

Abstract
β-Thalassemia is a common monogenic disorder due to mutations in the β-globin gene and gene therapy, based on autologous transplantation of genetically corrected haematopoietic stem cells (HSCs), holds the promise to treat patients lacking a compatible bone marrow (BM) donor. We recently showed correction of murine β-thalassemia by gene transfer in HSCs with the GLOBE lentiviral vector (LV), expressing a transcriptionally regulated human β-globin gene. Here, we report successful correction of thalassemia major in human cells, by studying a large cohort of pediatric patients of diverse ethnic origin, carriers of different mutations and all candidates to BM transplantation. Extensive characterization of BM-derived CD34+ cells before and following gene transfer shows the achievement of high frequency of transduction, restoration of haemoglobin A synthesis, rescue from apoptosis and correction of ineffective erythropoiesis. The procedure does not significantly affect the differentiating potential and the relative proportion of haematopoietic progenitors. Analysis of vector integrations shows preferential targeting of transcriptionally active regions, without bias for cancer-related genes. Overall, these results provide a solid rationale for a future clinical translation.
Keywords: gene therapy, haemoglobin, haematopoietic stem cells, lentiviral vectors, locus control region
→See accompanying Closeup by Milsom & Williams: http://dx.doi.org/10.1002/emmm.201000086
INTRODUCTION
β-Thalassemia is the most frequent monogenic disease with a global estimated annual birth incidence of 40,000/year, mainly in the Mediterranean, Middle East and Southern Asia countries (Modell & Darlison, 2008). The reduced or absent production of haemoglobin β-chains leads to severe anaemia, requiring regular blood transfusions (Weatherall & Clegg, 2001). As a consequence, the increased risk of viral infections and progressive iron accumulation can cause organs failure. Iron overload, although reduced by chelation, is not abolished and vital organs complications still occur affecting quality of life and representing the principal cause of death (Borgna-Pignatti et al, 2005). At present, the only definitive cure is allogeneic bone marrow transplantation (BMT), which is, however, available for a minority of patients. For all patients lacking a suitable bone marrow (BM) donor, gene therapy, as transplantation of autologous genetically corrected haematopoietic progenitor/stem cells, represents an attractive alternative to BMT, since is not limited by the histocompatibility barrier and does not require immunosuppression.
The clinical history of the disease and over 20 years of BMT experience indicates that even a mild correction of the globin chain imbalance in a fraction of maturing erythroblasts is sufficient to reduce the morbidity caused by ineffective erythropoiesis, to improve the clinical management of the disease and to increase the patients' life expectancy (Andreani et al, 2000; Lucarelli & Gaziev, 2008). Long term follow-up of transplanted patients characterized by persistent mixed chimerism demonstrates that a proportion of normal cells between 15 and 30% is sufficient to provide good quality of life and transfusion independence (Andreani et al, 2008). Our results in the murine model of thalassemia indicate that the in vivo selection of genetically modified erythroblasts results in long-term correction of the pathology in the presence of a limited number of transduced haematopoietic stem cells (HSCs) (Miccio et al, 2008). Therefore, it is predictable that even partial engraftment of genetically modified stem/progenitor cells would be therapeutic. So far, one report showed correction of the thalassemia major phenotype in human cells by a lentiviral vector (LV) carrying a transcriptionally regulated β-globin gene flanked by sequences from the chicken β-globin DNaseI hypersensitive site 4 insulator (cHS4) (Puthenveetil et al, 2004). Disappointingly, the inclusion of a large insert, such as the 1.2-kb cHS4, in 3′-long terminal repeat (LTR), causes inefficient viral RNA processing thus affecting the production of high titer viral stocks, required for clinical application (Hanawa et al, 2009; Urbinati et al, 2009). Recently, the results of the first trial of gene therapy in one patient were disclosed and the unexpected observation of a relative dominant haematopoietic clone, apparently as result of the vector integration, raised a notion of alarm and caution (Kaiser, 2009; Williams, 2009). At this time, it is too early to predict if this event would turn out to be a serious adverse event. Nevertheless, extensive preclinical studies of biology, efficacy and safety of LV-mediated globin gene transfer in human cells are mandatory to make a rigorous evaluation of a predictable successful trial.
CD34+ cells are the target of gene transfer and transplantation in gene therapy clinical protocols. These cells are a heterogenous population covering not only stem cells but also earlier multipotent progenitors and later lineage-restricted progenitors, and their relative proportion is related to the haematopoietic state (steady or stressed) (Bradford et al, 1997; Cheshier et al, 1999), the source (cord blood, BM and mobilized peripheral blood) (Fritsch et al, 1996; Kinniburgh & Russell) and the age (van Lochem et al, 2004). Thus, the effect of gene transfer on progenitors subsets equilibrium has to be evaluated to avoid skewing to specific cell types. Importantly, the investigation of the transcriptional response of CD34+ cells to gene transfer would allow both to predict biological and functional outcome and to define the best culture conditions to preserve the original features.
We developed the novel LV GLOBE, harbouring the human β-globin gene under the control of the minimal promoter and two elements from the locus control region (LCR), and demonstrated its therapeutic efficacy in long-term correction of murine β-thalassemia (Miccio et al, 2008). In the present study, we analysed the efficacy and safety of GLOBE-mediated gene transfer in haematopoietic progenitors isolated from BM aspirates of a large cohort (n = 44) of pediatric patients affected by β-thalassemia major, characterized by different genetic mutations. Our study includes an extensive molecular and biological characterization of the target cells, the optimization of the transduction protocol and the impact of this procedure on progenitor cells, the evaluation of gene transfer efficiency and efficacy and the mapping of proviral integrations in thalassemic CD34+ cells. To date this represents the most comprehensive preclinical analysis performed in thalassemia major patients' cells, whose results will pave the way forward the proposal of the clinical application of gene therapy using GLOBE LV.
RESULTS
Characterization of BM-derived CD34+ cells isolated from patients affected by thalassemia major
Patients affected by β-thalassemia major were enrolled starting from 2005 in the BM transplantation programme at H.S. Raffaele (HSR, Milan) and Mediterannean Institute of Hematology (IME Foundation, Rome). Pre-transplantation BM samples from a group of patients (n = 44, Table 1) were donated for this research study. The patients were children (age range 2–15 years, median = 8, 24 males and 20 females) of different geographic and ethnic origin, from countries in the Mediterranean area. Twenty-seven were β0-thalassemia patients (carrying the same or different allelic mutations), nine were homozygous for β+ mutations and eight compound heterozygous for β0 and β+ mutations.
Table 1.
