

Abstract
Neuroblastoma (NB) is the most deadly extra-cranial solid tumour in children necessitating an urgent need for effective and less toxic treatments. One reason for the lack of efficacious treatments may be the inability of existing drugs to target the tumour-initiating or cancer stem cell population responsible for sustaining tumour growth, metastases and relapse. Here, we describe a strategy to identify compounds that selectively target patient-derived cancer stem cell-like tumour-initiating cells (TICs) while sparing normal paediatric stem cells (skin-derived precursors, SKPs) and characterize two therapeutic candidates. DECA-14 and rapamycin were identified as NB TIC-selective agents. Both compounds induced TIC death at nanomolar concentrations in vitro, significantly reduced NB xenograft tumour weight in vivo, and dramatically decreased self-renewal or tumour-initiation capacity in treated tumours. These results demonstrate that differential drug sensitivities between TICs and normal paediatric stem cells can be exploited to identify novel, patient-specific and potentially less toxic therapies.
Keywords: cancer stem cells, dequalinium, high-throughput screen, neuroblastoma, tumour initiating cells
INTRODUCTION
Neuroblastoma (NB) is a paediatric cancer hypothesized to develop from primitive neural crest cells that normally differentiate to form the sympathetic nervous system (Maris et al, 2007). Patients with advanced disease are treated with aggressive multimodal therapy that includes intensive chemotherapy, radiation, surgery, bone marrow transplant and immunotherapy. However, the survival of patients older than 1 year with metastatic disease is less than 40% due to relapse and extensive metastatic disease (Matthay et al, 1999). For those patients who survive their disease, there is significant morbidity due to treatment-related toxicities (Laverdiere et al, 2009). The dismal survival statistics as well as the significant side effects of available treatments led us to investigate new NB treatments.
Recent studies suggest that many solid tumours contain a population of cancer stem cells or tumour-initiating cells (TICs) that fuel tumour growth and seed metastases (reviewed in Dalerba et al, 2007; Frank et al, 2010; Ward & Dirks, 2007). While many tumours regress after treatments that target proliferating tumour cells, durable cures may be rare if therapeutic agents do not also eliminate TICs (Graham et al, 2002; Konig et al, 2008). We recently described a highly enriched TIC population from the bone marrow of patients with often-fatal relapse NB that forms aggressive metastatic tumours when as few as 10 cells are orthotopically transplanted into immune compromised mice (Hansford et al, 2007). These NB TICs have several properties of cancer stem cells including the expression of neural crest progenitor cell markers, self-renewal in vitro and in vivo and the ability to differentiate into cell types characteristic of the primary NB tumour. The NB TICs were isolated and propagated in serum-free sphere-forming conditions optimized for neural stem cells growth and maintenance and are used with minimal in vitro passaging. Therefore, NB TICs may more closely reflect the cells from bone marrow metastases as compared to NB cell lines established in serum and selected for adherent culture. We hypothesize that drugs that induce the death of NB TICs but that do not harm normal paediatric progenitor cells may be inherently less toxic than present therapies. Dermal stem cells, termed skin-derived progenitors or SKPs, have been isolated and cultured from neonatal skin and exhibit properties similar to neural crest stem cells (Biernaskie et al, 2009; Fernandes et al, 2004; Hansford et al, 2007; Toma et al, 2001). Like NB TICs, they are maintained as spheres in serum-free media, self-renew in vitro, express neural crest progenitor cell markers and differentiate into neural crest lineage cells, but are non-tumourigenic. The availability of two different neural crest-like stem cell populations, highly enriched TICs from NB and normal non-transformed paediatric stem cells, provides a powerful tool for drug discovery.
In this study, we developed a high-throughput cell-based screening assay to identify compounds that are preferentially active against NB TICs. NB TICs, normal paediatric SKPs and established NB cell lines grown in serum-free sphere-forming conditions were tested in parallel against small molecule libraries. Fifty-one compounds that selectively targeted NB TICs were identified. Two compounds, DECA-14 and rapamycin, were characterized in greater detail and shown to selectively inhibit NB TIC survival in vitro and significantly reduce both NB xenograft tumour volume and self-renewal or tumour-initiating capacity in vivo.
