
WT or mutant HA-VHL proteins were in vitro translated in the presence of [35S]methionine using the TNT reticulocyte in vitro transcription/translation system (Promega, Madison, WI) and then mixed with 1 µg of biotinylated HIF1α[ODD-OH] or HIF1α[ODD(PA)] peptide for 30 min at 37°C. Mixtures were then incubated with Streptavidin A beads (Pierce Biotechnology, Rockford, IL) and rocked for 90 min at 4°C. Beads were washed 5× with NETN and bound HA-VHL proteins were resolved by SDS–PAGE and visualized by autoradiography.
Mouse dorsal skin-fold window chamber assay
All animal experiments were performed in accordance with the institutional animal care guidelines (University of Ottawa, ON, Canada). SCID mice were anaesthetized by intraperitoneal injection of a mixture ketamine and xylazine (80 and 5 mg/kg, respectively). Dorsal window chambers were installed as described previously (Algire & Legallais, 1949). 786-HRE-Luc cells (2.5 × 105) were implanted in the dermis on the right dorsal side of the mouse using an 18G syringe. A circular glass coverslip was positioned over the incision allowing visualization of the tumour. Intratumoural injections of EMCV or irradiated EMCV (300 J/cm2 for 90 s in a Spectrolinker chamber) were performed by injecting 5 µl of PBS-based virus suspension (2 × 108 virus particles/tumour).
Bioluminescence imaging
Bioluminescent imaging was performed with the IVIS™ Imaging System, composed of a high sensitivity cooled charge-coupled camera mounted in a light-tight box (IVIS 100, Caliper Life Sciences, Alameda, CA). For each imaging procedure, the mice were injected with a solution of D-luciferin potassium salt (Caliper Life Sciences) at a concentration of 5 mg/ml in saline injected intraperitoneally at a dose of 50 mg/kg at 10 min before the start of imaging. The animals were then placed in the light-tight imaging chamber 20 cm below the imaging optics. During image acquisition, a 2% isofluorane–air mixture was delivered through a nose cone, and the body temperature was maintained at 37°C by a heated stage. A white-light image was obtained first to provide an anatomical reference for the BLI image, which was then acquired with 10 s integration time. A false-colour image of the BLI photon counts was overlaid on the white-light image, and the total BLI signal from a region of interest over the window chamber including the tumour were determined.
Immunohistochemical staining
Sample preparation and immunohistochemical staining were performed as described previously (Evans et al, 2007).
Acknowledgments
This work was supported by the Canadian Cancer Society (18460). F.C.R. is a recipient of the German Research Foundation (DFG, Ro 3750/1-1). A.M.R. is a recipient of the Canadian Cancer Society Harold E. Johns Studentship Award. I.R.W. is a recipient of the Canadian Institutes of Health Research (CIHR) Canada Graduate Scholarship. C.G. is supported by a Royal Australasian College of Physicians CSL Fellowship and a National Health and Medical Research Council Overseas Biomedical Postdoctoral Fellowship. M.O. is a Canada Research Chair in Molecular Oncology.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author Contributions
Frederik C. Roos: Study design, acquisition of data, analyses and interpretation of data, manuscript preparation.
Andrew M. Roberts: Study design, acquisition of data, analyses and interpretation of data, manuscript preparation.
Irene I. L. Hwang: Study design, acquisition of data, analyses and interpretation of data, manuscript preparation.
Eduardo H. Moriyama: Acquisition of data, analyses and interpretation of data.
Andrew J. Evans: Acquisition of data, analyses and interpretation of data.
Stephanie Sybingco: Analyses and interpretation of data.
Ian R. Watson: Acquisition of data, analyses and interpretation of data.
Leticia A. M. Carneiro: Acquisition of data, analyses and interpretation of data.
Craig Gedye: Acquisition of data.
Stephen E. Girardin: Analyses and interpretation of data.