Thalassemia major samples
| Sample | Sex | Age | Origin | Phenotype | Mutation | Sourcea |
|---|---|---|---|---|---|---|
| THAL 15 | F | 4 | Qwait | β0 | cod39/cod39 (C > T) | IME |
| THAL 20 | M | 6 | Palest. terr. | β0 | cod5/cod5 (−CT) | HSR |
| THAL 22 | M | 4 | Palest. terr. | β+ | IVS1-110/IVS1-110 (G > A) | HSR |
| THAL 23 | M | 5 | Palest. terr. | β+ | IVS1-110/IVS1-110 (G > A) | HSR |
| THAL 27 | M | 13 | Iraq | β0 | IVS1-1/IVS1-1 (G > A) | HSR |
| THAL 28 | F | 11 | Lebanon | β0 | cod5/cod5 (−CT) | HSR |
| THAL 33 | M | 11 | Iraq | β+ | IVS1-5/IVS1-5 (G > C) | IME |
| THAL 34 | F | 11 | Iran | β+ | IVS1-1 (G > A)/cod30 (G > C) | IME |
| THAL 35 | M | 5 | Iraqi Kurd | β0 | IVS1-1/IVS1-1 (G > A) | HSR |
| THAL 36 | F | 6 | Iraqi Kurd | β0 | IVS1-1/IVS1-1 (G > A) | HSR |
| THAL 37 | F | 10 | Iraqi Kurd | β0 | IVS2-1/IVS2-1 (G > A) | HSR |
| THAL 38 | F | 3 | Iraqi Kurd | β0 | IVS1-1 (G > A)/cod8/9 (+G) | HSR |
| THAL 39 | M | 9 | Iraqi Kurd | β0 | cod8/9/cod8/9 (+G) | HSR |
| THAL 40 | M | 13 | Iraq | β0 | IVS1-1/IVS1-1 (G > A) | HSR |
| THAL 41 | M | 6 | Kuwait | β+ | IVS1-5 (G > C)/cod36/37 (−T) | IME |
| THAL 42 | F | 14 | Syria | β+ | cod39 (C > T)/IVS1-6 (T > C) | IME |
| THAL 43 | F | 8 | Lebanon | β+ | IVS1-1 (G > A)/Lepore | IME |
| THAL 44 | F | 10 | Kuwait | β0 | cod44/cod44 (−C) | IME |
| THAL 45 | F | 10 | Palest. terr. | β+ | IVS1-6/IVS1-6 (T > C) | HSR |
| THAL 47 | M | 12 | Lebanon | β0 | IVS1-1/IVS1-1 (G > A) | HSR |
| THAL 48 | M | 8 | Lebanon | β+ | cod29/cod29 (C > T) | HSR |
| THAL 49 | M | 13 | Lebanon | β+ | IVS1-110/IVS1-110 (G > A) | HSR |
| THAL 51 | M | 15 | Lebanon | β+ | IVS1-110 (G > A)/cod44 (−C) | IME |
| THAL 52 | M | 12 | Lebanon | β+ | IVS1-110/IVS1-110 (G > A) | HSR |
| THAL 53 | F | 7 | Iraqi Kurd | β0 | cod8/cod8 (−AA) | HSR |
| THAL 54 | F | 3 | Iraqi Kurd | β0 | IVS1-1 (G > A)/cod8/9 (+G) | HSR |
| THAL 55 | F | 5 | Iraqi Kurd | β0 | IVS1-1 (G > A)/cod8/9 (+G) | HSR |
| THAL 56 | F | 4 | Iraqi Kurd | β+ | IVS1-6 (T > C)/IVS2-1 (G > A) | HSR |
| THAL 57 | M | 2 | Iraqi Kurd | β0 | IVS2-1 (G > A)/cod 8 (−AA) | HSR |
| THAL 58 | M | 10 | Iraqi Kurd | β0 | IVS2-1 (G > A)/cod 8 (−AA) | HSR |
| THAL 65 | F | 15 | Iraqi Kurd | β0 | IVS2-1 (G > A)/cod 8/9 (+G) | HSR |
| THAL 66 | F | 6 | Iraq | β+ | IVS1-110 (G > A)/cod8 (−AA) | IME |
| THAL 68 | M | 3 | Maldives | β0 | cod30/cod30 (G > C) | IME |
| THAL 75 | M | 9 | Iraqi Kurd | β0 | IVS2-1/IVS2-1 (G > A) | HSR |
| THAL 84 | F | 3 | Egypt | β0 | IVS1-1/IVS1-1 (G > A) | IME |
| THAL 87 | M | 8 | Egypt | β+ | IVS1-110/IVS1-110 (G > A) | IME |
| THAL 92 | M | 8 | Syria | β0 | cod39/cod39 (C > T) | HSR |
| THAL 94 | M | 5 | Syria | β0 | cod15 (G > A)/cod8 (−AA) | HSR |
| THAL 101 | M | 3 | Syria | β+ | cod39 (C > T)/IVS1-6 (T > C) | HSR |
| THAL 103 | F | 3 | Syria | β+ | −30/−30 (T > T) | HSR |
| THAL 104 | F | 4 | Syria | β0 | cod39/cod39 (C > T) | HSR |
| THAL 105 | F | 8 | Syria | β0 | IVS1-1/IVS1-1 (G > A) | HSR |
| THAL 106 | M | 7 | Syria | β0 | IVS1-1/IVS1-1 (G > A) | HSR |
| THAL 113 | M | 7 | Syria | β0 | cod8/cod8 (−AA) | HSR |
Palest. terr., Palestinian territories; Iraqi Kurd, Iraqi Kurdistan.
Source of the BM aspirates: HSR (S.Raffaele Hospital) or IME (Mediterranean Institute of Hematology).
We isolated CD34+ cells from the mononuclear cells (MNC) fraction of thalassemic BM samples (THAL, n = 30), to a yield of 0.22 ± 0.03 × 106 cells/ml (2.72 ± 0.36%/total MNC) that was comparable (p > 0.05) to that of normal donor samples (ND, n = 9, 0.16 ± 0.03 × 106 cells/ml; 1.44 ± 0.24%/total MNC). Fluorescence-activated cell-sorter scanner (FACS) analysis for the expression of surface markers, characterizing the multipotent, lymphoid and myeloid progenitors (Olweus et al, 1996; Terstappen et al, 1991; Tjonnfjord et al, 1994) shows that CD34+CD38−CD50+, CD34+CD10+ and CD34+CD33+ are 2.4 ± 1.1, 61 ± 4 and 35 ± 3.2%, respectively (Fig 2B). Erythroid hyperplasia is present in thalassemic BM as revealed by the increased percentages of Glycophorin A (GpA)+ cells in comparison to the ones observed in normal donor samples (59 ± 9% of total MNC, n = 7 (vs.) 8.2 ± 1.9%, n = 4, p < 0.01). Because of this erythroid expansion, we investigated the existence of an increased proportion of erythroid committed progenitors by analyzing CD34+GpA+, CD34+CD36+ and CD34+CD71+ cells. Figure 1A shows no significant differences (p > 0.05) between normal (0.7 ± 0.2%) and thalassemic (0.9 ± 0.1%) samples in the proportion of CD34+ cells co-expressing GpA. CD34+CD36+ cells are more abundant in normal than in thalassemic samples (8.6 ± 1.6% vs. 4.7 ± 1%, p = 0.05), without significant impact on the relative frequency of clonogenic progenitors (see below). CD71+ cells are significantly decreased (p < 0.001, 28 ± 3% vs. 64 ± 4%) in the CD34+ cell population of thalassemic samples in comparison to normal samples, suggesting a down-regulation of the transferrin receptor due to iron overload.
Figure 2. Effect of cytokines treatment on clonogenic activity and progenitors subpopulations of CD34+ cells.

- CFU activity of CD34+ cells and proportion of the different type of colonies (BFU-E, CFU-GM and CFU-GEMM) in thalassemia major samples untreated (THAL, n = 20), activated with cytokines (THAL-act, n = 19) and transduced with the GLOBE vector (THAL-GLOBE, n = 18).
- Cumulative data from FACS analysis of multipotent (CD34+CD38−CD50+), lymphoid (CD34+CD10+) and myeloid (CD34+CD33+) progenitors subpopulations in thalassemia major untreated (THAL, black bars, n = 3, 6 and 6, respectively) and activated (THAL-act, white bars, n = 3) samples. Values represent the mean ± SEM. Asterisks indicate parameters significantly different between groups (*p < 0.05; **p < 0.01).
Figure 1. Characterization of thalassemia major BM-CD34+ cells.

- Expression of erythroid lineage markers in BM-CD34+ progenitors: cumulative FACS data of the percentage of CD34+GpA+, CD34+CD36+ and CD34+CD71+ cells plotted for normal (ND, n = 5, 5 and 3, respectively) and thalassemic (THAL, n = 17, 8 and 9, respectively) samples.
- CFU activity of CD34+ cells analysed as total number of colonies/1000 cells plated and as proportion of the different type of colonies (BFU-E, CFU-GM and CFU-GEMM) in ND (n = 12) and THAL (n = 20) samples. Values are given as mean ± SEM. Asterisks indicate values significantly different between groups (***p < 0.001).
The property of individual early progenitors to give rise to burst-forming unit erythroid (BFU-E), colony-forming unit granulo-monocyte (CFU-GM) and colony-forming unit granulocyte–erythrocyte–monocyte–megakaryocyte (CFU-GEMM) colonies was analysed by colony-forming unit (CFU) assay. An equal number of clonogenic cells (52 ± 5 CFU/1 × 103 cells plated) is present in normal (n = 12) and thalassemic (n = 20) samples. Percentages of BFU-E, CFU-GM and CFU-GEMM in patients' samples are comparable to normal ones (49 ± 4%, 48 ± 4%, 0.9 ± 0.4% vs. 49 ± 5%, 50 ± 5% and 1.2 ± 0.4%, Fig 1B) indicating no significant alterations associated with the disease in the phenotype and differentiating potential of CD34+ cells.