RESULTS
Small molecule screens identify novel chemotherapeutics
NB TICs cultured from bone marrow metastases of a multiple-relapse NB patient (NB12) were treated with 4383 compounds from the LOPAC1280™ compound library, the Prestwick Chemical Library and the Spectrum Collection. Normal human paediatric SKP cells (FS90) were tested in parallel as a counterscreen to identify compounds that are selectively cytotoxic or cytostatic against NB TICs. Cells were treated with test compound for 30 hours prior to a further 24 hour incubation in the presence of alamarBlue® and subsequent fluorometric reading (Fig 1A). Figure 1B shows primary screen data for the three compound libraries. Primary hits were defined as the compounds whose B-scores were shifted by at least three standard deviations (99.73% confidence interval, indicated by coloured lines) from the mean of the general sample population. Eighty-seven percent of primary hits were confirmed when compounds were retested in a 3-point dilution series. Primary hits were then tested against NB TICs (NB12) or SKPs (FS90) in a 10-point dilution series and cell survival assessed by alamarBlue® signal to determine IC50 values. Selective cytotoxicity against NB TICs was defined as a reduced IC50 value when compared to treatment of normal stem cells (SKPs). Based on both primary screen results (B-score shifted more than three standard deviations from the mean) and confirmatory tests (greater than three-fold shift in IC50 by alamarBlue® assay), we identified 51 compounds that were selectively cytotoxic or cytostatic against NB TICs and 51 compounds with activity against both NB TICs and normal non-transformed precursor cells (SKPs) (Fig 1C, Supporting Information Fig (SI) 1). In addition to known anticancer agents, we identified several classes of compounds including antihistamines, antimicrobials and antimalarial agents as well as specific antagonists to acetaldehyde dehydrogenase, PKC, NF-κB and nicotinic acetylcholine receptor subunits (Fig 1D, SI1).
Figure 1. Primary screen identified NB TIC-selective compounds.

- NB TICs (NB12), SKPs (FS90) or SMS-KCNR cells were treated in parallel with compounds from LOPAC1280™ library, Prestwick Chemical Library or Spectrum Collection as described in Materials and Methods.
- Plot of B-scores from the LOPAC1280™, Prestwick and Spectrum compound libraries screened against FS90 or NB12 cells. Compounds with B-scores greater than three standard deviations were considered primary hits. Black line, population mean. Coloured line, three standard deviations from the mean.
- Venn diagram of confirmed primary hits. Cell selectivity was determined by both primary screen and confirmatory assay results. Hit compounds are listed in SI1.
- Confirmed primary hits that preferentially target NB TICs (red) or that have activity against both TICs and SKPs (blue) classified by known or suspected mechanism of action.
To evaluate the value of screening NB TICs in comparison to established NB cell lines, we tested the three compound libraries against an established NB cell line, SMS-KCNR, that had been derived under adherent growth conditions in the presence of serum (Reynolds et al, 1986). SMS-KCNR was derived from NB bone marrow metastases, represents a primitive neuroblast phenotype and can grow robustly under the same serum-free neurosphere culture conditions as NB TICs and SKPs. Despite these similarities, there was only 28.0% overlap in the primary hits observed with NB TICs and SMS-KCNR cells (Fig 1C). Unlike the NB TICs used in this study, SMS-KCNR carries an MYCN amplification. To ensure the differential drug sensitivities we observed were not due to MYCN amplification, we examined two established cell lines lacking MYCN amplification (SK-N-AS and SH-SY5Y). As with SMS-KCNR, SK-N-AS and SH-SY5Y cells were screened in parallel with NB12 TICs in serum-free neurosphere culture conditions (SI2A). There was approximately 50% overlap in the primary hits between the lines (50.88% SK-N-AS vs. NB12, 50.00% SH-SY5Y vs. NB12, 45.69% SK-N-AS vs. SH-SY5Y). This is in contrast to a comparison of TICs from bone marrow metastases of three unfavourable prognosis NB patients where there was 78% overlap between NB12 and NB88R2, 81.3% overlap between NB12 and NB122R and 76.5% overlap between NB122R and NB88R2 (SI2B). Thus, NB TICs from different patients show greater similarity to each other regarding drug sensitivity than they do to established NB cell lines.
