

Abstract
The role for cellular prion protein PrPc in β-amyloid (Aβ) oligomer-induced synaptic impairment is a topic of great interest and some controversy. In this issue of EMBO Molecular Medicine Aguzzi and co-workers explore the contribution of PrPc to deficient long term potentiation (LTP) and soluble Aβ levels in an Alzheimer's disease mouse model and show that the role of prions in Aβ related toxicity is far from ‘black and white’ suggesting complex interpretations of the data available thus far.
Keywords: Amyloid-beta (Aβ), oligomers, Aβ receptor, prion protein (PrP), neurotoxicity
See related article in EMBO Mol Med (Calella AM et al (2010) EMBO Mol Med 2: 306–314)
Pathogenic amyloid formation is characteristic of several neurodegenerative disorders including Alzheimer's and Parkinson's disease, transmissible spongiform encephalopathies and others (Aguzzi & O'Connor, 2010). The prion diseases are propagated via conversion of the cellular prion protein PrPc into an abnormal β-sheet enriched isoform PrPSc (Aguzzi & O'Connor, 2010). In Alzheimer's disease (AD), β- and γ-secretases cleave the amyloid precursor protein (APP), resulting in the generation of Aβ peptides that aggregate in β-sheet enriched Aβ fibrils (De Strooper, 2010) and form the characteristic amyloid plaques in the brain of AD patients. Recent insights suggest that small oligomeric assemblies of Aβ, in contrast to monomeric and fibrillar species, are toxic for neuronal synapses, but the molecular targets of these assemblies and the mechanism of toxicity remain very controversial topics (Ashe & Zahs, 2010, see also supplemental data there). The main problem is that oligomeric Aβ assemblies are in a dynamic equilibrium with monomeric and fibrillar Aβ assemblies, implying that various biophysical parameters determine the relative abundance of different aggregation states. The dynamic nature of this process makes the definition of such toxic assemblies elusive and probably also explains why so many various direct and indirect interactions of Aβ peptides with membrane bound and intracellular proteins have been described (Ashe & Zahs, 2010).
One of the most spectacular candidates in the series of candidate receptors for these toxic assemblies is, without doubt, the prion protein (Lauren et al, 2009). Indeed, an interaction between Aβ and the prion protein suggests a potential common molecular substratum for the neurotoxicity seen in both diseases. Prion protein (PrP) was identified in an unbiased screening for receptors that could bind Aβ42 oligomers prepared according to a particular protocol to yield Aβ-derived diffusible ligands (ADDL) (Lambert et al, 1998). Such ADDLs are neurotoxic, interfere with LTP and are considered a more or less stable form among several toxic species along the Aβ aggregation pathway. The interactions between ADDLs and cellular PrP along with other Aβ binding molecules that might mediate AD pathogenesis are depicted in Figure 1. These oligomers failed to impair LTP in mouse hippocampal slices lacking PrP (Lauren et al, 2009) and the same authors have recently demonstrated that in an AD transgenic mouse model (APPswe/Psen1ΔE9) characterized by amyloid plaques formation and learning and memory deficits, deletion of the endogenous PrP gene prevented the development of the functional deficits despite unchanged levels of Aβ generation and Aβ deposition in their brains (Gimbel et al, 2010). The temptation to extrapolate these interesting findings towards real AD is obvious but it requires some caution as other researchers (Balducci et al, 2010) did not observe any protection in prion deficient animals with regard to acute memory impairments when injecting different Aβ oligomer preparations. Calella et al have now revisited this issue and investigated the potential role of PrP in Aβ neurotoxicity in an extensive series of elegant genetic experiments, crossing loss- and gain-of function PrP mouse strains with a transgenic AD mouse model (APPKM670/671NL/Psen1L166P). These authors do not find any significant modulation of LTP formation by the presence or absence of PrPc. The study intelligently explorates any genetic confounders that could be blurring the effects and rules out the possibility that PrPc is the direct mediator of the synaptotoxicity caused by Aβ in this model. They conclude, appeasingly ‘The hypothesis of PrPC being a crucial mediator of Aβ synaptotoxicity might be not universal’.
Figure 1.