Cytokines treatment of CD34+ progenitors induces minor changes in their phenotype and activity
We optimized a short-term gene transfer procedure consisting of a pre-stimulation step in the presence of a cocktail of cytokines, followed by transduction with the vector stock. In order to assess the impact of this treatment on CD34+ cells, we compared progenitors' phenotype and activity of untreated samples (THAL, n = 20) to the ones of samples activated by cytokines for 42 h (as duration of pre-stimulation and transduction) (THAL-act, n = 19), or transduced with the vector (THAL-GLOBE, n = 18). No significant difference (p > 0.05) is found in the number of clonogenic progenitors in THAL, THAL-act and THAL-GLOBE samples (52 ± 5, 61 ± 5 and 60 ± 5/1000 cells plated, respectively). Similarly, the proportion of BFU-E (49 ± 4%, 58 ± 3% and 58 ± 4%), CFU-GM (48 ± 4%, 39 ± 3% and 40 ± 3%) and CFU-GEMM (0.9 ± 0.4%, 0.7 ± 0.2% and 0.5 ± 0.1%) is not significantly modified by the exposure to cytokines and to vector stock (Fig 2A). Control experiments performed with normal donor cells reveal no difference in response to cytokine treatment between patients' and normal cells (Fig S1 of Supporting Information).
Analysis of the effect of cytokines on progenitor subpopulations shows no significant difference in the proportion of multipotent progenitors (CD34+CD38−CD50+) before and after stimulation (2.4 ± 1.1% vs. 2.1 ± 1.4%). Differently, a significant decrease in the proportion of lymphoid committed progenitors CD34+CD10+ (61 ± 4% vs. 38 ± 3%, p < 0.05), associated with a marked increase in the frequency of myeloid committed progenitors CD34+CD33+ (35 ± 3% vs. 65 ± 5%, p < 0.01) is observed in thalassemic cells stimulated with cytokines (Fig 2B). Importantly, no reduction in the total number of cells expressing the CD34 marker is evident. In normal donor samples there is a similar, but less pronounced, shift in the subpopulations of committed progenitors after activation (data not shown). Notably, the change in the relative proportion of committed progenitors, induced by cytokine exposure, is not associated to a significant alteration in the number and activity of early progenitor cells, scored by CFU assay.
Genome expression profiling of thalassemic and normal CD34+ cells and molecular response to cytokine activation
To understand the global impact of cytokine stimulation on the gene expression programme of CD34+ cells from thalassemic patients, we determined the expression profile by Affymetrix microarray analysis on RNA extracted from 20 thalassemic and 22 normal samples, before and after in vitro stimulation. An unsupervised hierarchical clustering analysis identified two main branches corresponding to unstimulated and stimulated samples, indicating that cytokine treatment had the strongest effect in discriminating between samples, overcoming disease status (Fig 3A). Indeed, thalassemic and normal samples clustered together, within each main branch. However, when five samples derived from pediatric donors were added to the unstimulated control data set, thalassemic and healthy pediatric samples clustered together, suggesting that age is a major factor in driving sample clustering at least in untreated CD34+ cells (Fig 3B). To define the genes consistently over- or under-expressed in normal versus thalassemic samples before and after cytokine treatment, we used a supervised hierarchical ordering approach. Less than 25 genes (0.1%) were expressed at significantly different levels in thalassemic versus normal CD34+ cells in either condition, after Bonferroni correction (Fig S2 A and B of Supporting Information). Overall, these results indicate that cytokine treatment induces major changes in the gene expression programme of CD34+ cells, with no substantial difference between cells obtained from thalassemic and healthy donors.
Figure 3. Genome expression profiling of thalassemic CD34+ cells and molecular response to cytokine activation.
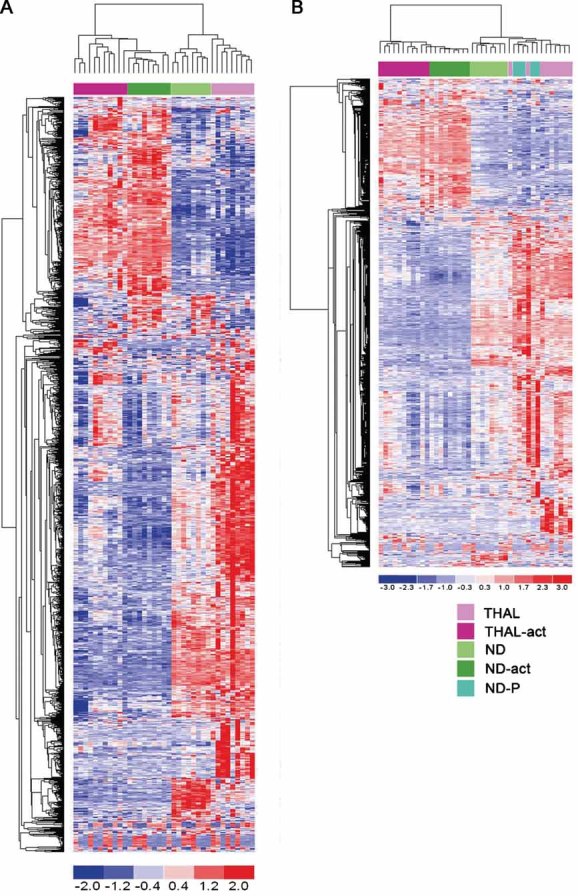
- Unsupervised cluster analysis of transcripts from thalassemic and normal cells untreated (THAL, n = 9; ND, n = 8) and activated with cytokines (THAL-act, n = 11; ND-act, n = 9).
- Unsupervised cluster analysis of transcripts from thalassemic and normal cells, untreated and activated with cytokines, including normal donor pediatric cells (ND-P, n = 5). In red overexpressed probe sets, in blue downregulated ones.
GLOBE transduces at high efficiency CD34+ cells from β0 and β+ patients leading to correction of haemoglobin A deficiency
Correction of β-thalassemia by gene therapy requires gene transfer in stem/progenitor cells and high level of β-globin gene expression in the differentiated erythroid progeny. To this aim, we utilized the GLOBE LV, recently described correcting thalassemia in the murine model (Miccio et al, 2008), and its derivative containing the woodchuck post-transcriptional regulatory element. BM-CD34+ cells from 22 thalassemia major patients (Table 1 of Supporting Information) were transduced with GLOBE (THAL-GLOBE) or maintained in medium containing the cytokines only (THAL). Gene transfer efficiency is variable, as expected from primary cells, but generally high, ranging from 37 to 95% with a mean value of 65%. Quantitative PCR (qPCR) performed on DNA extracted from bulk cultures at day 14 reveals an average vector copy number (VCN)/cell of 1.6 (normalized value 2.6) (Fig 4A). The same analysis, performed at clonal level in a high number of CFU (n = 213) grown in methylcellulose from transduced CD34+ cells, shows a Poisson-like distribution, with most of the colonies (83.6%) carrying a limited VCN (1–4) and a low frequency (2.35%) of colonies with high VCN (>9) (Fig S3 of Supporting Information). The association between VCN and the extent of disease correction, evaluated by FACS and high-performance liquid chromatography (HPLC) analysis of haemoglobin A (HbA) production, and morphological analysis of mature erythroblasts, is indicated for each sample in the Table S1 of Supporting information.
Figure 4. Transduction efficiency of thalassemia major CD34+ cells by GLOBE vector and correction of HbA deficiency in transduced erythroblasts.
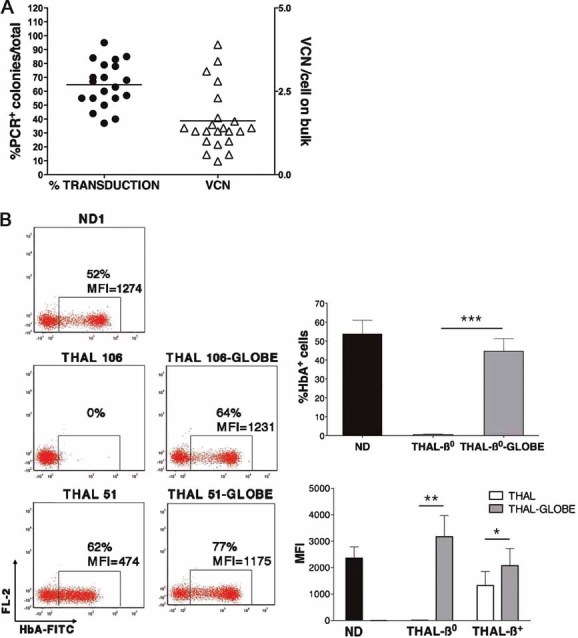
- Schematic representation of the percentage of transduction as determined by PCR on CFUs derived from transduced CD34+ cells, and of the average VCN/cell, as determined by qPCR on bulk erythroid culture. Single dots and triangles represent individual experiments.