Secondary screens identify compounds selective against TICs from multiple NB patients
Based on the primary screen B-score values as well as the results of confirmatory assays evaluated through IC50 values, we selected 38 compounds for secondary screening. In vitro sphere formation capacity was used as a surrogate measure of TIC self-renewal capacity (Reynolds & Rietze, 2005; Singh et al, 2004). NB TICs from multiple patients (described in SI3) as well as SKPs from several individuals were tested to determine whether compounds were generally toxic against NB TICs or whether effects were patient-specific. Sixteen compounds displayed a large difference in EC50 values for sphere formation between NB TICs and SKPs (>50-fold, indicated by *** in SI1 and SI4D) and included compounds such as crinamine (SI4A) and dequalinium analogue, C-14 linker (DECA-14, Fig 2A). Sixteen compounds had a modest difference in EC50 values (3-50-fold, indicated by * in SI1 and SI4D) and included compounds such as quinacrine and parthenolide (SI4B). Six compounds had equal potency on NB TICs and SKPs (indicated by = in SI1 and SI4D). Interestingly, a small number of patient-specific TIC-selective drugs were identified (colchicine, podophyllotoxin, vincristine and vinblastine, SI4C).
Figure 2. DECA-14 selectively targeted NB TICs in vitro and induced rapid apoptotic cell death.
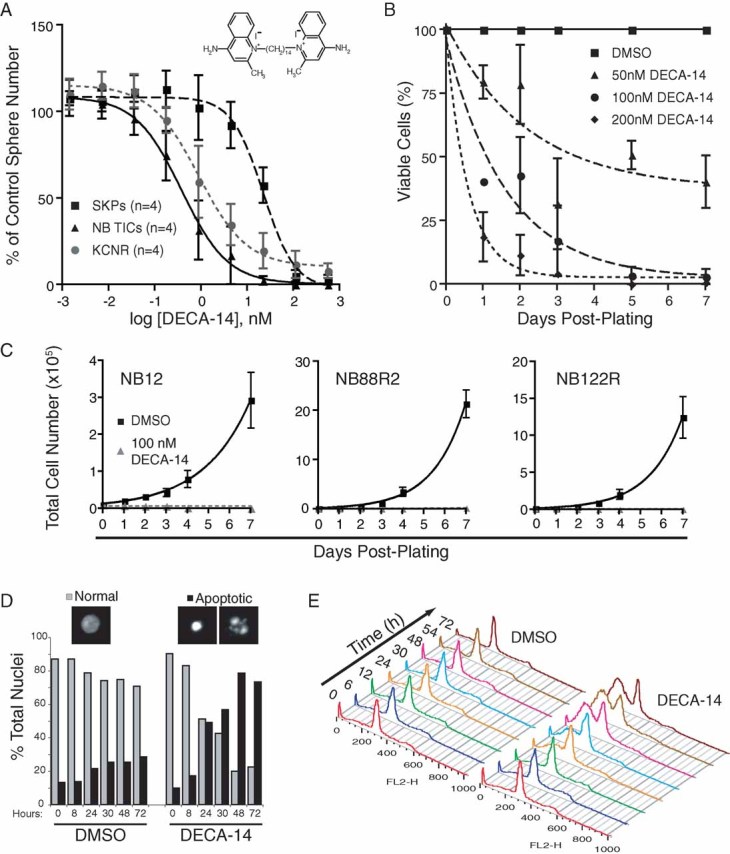
- DECA-14 was 59-fold more potent against NB TICs (EC50 = 0.38 nM) than SKPs (EC50 = 22.5 nM) in sphere formation secondary assays. Points, mean of 4 unique NB TICs or SKPs and 4 independent experiments with SMS-KCNR cells. Bars, SE.
- NB12 TICs were treated in triplicate with DECA-14 and cell viability measured by trypan blue exclusion.
- TICs from high-risk NB patients (NB12, NB88R2, NB122R) were treated with 100 nM DECA-14 or DMSO and viable cell number determined. Points, mean of triplicate experiment. Bars, SE.
- Condensed and fragmented nuclear morphology was observed in NB12 TICs after 100 nM DECA-14 treatment. Results from a representative experiment are shown.
- NB12 TICs were treated with 100 nM DECA-14 or DMSO for the indicated times and DNA content of propidium iodide-stained samples was assessed.