Laurie E. Ailles: Contributed important reagent.
Michael A. S. Jewett: Contributed important reagent.
Michael Milosevic: Analyses and interpretation of data.
Brian C. Wilson: Analyses and interpretation of data.
John C. Bell: Study design, analyses and interpretation of data.
Sandy D. Der: Study conception, study design, analyses and interpretation of data, manuscript preparation.
Michael Ohh: Study conception, study design, analyses and interpretation of data, manuscript preparation.
For more information
Array Express:
www.ebi.ac.uk/microarray-as/a/e
(accession nr E-MTAB-294)
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Aghi M, Martuza RL. Oncolytic viral therapies—the clinical experience. Oncogene. 2005;24:7802–7816. doi: 10.1038/sj.onc.1209037. [DOI] [PubMed] [Google Scholar]
- Algire GH, Legallais FY. Recent developments in the transparent-chamber technique as adapted to the mouse. J Natl Cancer Inst. 1949;10:225–253. incl 228 pl. [PubMed] [Google Scholar]
- An J, Rettig MB. Mechanism of von Hippel–Lindau protein-mediated suppression of nuclear factor kappa B activity. Mol Cell Biol. 2005;25:7546–7556. doi: 10.1128/MCB.25.17.7546-7556.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Ann Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, Wang GH, Senkevich TG, Alnemri ES, Moss B, et al. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:1172–1176. doi: 10.1073/pnas.94.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulakia CA, Chen G, Ng FW, Teodoro JG, Branton PE, Nicholson DW, Poirier GG, Shore GC. Bcl-2 and adenovirus E1B 19 kDA protein prevent E1A-induced processing of CPP32 and cleavage of poly(ADP-ribose) polymerase. Oncogene. 1996;12:529–535. [PubMed] [Google Scholar]
- Brewer LA, Lwamba HC, Murtaugh MP, Palmenberg AC, Brown C, Njenga MK. Porcine encephalomyocarditis virus persists in pig myocardium and infects human myocardial cells. J Virol. 2001;75:11621–11629. doi: 10.1128/JVI.75.23.11621-11629.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks MA, Ali AN, Turner PC, Moyer RW. A rabbitpox virus serpin gene controls host range by inhibiting apoptosis in restrictive cells. J Virol. 1995;69:7688–7698. doi: 10.1128/jvi.69.12.7688-7698.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bump NJ, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P, et al. Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science. 1995;269:1885–1888. doi: 10.1126/science.7569933. [DOI] [PubMed] [Google Scholar]
- Chiocca EA. Oncolytic viruses. Nat Rev. 2002;2:938–950. doi: 10.1038/nrc948. [DOI] [PubMed] [Google Scholar]
- Cohen HT, McGovern FJ. Renal-cell carcinoma. N Engl J Med. 2005;353:2477–2490. doi: 10.1056/NEJMra043172. [DOI] [PubMed] [Google Scholar]
- Demers GW, Johnson DE, Tsai V, Wen SF, Quijano E, Machemer T, Philopena J, Ramachandra M, Howe JA, Shabram P, et al. Pharmacologic indicators of antitumor efficacy for oncolytic virotherapy. Cancer Res. 2003;63:4003–4008. [PubMed] [Google Scholar]
- Evans AJ, Russell RC, Roche O, Burry TN, Fish JE, Chow VW, Kim WY, Saravanan A, Maynard MA, Gervais ML, et al. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol. 2007;27:157–169. doi: 10.1128/MCB.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Hershberger PA, Dickson JA, Friesen PD. Site-specific mutagenesis of the 35-kilodalton protein gene encoded by Autographa californica nuclear polyhedrosis virus: cell line-specific effects on virus replication. J Virol. 1992;66:5525–5533. doi: 10.1128/jvi.66.9.5525-5533.