- Left: Representative FACS analysis for HbA expression at 2 weeks of erythroid culture from normal (ND1), thalassemic (THAL 106, 51) and thalassemic GLOBE-transduced (THAL 106-GLOBE, 51-GLOBE) CD34+ cells after intracellular HbA staining. Right: Cumulative data from FACS analysis for HbA expression in erythroid cells. In the upper panel, the mean proportion ± SEM of HbA+ cells in normal (ND, black bar, n = 5), thalassemia-β0 (THAL-β0, white bar, n = 7) and thalassemia-β0-GLOBE-transduced (THAL-β0-GLOBE, grey bar, n = 7) cultures. In the lower panel, the MFI of HbA-expressing cells in the normal (ND, black bar, n = 5), thalassemic (THAL-β0 and -β+, white bars, n = 7 and 3) and thalassemic GLOBE-transduced (THAL-β0 and -β+, grey bars, n = 7 and 3) cultures. Asterisks indicate parameters significantly different between groups (*p < 0.05; **p < 0.01; ***p < 0.001).
To assess the efficacy of GLOBE to correct HbA deficiency, transduced and control cells (n = 10) were grown in erythroid unilineage culture, as in vitro modelling of erythropoiesis. The representative FACS dot-plot in Fig 4B (left panel) shows undetectable expression of HbA in a β0 sample (THAL 106) and residual HbA production, revealed by low mean fluorescence intensity (MFI), in a β+ sample (THAL 51). In normal erythroid cultures (ND, n = 5), the proportion of HbA+ cells reaches a maximum of 74% depending on the amount of mature cells. Following transduction with GLOBE the proportion of HbA+ cells in cultures from thalassemic patients achieves a level comparable to the normal ones (44.6 ± 6.7% vs. 53.8 ± 7.2%, MFI 3170 ± 802AU vs. 2360 ± 429AU) (Fig 4B, right panel). A statistically significant increase in the amount of HbA (2076 ± 647AU vs. 1323 ± 429AU, p < 0.05) is observed in transduced samples from β+ patients (THAL-β+). At clonal level, the HbA expression pattern in BFU-Es, isolated from five independent transduction experiments, is comparable to that observed in normal colonies (Fig S4 of Supporting Information).
Vector-derived β-globin synthesis provides correction of β/α chains imbalance in erythroid cells
Since the imbalance between α- and β-globin chains is the main cause of erythroid precursors death leading to ineffective erythropoiesis and anaemia, the evaluation of the newly synthesized globin chains provides the only quantitative measurement of the correction of the β/α ratio. Therefore, we performed reverse phase-HPLC analysis of radiolabelled protein extracts from erythroid cultures at day 14. As reported in Fig 5, this analysis shows absence of β-chain synthesis in β0 cells and low level of synthesis in β+ cells, resulting in a β/α ratio of 0.14. In these experiments, the β/α ratio in control normal cells (ND1 and 2) is 0.97 and 0.79, respectively. Restoration of β-chain synthesis in transduced β0 cells, revealed by the appearance of a distinct peak in Fig 5 (lower left panel), results in a β/α ratio of 0.30. Similarly, this value is increased up to 0.35 in genetically modified β+ cells (Fig 5, right panels). Both these values are in the range of those observed in β-thalassemia carriers (Giordano et al, 1999), indicating that gene transfer with GLOBE in thalassemic cells provides transgene expression at therapeutic level.
Figure 5. Reverse phase HPLC analysis of radiolabelled globin chains synthetized in erythroblastic culture.

HPLC profile of globin chains obtained from lysates of 15 × 106 erythroid cells after 14 days of liquide culture started from normal (ND1 and ND2), thalassemic (THAL 35 = THAL-β0 and THAL 51 = THAL-β+) and thalassemic GLOBE-transduced (THAL β0-GLOBE or THAL β+-GLOBE) BM CD34+ cells after metabolic incorporation of radiolabelled leucine. Neo-synthetized single globin chains (α, β, δ, Aγ and Gγ) are indicated on each corresponding peak.
Restoration of effective erythropoiesis by GLOBE-transduced cells
Differential counting of thalassemic cells at day 14 of the erythroid culture shows a block in the differentiation, indicated by reduced progression to the orthochromic normoblast stage and increased cell death, occurring particularly at the stage of polichromatophilic normoblast. In contrast, THAL-GLOBE cells differentiate to orthochromic erythroblasts and eventually to reticulocytes similarly to normal cells (Fig 6A). The relative proportion of proerythroblasts, basophilic, polichromatophilic, orthochromic normoblasts and reticulocytes in GLOBE-transduced samples are comparable to those of normal samples (1.0 ± 0.6%, 5.5 ± 1.6%, 17.3 ± 3.6%, 60.1 ± 4.4%, 6.9 ± 3.3% vs. 0.5 ± 0.5%, 4.8 ± 0.7%, 25.6 ± 2.2%, 55.2 ± 5.5%, 6.4 ± 4.6%, respectively). In thalassemia major control samples there are a high frequency of dead cells (21.7 ± 4.4%), few reticulocytes (0.4 ± 0.2%) and a significantly reduced (p < 0.01) proportion of late normoblasts (orthochromic, 44.8 ± 6.3%) with respect to both normal and transduced samples (Fig 6B). According to these data, analysis of apoptosis and cell death in thalassemic cultures reveals a high proportion of GpA+ cells co-expressing the apoptotic marker Annexin-V (33.5 ± 5.4%) and stained positive after PI exposure (18.1 ± 3.1%). Differently, the frequency of apoptotic and late apoptotic/dead cells in transduced samples is significantly reduced (17.3 ± 3.1% and 8.9 ± 2.9%, p < 0.05) and comparable to that observed in normal control (13.80 ± 0.5% and 7.6 ± 1.6%) (Fig 6C and D). These results indicate that thalassemic cells transduced with GLOBE are able to overcome the arrest in erythroid maturation and progress to normal erythropoiesis.
Figure 6. Correction of ineffective erythropoiesis.

- Photographs of May Grünwald–Giemsa-stained cytospin of erythroid cells from normal donor (ND), β-thalassemia (THAL) and GLOBE-transduced β-thalassemia (THAL-GLOBE) samples after 2 weeks of erythroid liquid culture (Microscope: Olympus Provis AX70. Objective: 100×/1.30 oil).
- Percentage cell number for each erythroblasts population after differential counting. ND n = 4, THAL n = 7, THAL-GLOBE n = 7.
- Representative FACS analysis of GpA+ erythroid cells for apoptotic/dead cells using Annexin V or propidium iodide labelling. Percentage of double positive cells on the total is reported in the panel.
- Percentage of apoptotic (GpA+ Annexin V+) or late apoptotic/dead (GpA+ PI+) erythroid cells at day 14 of culture. ND n = 2, THAL n = 3, THAL-GLOBE n = 3 THAL vs. THAL-GLOBE; *p < 0.05.