Dequalinium analogue, C-14 linker potently induces apoptosis in NB TICs
Dequalinium analogue, C-14 linker (DECA-14) was selected for more detailed in vitro and in vivo analyses due to its greatly increased toxicity against NB TICs as compared to SKPs and its potentially novel mechanism of action. DECA-14 is an analogue of dequalinium (DECA-10), an antimicrobial agent used in mouthwashes and throat lozenges. DECA-14 decreased the alamarBlue® signal of NB12 TICs by 74% in the primary screen and affected NB TICs in secondary sphere assays at significantly lower doses than SKPs with EC50 values for sphere formation of 0.38 nM for NB TICs versus 22.53 nM for SKPs (Fig 2A). SMS-KCNR are 2.4-fold less sensitive to DECA-14 than NB TICS with an EC50 of 0.9 nM for sphere formation while SK-N-AS and SH-SY5Y cells have a sensitivity similar to SKPs using an alamarBlue® assay (SI5). NB TICs treated with DECA-14 exhibited decreased cell viability within 24 hours as measured by trypan blue exclusion with the majority of cells dead after 72 hours (Fig 2B). TICs from multiple NB patient bone marrow metastases (NB12, NB67, NB88R2, NB122R) were equally susceptible to DECA-14 (Figs 2A and C). DECA-14 treatment induced apoptosis in NB TICs as determined by the appearance of fragmented or condensed nuclei (Fig 2D), increased sub-2n DNA content (Fig 2E, SI6) and cleaved PARP and cleaved caspase 7 in DECA-14-treated cells (SI6).
DECA-14 treatment affects genes that regulate mitochondria electron transport
To gain insight into the mechanism of DECA-14-induced death, we performed global gene expression analysis comparing NB12, NB88R2 and NB122R cells treated with 100 nM DECA-14 or DMSO for 24 hours. RNA samples were analysed on Affymetrix GeneChip Human Gene 1.0 ST Array. Genes with evidence of differential expression between DECA-treated and untreated cells were identified using the LIMMA Bioconductor package as described in the Methods (Fig 3A). Twenty-four genes were found to be downregulated in DECA-14-treated cells (uncorrected p-value < 0.01), while thirty-two genes were found to be upregulated (uncorrected p-value < 0.01). Four genes, ND3, ND4, ND5, LOC44055, were most significantly downregulated following DECA treatment, while three genes, ND6, CHAC1, STC2, were most significantly upregulated (BH-corrected p-value < 0.05). To characterize the differentially expressed genes further, we conducted a canonical pathway enrichment analysis using the Ingenuity Software. We found that oxidative phosphorylation was one of the key pathways enriched among the downregulated genes, while upregulated genes participated in a variety of metabolic and signalling pathways (Figs 3B, C). Semi-quantitative multiplex PCR (SI7) confirmed alteration of mRNA levels. Four of the seven most significantly altered genes were mitochondria-encoded subunits of NADH dehydrogenase, which make up complex I of the mitochondrial electron transport chain. Inhibition of complex I can interfere with energy/ATP generation, increase free radical production and induce apoptotic cell death (Li et al, 2003).
Figure 3. Gene expression analysis suggests that DECA-14 targets metabolic pathways.
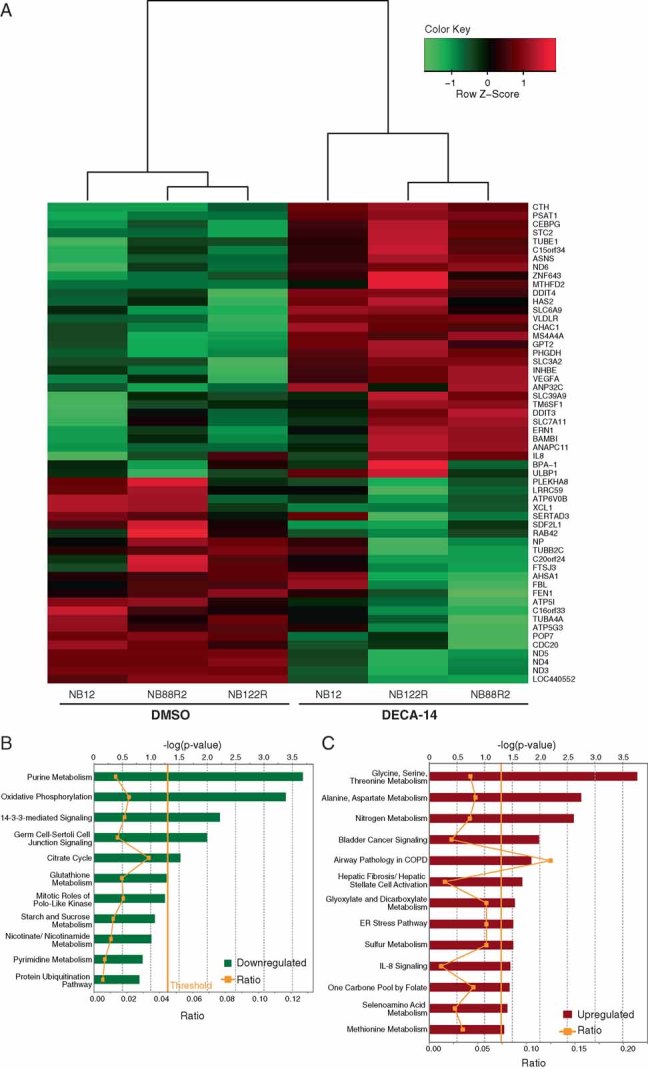
- Genes with most significant evidence of differential expression between DECA-treated and untreated cells; 24 genes were found to be downregulated in DECA-14-treated cells while 32 genes were found to be upregulated.