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang II, Watson IR, Der SD, Ohh M. Loss of VHL confers HIF-dependent resistance to vesicular stomatitis virus: role of HIF in antiviral response. J Virol. 2006;80:10712–10723. doi: 10.1128/JVI.01014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev. 2002;2:673–682. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neurooncology. 2005;7:134–153. doi: 10.1215/S1152851704001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Jang SK. Continuous heat shock enhances translational initiation directed by internal ribosomal entry site. Biochem Biophys Res Commun. 2002;297:224–231. doi: 10.1016/s0006-291x(02)02154-x. [DOI] [PubMed] [Google Scholar]
- Kim S, La Motte-Mohs RN, Rudolph D, Zuniga-Pflucker JC, Mak TW. The role of nuclear factor-kappaB essential modulator (NEMO) in B cell development and survival. Proc Natl Acad Sci USA. 2003;100:1203–1208. doi: 10.1073/pnas.0337707100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 2004;6:33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Laguna MP, Kummerlin I, Rioja J, de la Rosette JJ. Biopsy of a renal mass: where are we now? Curr Opin Urol. 2009;19:447–453. doi: 10.1097/MOU.0b013e32832f0d5a. [DOI] [PubMed] [Google Scholar]
- Laurie SA, Bell JC, Atkins HL, Roach J, Bamat MK, O'Neil JD, Roberts MS, Groene WS, Lorence RM. A phase 1 clinical study of intravenous administration of PV701, an oncolytic virus, using two-step desensitization. Clin Cancer Res. 2006;12:2555–2562. doi: 10.1158/1078-0432.CCR-05-2038. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Miao Y, Ni Y, Bosmans H, Yu J, Vaninbroukx J, Dymarkowski S, Zhang H, Marchal G. Radiofrequency ablation for eradication of renal tumor in a rabbit model by using a cooled-tip electrode technique. Ann Surg Oncol. 2001;8:651–657. doi: 10.1007/s10434-001-0651-y. [DOI] [PubMed] [Google Scholar]
- Moran JM, Moxley MA, Buller RM, Corbett JA. Encephalomyocarditis virus induces PKR-independent mitogen-activated protein kinase activation in macrophages. J Virol. 2005;79:10226–10236. doi: 10.1128/JVI.79.16.10226-10236.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins TA, Harder ZM, Korneluk RG, Holcik M. Distinct regulation of internal ribosome entry site-mediated translation following cellular stress is mediated by apoptotic fragments of eIF4G translation initiation factor family members eIF4GI and p97/DAP5/NAT1. J Biol Chem. 2003;278:3572–3579. doi: 10.1074/jbc.M206781200. [DOI] [PubMed] [Google Scholar]
- Ngoi SM, Chien AC, Lee CG. Exploiting internal ribosome entry sites in gene therapy vector design. Curr Gene Ther. 2004;4:15–31. doi: 10.2174/1566523044578095. [DOI] [PubMed] [Google Scholar]
- Norman KL, Lee PW. Not all viruses are bad guys: the case for reovirus in cancer therapy. Drug Discov Today. 2005;10:847–855. doi: 10.1016/S1359-6446(05)03483-5. [DOI] [PubMed] [Google Scholar]
- O'Shea CC. Viruses: tools for tumor target discovery, and agents for oncolytic therapies—an introduction. Oncogene. 2005;24:7636–7639. doi: 10.1038/sj.onc.1209035. [DOI] [PubMed] [Google Scholar]
- Park JG, Ku JL, Park SY. Isolation and culture of renal cancer cell lines. Methods Mol Med. 2004;88:111–119. doi: 10.1385/1-59259-406-9:111. [DOI] [PubMed] [Google Scholar]