GLOBE integration preferences in CD34+ cells from thalassemic patients
To gain insight on the integration preferences of the GLOBE vector, we transduced 1 × 106 CD34+ cells from four thalassemic patients at an MOI of 100, after 30 h of pre-stimulations with cytokines, as described above. Cells were cultured for 14 days (five to six cell doublings) to dilute unintegrated lentiviral genomes, and vector-genome junctions cloned and sequenced by linker-mediated (LM) PCR, as previously described (Cattoglio et al, 2007). No cell selection was expected to occur in this short culture period. A total of 403 unique integration sites (106 from THAL 20, 136 from THAL 37, 38 from THAL-40 and 123 from THAL 41; Table S2 of Supporting Information) were mapped on the human genome and annotated as intergenic, intragenic and transcription start site (TSS) proximal as indicated in Fig 7A. A collection of 438 integration sites of a LV expressing green fluorescent protein (GFP) under a CMV promoter (Felice et al, 2009) and of 385 random control sequences (Cattoglio et al, 2007) were used as comparison. The overall distribution of GLOBE and CMV-GFP integrations showed the same over-representation of intragenic (57.3% vs. 58.9%, p > 0.05) and under-representation of intergenic (32.0% vs. 31.8%, p > 0.05) sites compared to the random controls (35.1% intragenic and 59.5% intergenic, p < 0.001 in all comparisons), which essentially reflect the gene content of the human genome (Fig 7A). For both vectors, TSS-proximal integrations were only slightly over-represented compared to random controls (10.9 and 9.1% vs. 5.5%, p < 0.01 and p > 0.05, respectively). Importantly, GLOBE showed the same low tendency of CMV-GFP to integrate at recurrent sites (hot spots) in the CD34+ cell genome (6.4% vs. 5.9%, p > 0.05), identifying 12 hot spots in both cases (Table S3 of Supporting Information). Hot spots were defined according to standard criteria, i.e. at least two integrations in <30 kb, three in <50 kb and four in <100 kb (Wu et al, 2006). It is worth noting that 1.6% of random sites met the same criteria (three hot spots), defining a background level of false positivity. The hot spots lists contain three cancer-related genes for GLOBE (NBN, NF1 and RFX2) and one for CMV-GFP (UHRF2) and random controls (ESR1). Although these numbers are too small to draw statistically significant conclusions, there appears to be no tendency for either LV to integrate into proto-oncogenes, as observed for retroviral vectors derived from the Moloney murine leukaemia virus (Cattoglio et al, 2007).
Figure 7. Distribution of the GLOBE vector integration sites in the human genome and correlation with gene activity in CD34+ cells.
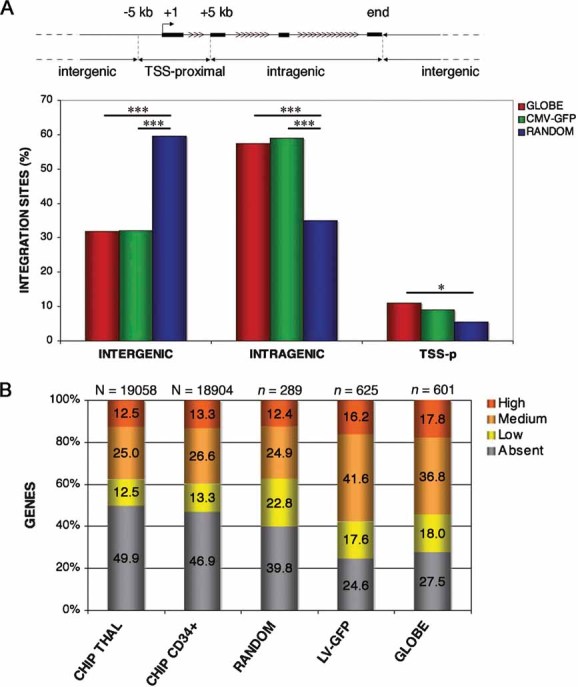
- GLOBE vector integration sites (red bars) in the genome of BM-CD34+ cells from thalassemic patients. LV-CMV-GFP integrations (LV-GFP) and random genomic sequences originated by LM-PCR (RANDOM), are used as control (green and blue bars, respectively). Intergenic, intragenic and TSS-proximal integrations are defined as represented in the scheme. Asterisks indicate frequency significantly different between groups (*p < 0.05; ***p < 0.001).
- Distribution of target genes in the different expression level categories. The first two bars (CHIP THAL and CHIP CD34+) show the distribution of the genes on the microarray of thalassemic and normal CD34+ cells activated with cytokines, the third bar (RANDOM) represents random control sequences, the fourth and fifth bars (LV-GFP and GLOBE) report the expression values of genes targeted by control LV-CMV-GFP and GLOBE integrations, respectively. The N-value indicates the number of probesets, the n values indicate the number of genes analysed.
To correlate vector integration with gene activity, we determined the expression level of all genes located in an arbitrary ±60-kb window around each GLOBE, CMV-GFP and random integration site. For this analysis, we used Affymetrix expression profiles of CD34+ cells isolated from three different thalassemic patients (for GLOBE integration sites) or from three normal donors (for LV-GFP and random sites) and activated in culture in the same conditions used for transduction. Average gene expression values were divided in four classes, i.e. absent, low, intermediate and high (see Materials and Methods Section). The analysis showed that the GLOBE vector integrates preferentially within or close to genes active in CD34+ cells at the time of transduction (73%), with a significant over-representation compared to random controls (60.2%, p < 0.001) or to the overall distribution of gene expression values in thalassemic CD34+ cells (53.1%, p < 0.001) (Fig 7B). The control LV-GFP vector showed a virtually identical preference for expressed genes (75.4% vs. 72.5%, p > 0.05) and a very similar distribution in the four gene expression classes, with no specific preference for highly expressed genes (16.2% vs. 17.8%, p > 0.05). Overall, these results indicate that the presence of β-globin transcriptional regulatory elements in the GLOBE vector does not influence its general integration preferences when compared to a conventional SIN-LV vector.
DISCUSSION
The development of HIV-derived vectors and the optimization of HSC transduction conditions has provided a significant contribution to the field of gene therapy for β-thalassemia, leading to the application of LVs expressing the human β-globin gene in preclinical murine models and in human thalassemic cells (Malik et al, 2005; Sadelain et al, 2007). The unique feature of β-globin LVs, carrying the transgene expressed by a combination of large regulatory elements derived from the globin locus, greatly affects the production of high-titer viral stocks and the transduction efficiency of target cells. Moreover, a delicate balance must be achieved to reconcile the needs for high efficiency of transduction of a considerable number of stem/progenitor cells, obtainable by cytokine-mediated stimulation and expansion and the maintenance of their biological features. Therefore, the characterization of transduced cells is crucial to predict a favourable outcome of the overall procedure. We characterized the BM-derived CD34+ cell population, which represents the preferential target of gene therapy clinical trials for genetic diseases involving children (Aiuti et al, 2009; Gaspar et al, 2004; Hacein-Bey-Abina et al, 2002), by analyzing the phenotype, clonogenic capacity and changes induced by the transduction procedure. BM will represent the most suitable source of haematopoietic progenitors for autologous transplantation in pediatric thalassemic patients in future gene therapy trials. Indeed, BM harvest is a safe procedure in children (Buckner et al, 1984), while cytokine-mobilization in thalassemic patients poses some safety concerns, due to the chronic hypercoagulable state (Taher et al, 2008) and the condition of splenomegaly often associated with the disease. We found that the frequency of multipotent progenitors and lineage-committed precursors in thalassemic BM is comparable to that in normal samples, and is not affected by the transduction procedure. The phenotype analysis for expression of surface markers characterizing the multipotent, common lymphoid and myeloid progenitors reveals that short exposure to cytokines, present in the preactivation/transduction medium, favours the expansion of the myeloid subpopulation against the lymphoid one. Experiments in normal donor samples give comparable results and the change in the relative proportion of committed progenitors is not reflecting a significant alteration in the number and quality of early progenitors, scored by clonogenic assay. Considering that, in the setting of gene therapy, the haematopoietic reconstitution will occur in the absence of immunosuppression, a decreased number of lymphoid progenitors in the transplant is not likely to compromise the outcome. Indeed, no evidence of immunological failure was reported in gene therapy clinical trials for genetic diseases. Moreover, in the presence of stressed erythropoiesis and erythroid expansion, like in thalassemia major BM, it is relevant to test the proportion of erythroid committed progenitors. Differently from previous results (Mathias et al, 2000), by analyzing a large number of samples we show normal expression of the erythroid differentiation markers and clonogenicity. Differences in patients' age and clinical status at the time of marrow harvest could explain these findings. Moreover, gene expression profiling of cytokine-treated and -untreated CD34+ cells suggests that the effect of the transduction is similar between patients and healthy donors. Nevertheless, the nature of specific genes that are differentially expressed upon cytokine treatment might suggest a specific reponse in thalassemic cells. Indeed, analysis of specific pathways relevant for HSC biology and activity is in progress. However, the best prediction for repopulating activity of human cells comes from transplantation of high number of CD34+ cells in immunodeficient mice. The source of cells for this kind of experiment would come only from back-up, harvested from patients before transplantation, and we are planning these experiments for the future.