- Canonical pathways enriched among the 24 genes downregulated in DECA-treated cells.
- Canonical pathways enriched among the 32 genes upregulated in DECA-treated cells. The threshold line corresponds to the enrichment p-value of 0.05 computed by the Ingenuity Software.
DECA-14 targets NB TICs in vivo
To examine whether DECA-14 was effective against NB TICs in vivo, we established xenograft tumours by subcutaneous injection of NB12 cells into NOD/SCID mice. Once tumours reached 0.5 cm3, animals were treated with intraperitoneal injections of DECA-14 every other day. Two doses of DECA-14 were tested: 4.5 or 2.5 mg/kg. With the higher dose, significant constipation was noted and as a result, dosing was limited to 10 days. For subsequent experiments, the dose was reduced to 2.5 mg/kg DECA-14 for 14 days and mice received supplemental hydration with daily subcutaneous injection of lactated Ringer's solution. Treatment with either dose resulted in significantly reduced tumour weight relative to vehicle-treated controls (65% reduction at 4.5 mg/kg and 50.6% reduction with 2.5 mg/kg) (Fig 4A). Treated animals had ∼13% reduced body weight (20.89 ± 0.32 g for 2.5 mg/kg DECA-14-treated animal vs. 24.22 ± 0.48 g for vehicle-treated animal, p < 0.0001). No other significant toxicities were noted upon necropsy and pathological examination of liver, lungs, kidney and heart of treated animals.
Figure 4. DECA-14 selectively targeted NB TICs in vivo.
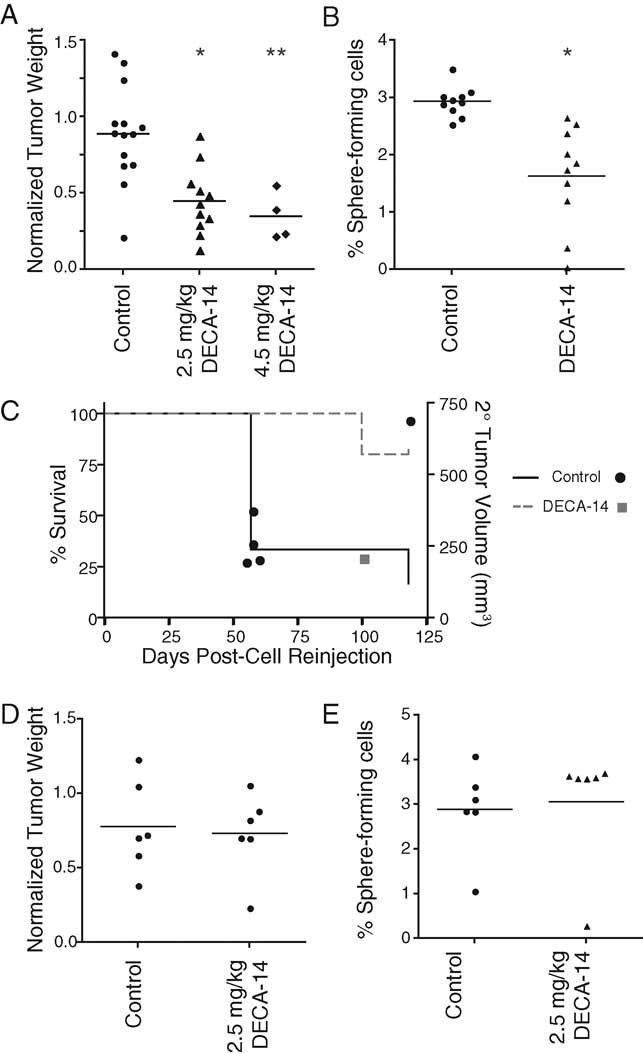
- NB12 xenograft tumours were established and treated with vehicle, 4.5 mg/kg DECA-14 or 2.5 mg/kg DECA-14 as described in Material and Methods. Point, tumour weight at autopsy. The median for each group is indicated. *, p = 0.0007, and **, p = 0.0014 compared to vehicle-treated control group.