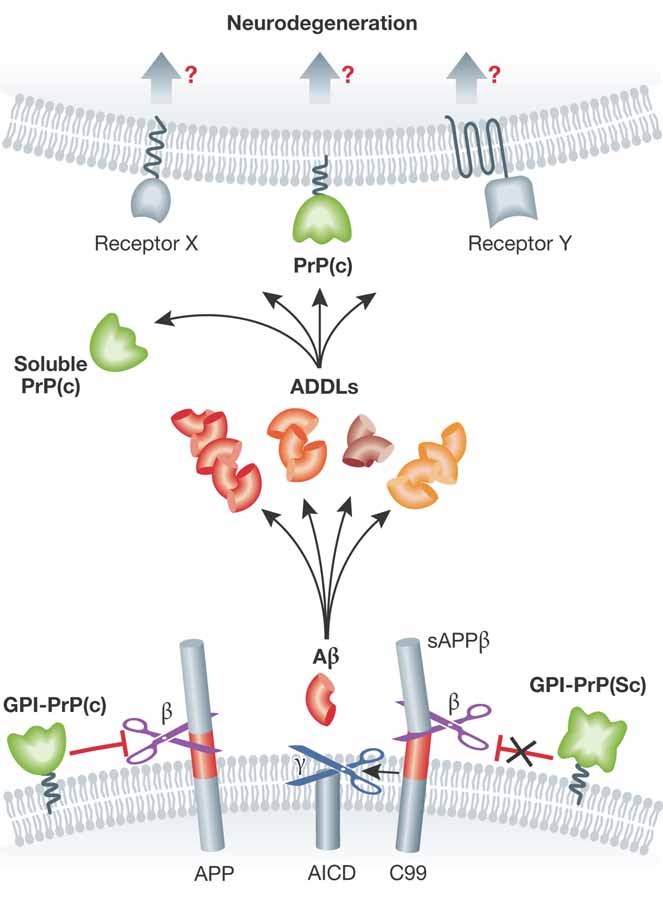
Cellular PrP in amyloid-beta induced neurodegeneration.
»Calella et al have (…) investigated the potential role of PrP in Aβ neurotoxicity in a series of elegant genetic experiments…«
»…PrP has a remarkable good affinity for Aβ peptides…«
The story is however far from finished. All groups involved agree that PrP has a remarkable good affinity for Aβ peptides tested in various conformations (Balducci et al, 2010; Calella et al, 2010; Lauren et al, 2009). Callela et al investigated the effects of expressing a soluble form of PrP (without its GPI-anchor) in their AD mouse model. In this case, whereas the levels of soluble and insoluble Aβ remain unchanged, LTP is less affected. Although seemingly contradictory with Lauren et al, the finding suggests that secreted PrP might interfere with Aβ mediated toxic pathways by directly binding to the peptide, not unlike the effect of Aβ antibodies in similar experiments. Whether such a protective effect is also observed with endogenously expressed (soluble) PrP remains obviously unaddressed.
»Other aspects of PrP and APP biology also suggest that the situation might be more complicated.«
Other aspects of PrP and APP biology also suggest that the situation might be more complicated. Parkin et al, 2007 showed for instance that cellular PrP can inhibit β-secretase-mediated cleavage of APP. The prediction that the lack of functional PrPc would lead to a rise in Aβ levels was confirmed by analysing the brain of PrP knock out mice (Parkin et al, 2007). However, the PrP gene is located close to a quantitative trait locus (QTL) for Aβ levels (Ryman et al, 2008) and comparing Aβ levels between inbred wild type and knock out strains might still be confounded by such genes closely linked to the targeted locus. Calella et al (2010) demonstrate in their paper how such QTL can dramatically alter Aβ levels over various generations.
Finally, while highlighting the PrPc interaction, Lauren et al (2009) have clearly shown that PrPc is not the only cell-surface molecule binding Aβ oligomers, as a high level of Aβ binding signals was still observed in Prnp−/− hippocampal neurons (50% compared to wild type). Furthermore, as discussed above, the in vivo generated Aβ oligomer pool is likely more complex than any in vitro generated Aβ oligomer mixture, and may therefore contain several ‘strains’ of toxic and less toxic conformers, somewhat resembling PrPSc (Aguzzi, 2008). Each of these conformations might act via different pathways. Therefore, and in conclusion, one cannot exclude that a remarkably high affinity of PrP to Aβ could be ascribed to a sub-pool of amyloid species, which is not necessarily the (most) toxic one.
Acknowledgments
The authors declare that they have no conflict of interest.
References
- Aguzzi A. Proc Natl Acad Sci USA. 2008;105:11–12. doi: 10.1073/pnas.0710824105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A, et al. Nat Rev Drug Discov. 2010;9:237–248. doi: 10.1038/nrd3050. [DOI] [PubMed] [Google Scholar]
- Ashe KH, et al. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balducci C, et al. Proc Natl Acad Sci USA. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calella AM, et al. EMBO Mol Med. 2010;2:306–314. doi: 10.1002/emmm.201000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Physiol Rev. 2010;90:465–494. doi: 10.1152/physrev.00023.2009. [DOI] [PubMed] [Google Scholar]
- Gimbel DA, et al. J Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, et al. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauren J, et al. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin ET, et al. Proc Natl Acad Sci USA. 2007;104:11062–11067. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman D, et al. Neurobiol Aging. 2008;29:1190–1198. doi: 10.1016/j.neurobiolaging.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]