Recently, we achieved long-term correction of thalassemia in the th3 murine model using the novel GLOBE vector (Miccio et al, 2008). Following on from this successful result, the exploitation of the therapeutic efficacy of GLOBE was investigated in BM-derived CD34+ cells. We have shown correction of hallmark features of the β-thalassemia phenotype, such as synthesis of HbA and ineffective erythropoiesis. Analyses of erythroid cultures demonstrate that GLOBE is able to efficiently transduce CD34+ cells, providing physiological levels of HbA in the erythroid progeny with a relatively low number of integrants per cell, in line with our results in the murine model (Miccio et al, 2008). The beneficial effect of transgene expression results in the increased proportion of mature erythroblasts in comparison to untransduced controls. Importantly, HPLC analysis of newly synthesized globin chains reveals that α/β chain ratio is in the range of that reported in literature by the analysis of β-thalassemia carriers (Giordano et al, 1999), indicating that gene transfer with GLOBE provides transgene expression at therapeutic level. Successful results in patients' cells were previously achieved by transduction with a β-globin LV contained almost all LCR sites (HS 2, 3 and 4), in addition to the 3′ enhancer and the 1.2 kb cHS4 insulator (Puthenveetil et al, 2004). However, as reported in recent studies, large insulator elements negatively affect vector titer and stability of viral particles, thus limiting the scale-up production for clinical application (Hanawa et al, 2009; Urbinati et al, 2009). Recent work on smaller sequences from cHS4 gave promising results in murine cells (Arumugam et al, 2009). So far, efficacy data obtained with GLOBE in a large and heterogenous group of patients' samples strengthen the therapeutic potential of transcriptionally regulated globin LVs for future clinical application.
An important issue to address is the prediction of the safety of a gene therapy approach (Nienhuis et al, 2006). In contrast to RVs, LVs appear to integrate in the host genome throughout the transcriptional unit without preference for TSSs or promoters (Bushman et al, 2005), and are therefore associated to a lower risk of insertional activation of cellular genes by transcriptional mechanisms. Data from the first trial using LV in two patients affected by adrenoleukodystrophy are reassuring in terms of safety, with no evidence of expansion of specific transduced clones (Cartier et al, 2009). Moreover, vectors carrying β-globin promoters and LCR elements restrict transgene expression to the differentiated progeny within a single lineage, thereby reducing the risk of activating oncogenes in haematopoietic stem and progenitor cells. Our integration site analysis shows that GLOBE has integration preferences virtually indistinguishable from those of any other LV. GLOBE integration sites were associated with transcriptionally active genes, evenly distributed among low to high expression categories. Integrations hot spots are less frequent compared to RVs, and cancer-related genes do not appear to be over-represented compared to controls. We found that most of the target genes are maintained transcriptionally active in the genome of differentiating erythroblasts at day 7 and day 14 (unpublished results). Overall, these findings suggest the presence of a favourable chromatin context for transgene expression around GLOBE integration sites and no specific risk for the GLOBE vector. Notably, it was recently published that the epigenetic changes in the transgene promoter can be modulated by the presence of insulator elements in the vector (Arumugam et al, 2009), thus representing a tool for further improvement of transgene expression.
In conclusion, our results demonstrate the efficacy of a gene therapy approach for β-thalassemia by transduction of human progenitor cells with GLOBE vector and set the basis for a future clinical trial.
MATERIALS AND METHODS
Human subjects
Patients affected by thalassemia major or transfusion-dependent thalassemia intermedia were enrolled in the BMT programmes at H.S. Raffaele and IME Foundation. The diagnosis was based on family history, transfusion dependence, analysis of globin chains synthesis by HPLC and molecular analysis of mutations. BM aspirates performed for the clinical purposes of pre-transplant marrow evaluation were collected, following an hypertransfusion regimen, under general anesthesia. BM and blood samples (5–10 ml) were obtained from 32 patients at HSR and 12 patients at IME Foundation (age range 2–15 years). Normal CD34+ cells were either isolated from BM of healthy individuals or purchased from Lonza Inc (Walkersville, MD). All samples were obtained after informed consent from patients or legal guardians and with the approval of Institutional Ethical Committees.
Genetic screening for globin mutations
Genomic DNA was isolated from peripheral blood leukocytes using the Gentra Puregene Blood Kit (Qiagen). Characterization of mutations was performed by DNA sequencing of the β-globin gene on the ABI Prism 310 Genetic Analyzer (PE Applied Biosystems).
Isolation and characterization of BM-CD34+ cells
Mononuclear cells were isolated from BM aspirates by Ficoll density separation and CD34+ cells were selected using anti-CD34 microbeads (Miltenyi Biotech). CD34+ cells labelled with fluorescein isothiocyanate (FITC) or phycoerythrin (PE)-coniugated anti-CD34 antibodies (BD Biosciences—Pharmingen) were analysed for the expression of other markers using the following antibodies: PE-conjugated anti-GpA (DakoCytomation), anti-CD71 (Immunotech), anti-CD33 (CALTAG), FITC-conjugated anti-CD36, anti-CD50 (BD Biosciences—Pharmingen), anti-CD10 and tri-color anti-CD38 (CALTAG). Analyses by FACS were performed using FACSCanto flow cytometer (Becton Dickinson).
CFU assay
CD34+ cells were plated at 1 × 103 cells/ml in methylcellulose medium (GFH4434, Stem Cell Technologies). BFU-E, CFU-GM and CFU-GEMM colonies were scored after 14 days.
Virus production
Viral stocks were produced and titered as described (Miccio et al, 2008) by transduction of HEL cells with serial dilution of vector stocks followed by qPCR after 3 weeks of culture to allow dilution of unintegrated vector below detection level. VCN/cell was measured by qPCR on genomic DNA, using primers and probe annealing to RRE region: forward primer 5′-TGAAAGCGAAAGGGAAACCA-3′, reverse primer 5′-CCGTGCGCGCTTCAG-3′ and probe 5′-VIC-AGCTCTCTCGACGCAGGACTCGGC-MGB-3′. Titers were espressed as transforming units (TU)/ml and calculated multiplying the VCN/cell to the number of transduced cells and then dividing for the viral vector dilution. Vector particle was measured by HIV-1 gag p24 test (NEN Life Science Products) and vector infectivity was calculated as the ratio between titer and vector particle.
Transduction of human CD34+ cells
CD34+ cells were seeded at 0.5–1 × 106 cells/ml and pre-stimulated for 30 h in CellGro medium (CellGenix) supplemented with 300 ng/ml, human stem cell factor (hSCF) 300 ng/ml, human Flt3-ligand (hFlt3-l), 100 ng/ml human trombopoietin (hTPO) and 60 ng/ml human Interleukin-3 (hIL-3) (all PeproTech) on plates coated with retronectin (Takara Shuzo). Transduction was performed overnight at multiplicity of infection (MOI) of 50–100 and at a viral concentration of 5 × 107 trasforming units (TU)/ml.
The paper explained
PROBLEM
β-Thalassemia major or Cooley's anaemia, leads to a profound anaemia and to death in the first year of life, unless regular transfusions are administered. So far, allogeneic bone marrow transplantation (BMT) from HLA-matched donors is the only curative treatment, but it is limited to less than 25% of patients. Gene therapy, based on autologous transplantation of genetically corrected hematopoietic stem cells (HSCs), represents a promising alternative for patients lacking a suitable donor. We have previously developed a lentivirus vector (LV), GLOBE, coding for the human beta globin gene and showed that transplantation of GLOBE-transduced HSCs corrects thalassemia in murine models. To translate these results into the clinics, it is now essential to demonstrate the feasibility, safety and therapeutic efficacy of this approach by testing the GLOBE LV in the context of human thalassemic cells.
RESULTS
CD34+ cells purified from BM aspirates of thalassemia major patients were transduced with GLOBE and induced to differentiate to the erythroblastic lineage. The procedure restored haemoglobin production and erythropoiesis overcoming the β-thalassemia phenotype. We sequenced and mapped the GLOBE integration sites in the human cells to assess the risk of integration in potentially dangerous loci and showed that GLOBE integrates at low copy number into the human genome without preferential targeting of proto-oncogenes, tumor-supressor or cell cycle related genes.