- Dissociated DECA-14- or vehicle-treated NB12 tumours were plated in methylcellulose-containing media. Percentage of sphere-forming cells per total cell number was calculated from duplicate wells after 10 days. *, p = 0.0003 compared to vehicle-treated control group.
- Kaplan–Meier survival curve for recipient NOD/SCID mice re-injected with dissociated NB12 tumour cells from control- (black, circle) or 2.5 mg/kg DECA-14-treated (grey, square) tumours. Survival percentage is depicted on the left axis and individual tumour volume at autopsy is depicted on the right axis. Wilcoxon: p = 0.0455, Log-Rank: p = 0.0455.
- SK-N-AS xenograft tumours were established and treated with vehicle or 2.5 mg/kg DECA-14 as described in Material and Methods. Point, tumour weight at autopsy. The median for each group is indicated.
- Dissociated DECA-14- or vehicle-treated SK-N-AS tumours were plated in methylcellulose-containing media. Percentage of sphere-forming cells per total cell number was calculated from duplicate wells after 18 days.
To determine whether DECA-14 treatment specifically targeted the self-renewing and tumour-initiating population in vivo, tumours were dissociated to single cell suspensions and viable cells were re-cultured to quantify sphere-formation capacity. DECA-14 treatment of tumours significantly reduced the percentage of sphere-forming cells suggesting that DECA-14 targeted NB TICs in vivo (Fig 4B). In a separate experiment, we examined whether cells from DECA-14-treated tumours had a reduced ability to initiate tumours in vivo. Fifty-thousand viable dissociated tumour cells were re-transplanted into the inguinal fat pad of NOD/SCID mice. Cells from three randomly selected vehicle-treated tumours and two DECA-14-treated tumours were injected into two recipient NOD/SCID mice per original tumour. Five of six vehicle-treated tumour cell recipients developed secondary tumours with a median tumour volume of 28.74 ± 94.67 mm3 that developed at a rate similar to those generated from NB TICs maintained in tissue culture. Only one of four DECA-14-treated tumour cell recipients developed a tumour and this was observed at significantly later time points compared to control-treated tumour cell recipients (Fig 4C). Unlike NB TIC xenograft tumours, DECA-14 had no effect on SK-N-AS xenograft tumours. There was no difference in either tumour weight or the ability of treated tumour-cells to grow in methylcellulose-containing culture (Figs 4D, E). Thus, DECA-14 treatment significantly reduced both tumour growth and the tumour-initiating capacity of NB TICs in vivo.
Rapamycin targets NB TICs in vitro
Our successful identification of NB TIC-selective agents from a collection of drugs and drug-like molecules suggests that that this system can be used to evaluate additional compounds that may be selectively toxic against TICs in other tumours. Several compounds predicted to be active against TICs were either not bioactive in our libraries or were subsequently found to be cytotoxic in 96 hour rather than the 54 hour assays used here. Rapamycin, a mammalian target of rapamycin (mTOR) inhibitor that was not present in our chemical libraries but was found in subsequent library screening to be cytotoxic in 96 hour assays (N. G. and D. R. K., unpublished data) has been identified as a leukaemic stem cell-selective agent (Yilmaz et al, 2006) and is currently being tested in clinical trials for various solid tumours. Therefore, we asked whether rapamycin could preferentially target NB TICs. In the sphere formation assay described above, rapamycin had little effect on SKPs or SMS-KCNR cells up to doses of 1 µM (Fig 5A). Likewise, rapamycin was not effective against SK-N-AS or SH-SY5Y cells (SI8A). In contrast, NB TICs from four patients were sensitive to rapamycin with a calculated EC50 value of 0.11 nM (Fig 5A). Rapamycin treatment resulted in a decrease of cell viability as early as 24 hours with maximal effects detected between 72 and 96 hours (SI8B). Rapamycin treatment was also associated with decreased proliferation of NB TICs isolated from multiple