- Pilder S, Logan J, Shenk T. Deletion of the gene encoding the adenovirus 5 early region 1b 21,000-molecular-weight polypeptide leads to degradation of viral and host cell DNA. J Virol. 1984;52:664–671. doi: 10.1128/jvi.52.2.664-671.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Ohh M. The von Hippel–Lindau tumor suppressor protein sensitizes renal cell carcinoma cells to tumor necrosis factor-induced cytotoxicity by suppressing the nuclear factor-kappaB-dependent antiapoptotic pathway. Cancer Res. 2003;63:7076–7080. [PubMed] [Google Scholar]
- Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, Salvesen GS, Pickup DJ. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme. Cell. 1992;69:597–604. doi: 10.1016/0092-8674(92)90223-y. [DOI] [PubMed] [Google Scholar]
- Roberts AM, Ohh M. Beyond the hypoxia-inducible factor-centric tumour suppressor model of von Hippel–Lindau. Curr Opin Oncol. 2008;20:83–89. doi: 10.1097/CCO.0b013e3282f310de. [DOI] [PubMed] [Google Scholar]
- Schepens B, Tinton SA, Bruynooghe Y, Beyaert R, Cornelis S. The polypyrimidine tract-binding protein stimulates HIF-1alpha IRES-mediated translation during hypoxia. Nucleic Acids Res. 2005;33:6884–6894. doi: 10.1093/nar/gki1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz EM, Badorff C, Hiura TS, Wessely R, Badorff A, Verma IM, Knowlton KU. NF-kappaB-mediated inhibition of apoptosis is required for encephalomyocarditis virus virulence: a mechanism of resistance in p50 knockout mice. J Virol. 1998;72:5654–5660. doi: 10.1128/jvi.72.7.5654-5660.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin CI, Horvat B. Current animal models: transgenic animal models for the study of measles pathogenesis. Curr Top Microbiol Immunol. 2009;330:111–127. doi: 10.1007/978-3-540-70617-5_6. [DOI] [PubMed] [Google Scholar]
- Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- Shen Y, Shenk TE. Viruses and apoptosis. Curr Opin Genet Dev. 1995;5:105–111. doi: 10.1016/s0959-437x(95)90061-6. [DOI] [PubMed] [Google Scholar]
- Shin HM, Kim MH, Kim BH, Jung SH, Kim YS, Park HJ, Hong JT, Min KR, Kim Y. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett. 2004;571:50–54. doi: 10.1016/j.febslet.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- Sufan RI, Moriyama EH, Mariampillai A, Roche O, Evans AJ, Alajez NM, Vitkin IA, Yang VX, Liu FF, Wilson BC, et al. Oxygen-independent degradation of HIF-alpha via bioengineered VHL tumour suppressor complex. EMBO Mol Med. 2009;1:66–78. doi: 10.1002/emmm.200900004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodoro JG, Branton PE. Regulation of apoptosis by viral gene products. J Virol. 1997;71:1739–1746. doi: 10.1128/jvi.71.3.1739-1746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- Wakimoto H, Fulci G, Tyminski E, Chiocca EA. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide's enhancement of viral oncolysis. Gene Ther. 2004;11:214–223. doi: 10.1038/sj.gt.3302143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- Watson IR, Blanch A, Lin DC, Ohh M, Irwin MS. Mdm2-mediated NEDD8 modification of TAp73 regulates its transactivation function. J Biol Chem. 2006;281:34096–34103. doi: 10.1074/jbc.M603654200. [DOI] [PubMed] [Google Scholar]
- Williamson JD, Reith RW, Jeffrey LJ, Arrand JR, Mackett M. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J Gen Virol. 1990;71:2761–2767. doi: 10.1099/0022-1317-71-11-2761. [DOI] [PubMed] [Google Scholar]
- Yang H, Minamishima YA, Yan Q, Schlisio S, Ebert BL, Zhang X, Zhang L, Kim WY, Olumi AF, Kaelin WG., Jr pVHL acts as an adaptor to promote the inhibitory phosphorylation of the NF-kappaB agonist Card9 by CK2. Mol Cell. 2007;28:15–27. doi: 10.1016/j.molcel.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.