IMPACT
This study provides solid preclinical data of efficacy and safety of the GLOBE-based gene therapy approach to β-thalassemia for its future clinical translation in the context of an acceptable risk/benefit ratio.
Erythroid liquid culture
CD34+ cells were cultured for 14 days using a procedure modified from previous reports (Giarratana et al, 2005; Migliaccio et al, 2002). The cells were seeded at 105 cells/ml in StemSpan medium (Stem Cell Technologies) containing 20% foetal bovine serum (FBS) (Hyclone) and supplemented with hSCF (10 ng/ml), human erythropoietin (Epo) (1 U/ml), hIL-3 (1 ng/ml), 10−6 M dexamethasone (Sigma) and 10−6 M β-estradiol (Sigma). At day 8, cells were grown in medium with 10% FBS and 2 U/ml Epo, and at day 11 they were cultured with 10% FBS only. Globin production was tested by FACS and HPLC analysis. Differential counting was performed on cells stained with May Grünwald–Giemsa reagent. Apoptotic and dead cells were revealed by flow cytometry using the Apoptosis Detection Kit I (BD).
Analysis of HbA and globin chains production
The proportion of erythroid cells expressing HbA was assessed by flow cytometry as described (Bohmer, 2001). 15 × 106 cells from erythroid cultures were incubated for 1 h in 3H-leucine (Perkin Elmer) in depleted Dulbecco's Modified Eagle medium (DMEM) (MP Biomedicals) and lysed for reverse phase-HPLC analysis (Galanello et al, 1998).
DNA analysis
Genomic DNA was extracted using the QIAmp DNA mini Kit (Quiagen) or the ChargeSwitch Forensic DNA Purification Kit (Invitrogen) and the average VCN/cell was measured as described (Miccio et al, 2008). Transduction efficiency was determined by PCR on CFUs, amplifying the HS2-HS3 sequence (forward primer HS2F 5′-GTTGGAGGATACCCATTCTCTATCT-3′ and reverse primer HS3R 5′-TGGGTCAGTGGTCTCAATGTAGCA-3′; PRIMM, Italy).
Sequencing and mapping of integration sites
CD34+ cells were transduced with GLOBE and cultured for 14 days to dilute unintegrated vector. Integration sites were cloned as described (Cattoglio et al, 2007) and mapped onto the human genome (UCSC HG18, Mar.2006) using BLAT requiring a 95% identity. Sequences were annotated as reported (Felice et al, 2009). A genomic region was defined as a ‘hot spot’ for integration according to criteria developed for defining cancer-related common integration sites (CISs) with minor modifications (Cattoglio et al, 2007). We classified cancer-related genes referring to the Upenn databases (http://microb230.med.upenn.edu/protocols/cancergenes.html).
Gene expression profiling and microarray analysis
Transcriptional profiling was determined in 42 samples of BM-derived CD34+ cells, using Affymetrix HG-U133 Plus 2.0 GeneChip arrays. RNA was isolated using RNeasy Plus Mini kit (Quiagen). Two-cycle target labelling assays, Affymetrix HG-U133 Plus 2.0 GeneChip arrays hybridization, staining and scanning, were performed using Affymetrix standard protocols (Affymetrix, Santa Clara, CA). Fluorescence signals were recorded by an Affymetrix scanner 3000 and image analysis performed with the GeneChip Operating Software (GCOS) software. Preprocessing of the data and normalization was carried out in R (‘Affy’ package), unsupervised hierarchical clustering in dCHIP (Li & Wong, 2001) and supervised ANOVA with the Partec Genomic Suite software. To generate normal control microarrays, RNA was isolated from three samples of cord blood derived-CD34+ cells activated with cytokines. The raw gene expression data are available at http://www.ebi.ac.uk/microarray (Accession #E-MEXP-2757 and E-MEXP-2758).
To correlate vector integration and gene activity the arrays generated from patients' cells (THAL 22, 36, 37) and three healthy donors, activated by cytokines, we re-annotated the Affymetrix HG-U133 plus 2.0 probe sets with custom CDF files to include only probes unequivocally matching a transcript (Dai et al, 2005; Ferrari et al, 2007). The average of the expression values in each triplicate was considered. Expression values from microarrays were combined and divided into four classes, as absent, low (below the 25th percentile in a normalized distribution), intermediate (between the 25th and 75th percentile) and high (above the 75th percentile).
Statistical analyses
We used paired two-tailed t-tests for the comparisons of samples before and after transduction and unpaired two-tailed t-tests to compare other population means. All these statistical analyses were performed using GraphPad Prism Version 4.0b (GraphPad Software, San Diego, USA). For pairwise comparisons in integration analysis, we applied a 2-sample test for equality of proportions with continuity correction using the Rweb 1.03 statistical analysis package. To search for gene differentially expressed in normal and thalassemic samples, we used a one-way analysis of variance with a pre-set fold change of >1, followed by Bonferroni correction for false discovery rate at a level of significance of 0.05.
Acknowledgments
We acknowledge Dr. Maria Pia Cappabianca from ANMI-Onlus for genetic screening of IME samples. We thank Ms. Francesca Tiboni for vectors production and technical assistance. This work was supported by grants from the Italian Telethon Foundation (Core Grant TIGET), Fondazione IME and the European Commission (VI FP, CONSERT, LBSH-CT-2004-005242).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
E.A.R designed and performed research, analyzed data and wrote the paper; R.M. performed integrations studies; M.C.F. performed research; G.M. performed microarray experiments and analyzed data; E.B. analyzed data; F.M. contributed to hypothesis and study design; F. Mastropietro performed HPLC analysis; A.A. analyzed data; G.T. analyzed data; C.R. performed genetic screening; M.D.C. contributed to discussion; M.A. contributed to hypothesis and study design; G.L. was responsible for patients management at IME; M.G.R. was responsible for patients management at HSR; S.M. provided clinical data and contributed to hypothesis; G.F. contributed to the hypothesis and overall study design and experiments, preparation and editing of the paper.
For more information
Accompanying Cloeup:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, Scaramuzza S, Andolfi G, Mirolo M, Brigida I, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- Andreani M, Nesci S, Lucarelli G, Tonucci P, Rapa S, Angelucci E, Persini B, Agostinelli F, Donati M, Manna M. Long-term survival of ex-thalassemic patients with persistent mixed chimerism after bone marrow transplantation. Bone Marrow Transplant. 2000;25:401–404. doi: 10.1038/sj.bmt.1702151. [DOI] [PubMed] [Google Scholar]
- Andreani M, Testi M, Battarra M, Indigeno P, Guagnano A, Polchi P, Federici G, Lucarelli G. Relationship between mixed chimerism and rejection after bone marrow transplantation in thalassaemia. Blood Transfus. 2008;6:143–149. doi: 10.2450/2008.0051-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam PI, Urbinati F, Velu CS, Higashimoto T, Grimes HL, Malik P. The 3′ region of the chicken hypersensitive site-4 insulator has properties similar to its core and is required for full insulator activity. PLoS One. 2009;4:e6995. doi: 10.1371/journal.pone.0006995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohmer RM. Flow cytometry of erythropoiesis in culture: bivariate profiles of fetal and adult hemoglobin. Methods Cell Biol. 2001;64:139–152. doi: 10.1016/s0091-679x(01)64011-1. [DOI] [PubMed] [Google Scholar]
- Borgna-Pignatti C, Cappellini MD, De Stefano P, Del Vecchio GC, Forni GL, Gamberini MR, Ghilardi R, Origa R, Piga A, Romeo MA, et al. Survival and complications in thalassemia. Ann N Y Acad Sci. 2005;1054:40–47. doi: 10.1196/annals.1345.006. [DOI] [PubMed] [Google Scholar]
- Bradford GB, Williams B, Rossi R, Bertoncello I. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp Hematol. 1997;25:445–453. [PubMed] [Google Scholar]
- Buckner CD, Clift RA, Sanders JE, Stewart P, Bensinger WI, Doney KC, Sullivan KM, Witherspoon RP, Deeg HJ, Appelbaum FR, et al. Marrow harvesting from normal donors. Blood. 1984;64:630–634. [PubMed] [Google Scholar]
- Bushman F, Lewinski M, Ciuffi A, Barr S, Leipzig J, Hannenhalli S, Hoffmann C. Genome-wide analysis of retroviral DNA integration. Nat Rev Microbiol. 2005;3:848–858. doi: 10.1038/nrmicro1263. [DOI] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- Cattoglio C, Facchini G, Sartori D, Antonelli A, Miccio A, Cassani B, Schmidt M, von Kalle C, Howe S, Thrasher AJ, et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood. 2007;110:1770–1778. doi: 10.1182/blood-2007-01-068759. [DOI] [PubMed] [Google Scholar]
- Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA. 1999;96:3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felice B, Cattoglio C, Cittaro D, Testa A, Miccio A, Ferrari G, Luzi L, Recchia A, Mavilio F. Transcription factor binding sites are genetic determinants of retroviral integration in the human genome. PLoS One. 2009;4:e4571. doi: 10.1371/journal.pone.0004571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Bortoluzzi S, Coppe A, Sirota A, Safran M, Shmoish M, Ferrari S, Lancet D, Danieli GA, Bicciato S. Novel definition files for human GeneChips based on GeneAnnot. BMC Bioinformatics. 2007;8:446. doi: 10.1186/1471-2105-8-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch G, Stimpfl M, Kurz M, Printz D, Buchinger P, Fischmeister G, Hoecker P, Gadner H. The composition of CD34 subpopulations differs between bone marrow, blood and cord blood. Bone Marrow Transplant. 1996;17:169–178. [PubMed] [Google Scholar]
- Galanello R, Satta S, Pirroni MG, Travi M, Maccioni L. Globin chain synthesis analysis by high performance liquid chromatography in the screening of thalassemia syndromes. Hemoglobin. 1998;22:501–508. doi: 10.3109/03630269809071547. [DOI] [PubMed] [Google Scholar]
- Gaspar HB, Parsley KL, Howe S, King D, Gilmour KC, Sinclair J, Brouns G, Schmidt M, Von Kalle C, Barington T, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364:2181–2187. doi: 10.1016/S0140-6736(04)17590-9. [DOI] [PubMed] [Google Scholar]
- Giarratana MC, Kobari L, Lapillonne H, Chalmers D, Kiger L, Cynober T, Marden MC, Wajcman H, Douay L. Ex vivo generation of fully mature human red blood cells from hematopoietic stem cells. Nat Biotechnol. 2005;23:69–74. doi: 10.1038/nbt1047. [DOI] [PubMed] [Google Scholar]
- Giordano PC, Van Delft P, Batelaan D, Harteveld CL, Bernini LF. Haemoglobinopathy analyses in the Netherlands: a report of an in vitro globin chain biosynthesis survey using a rapid, modified method. Clin Lab Haematol. 1999;21:247–256. doi: 10.1046/j.1365-2257.1999.00197.x. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay JP, Thrasher AJ, Wulffraat N, Sorensen R, Dupuis-Girod S, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346:1185–1193. doi: 10.1056/NEJMoa012616. [DOI] [PubMed] [Google Scholar]
- Hanawa H, Yamamoto M, Zhao H, Shimada T, Persons DA. Optimized lentiviral vector design improves titer and transgene expression of vectors containing the chicken beta-globin locus HS4 insulator element. Mol Ther. 2009;17:667–674. doi: 10.1038/mt.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser J. β-Thalassemia treatment succeeds, with a caveat. Science. 2009;326:1468–1469. doi: 10.1126/science.326.5959.1468-b. [DOI] [PubMed] [Google Scholar]
- Kinniburgh D, Russell NH. Comparative study of CD34-positive cells and subpopulations in human umbilical cord blood and bone marrow. Bone Marrow Transplant. 1993;12:489–494. [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucarelli G, Gaziev J. Advances in the allogeneic transplantation for thalassemia. Blood Rev. 2008;22:53–63. doi: 10.1016/j.blre.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Malik P, Arumugam PI, Yee JK, Puthenveetil G. Successful correction of the human Cooley's anemia beta-thalassemia major phenotype using a lentiviral vector flanked by the chicken hypersensitive site 4 chromatin insulator. Ann N Y Acad Sci. 2005;1054:238–249. doi: 10.1196/annals.1345.030. [DOI] [PubMed] [Google Scholar]
- Mathias LA, Fisher TC, Zeng L, Meiselman HJ, Weinberg KI, Hiti AL, Malik P. Ineffective erythropoiesis in beta-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp Hematol. 2000;28:1343–1353. doi: 10.1016/s0301-472x(00)00555-5. [DOI] [PubMed] [Google Scholar]
- Miccio A, Cesari R, Lotti F, Rossi C, Sanvito F, Ponzoni M, Routledge SJ, Chow CM, Antoniou MN, Ferrari G. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia. Proc Natl Acad Sci USA. 2008;105:10547–10552. doi: 10.1073/pnas.0711666105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio G, Di Pietro R, di Giacomo V, Di Baldassarre A, Migliaccio AR, Maccioni L, Galanello R, Papayannopoulou T. In vitro mass production of human erythroid cells from the blood of normal donors and of thalassemic patients. Blood Cells Mol Dis. 2002;28:169–180. doi: 10.1006/bcmd.2002.0502. [DOI] [PubMed] [Google Scholar]
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:480–487. doi: 10.2471/BLT.06.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienhuis AW, Dunbar CE, Sorrentino BP. Genotoxicity of retroviral integration in hematopoietic cells. Mol Ther. 2006;13:1031–1049. doi: 10.1016/j.ymthe.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Olweus J, Thompson PA, Lund-Johansen F. Granulocytic and monocytic differentiation of CD34hi cells is associated with distinct changes in the expression of the PU.1-regulated molecules, CD64 and macrophage colony-stimulating factor receptor. Blood. 1996;88:3741–3754. [PubMed] [Google Scholar]
- Puthenveetil G, Scholes J, Carbonell D, Qureshi N, Xia P, Zeng L, Li S, Yu Y, Hiti AL, Yee JK, et al. Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood. 2004;104:3445–3453. doi: 10.1182/blood-2004-04-1427. [DOI] [PubMed] [Google Scholar]
- Sadelain M, Boulad F, Galanello R, Giardina P, Locatelli F, Maggio A, Rivella S, Riviere I, Tisdale J. Therapeutic options for patients with severe beta-thalassemia: the need for globin gene therapy. Hum Gene Ther. 2007;18:1–9. doi: 10.1089/hum.2006.151. [DOI] [PubMed] [Google Scholar]
- Taher AT, Otrock ZK, Uthman I, Cappellini MD. Thalassemia and hypercoagulability. Blood Rev. 2008;22:283–292. doi: 10.1016/j.blre.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Terstappen LW, Huang S, Safford M, Lansdorp PM, Loken MR. Sequential generations of hematopoietic colonies derived from single nonlineage-committed CD34+CD38-progenitor cells. Blood. 1991;77:1218–1227. [PubMed] [Google Scholar]
- Tjonnfjord GE, Steen R, Evensen SA, Thorsby E, Egeland T. Characterization of CD34+ peripheral blood cells from healthy adults mobilized by recombinant human granulocyte colony-stimulating factor. Blood. 1994;84:2795–2801. [PubMed] [Google Scholar]
- Urbinati F, Arumugam P, Higashimoto T, Perumbeti A, Mitts K, Xia P, Malik P. Mechanism of reduction in titers from lentivirus vectors carrying large inserts in the 3′LTR. Mol Ther. 2009;17:1527–1536. doi: 10.1038/mt.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lochem EG, van der Velden VH, Wind HK, te Marvelde JG, Westerdaal NA, van Dongen JJ. Immunophenotypic differentiation patterns of normal hematopoiesis in human bone marrow: reference patterns for age-related changes and disease-induced shifts. Cytometry B Clin Cytom. 2004;60:1–13. doi: 10.1002/cyto.b.20008. [DOI] [PubMed] [Google Scholar]
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704–712. [PMC free article] [PubMed] [Google Scholar]
- Williams DA. Gene therapy continues to mature and to face challenges. Mol Ther. 2009;17:1305–1306. doi: 10.1038/mt.2009.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Luke BT, Burgess SM. Redefining the common insertion site. Virology. 2006;344:292–295. doi: 10.1016/j.virol.2005.08.047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.