patients (Fig 5B). We confirmed rapamycin activity by examining the phosphorylation of two proteins in the mTOR signalling pathway, p70S6K and S6 ribosomal protein (S6RP). Both of the proteins were hyperphosphorylated in all NB TICs tested and the activity of the mTOR pathway, as assessed by phosphorylation of S6RP, was significantly upregulated in NB TICs as compared to the two SKPs lines, FS90 and FS99, and SMS-KCNR (Fig 5C), suggesting that this pathway is constitutively activated in NB TICs. We did not, however, observe overexpression or hyperphosphorylation of mTOR or PTEN in NB TICs when compared to SKPs, although there was increased phosphorylation of the mTORC2 substrate Akt at serine 473 (Fig 5C). Phosphorylation of p70S6K and S6RP was rapidly inhibited by rapamycin (Fig 5C). Rapamycin treatment induced features of apoptosis in NB12 and NB122R cells. These included increase in sub-2n DNA content and the appearance of cleaved PARP (SI8C, D). NB TICs (NB12, NB88R2, NB122R) infected with lentiviruses encoding shRNAs to mTOR or the mTOR binding partners raptor or rictor showed 70–80% growth inhibition compared to NB TICs infected with lentiviruses carrying mock shRNAs to GFP or LacZ (Fig 5D and data not shown). These data suggest that components of the mTOR signalling pathway are constitutively and highly active in NB TICs, and regulate TIC survival.
Figure 5. Rapamycin selectively targeted NB TICs in vitro in a patient-specific fashion.

- Rapamycin targeted NB TICs while having little effect on SKPs or SMS-KCNR cells in sphere formation secondary assays. Points, mean of 4 unique NB TICs, 4 SKPs and 6 independent experiments with SMS-KCNR. Bars, SE.
- TICs from high-risk NB patients (NB12, NB88R2, NB122R) were treated with 50 nM rapamycin or DMSO and viable cell number determined. Points, mean of triplicate experiment. Bars, SE.
- SKPs (FS90, FS99), NB TICs (NB12, NB88R2, NB122R) or SMS-KCNR cells were treated with DMSO (−) or 50 nM rapamycin (+) for 1 h. Immunoblots of rapamycin-treated cell lysates demonstrated decreased phosphorylation of mTOR target proteins, p70S6K and S6RP. Note that Akt as well as mTOR target proteins are hyperphosphorylated in NB TICs in comparison to SKPs.
- shRNA knockdown of mTOR, Rictor, or Raptor decreased NB12 cell viability compared to uninfected or mock shRNAs to GFP or LacZ. Points, mean of triplicate experiment. Bars, SE.
- qRT-PCR results for mTOR, Rictor and raptor mRNA 72 hours post-infection of NB12 TICs with indicated shRNAs.
Rapamycin targets NB TICs in vivo
Like DECA-14, rapamycin was found to be effective against established xenograft tumours in vivo. Daily rapamycin treatment reduced tumour weight by 82.6% for NB12 tumours and 74.0% for NB88 tumours (Fig 6A) while vinblastine, a chemotherapy used to treat NB, only reduced tumour weight 43.4% relative to vehicle-treated controls. Rapamycin treatment inhibited the mTOR pathway in xenograft tumours as expected. In comparison to vehicle-treated tumours, lysates from tumours treated with rapamycin exhibited a decrease in phosphorylation of p70S6K (data not shown) and S6RP (Fig 6C). These results were confirmed by immunohistochemistry of formalin-fixed tumour sections for phospho-S6RP (Fig 6B). There was also an increase in ERK1/2 phosphorylation in rapamycin-treated tumours that is similar to the increase of ERK1/2 phosphorylation reported in tumour biopsies of patients treated with RAD001, a rapamycin analogue (Carracedo et al, 2008) (Fig 6C). To determine whether rapamycin treatment targeted the NB TICs in vivo, four vehicle-treated tumours as well as four rapamycin-treated tumours were dissociated and sphere formation was examined (Fig 6D). Sphere-forming ability was reduced 7.75-fold in rapamycin-treated tumours demonstrating that rapamycin targeted the tumour cell population capable of self-renewal. The results suggest that rapamycin and rapamycin analogues may be ideal therapeutics for patients whose NB TICs that are found to be sensitive to this drug in vitro.
Figure 6. Rapamycin significantly inhibited tumour growth and targeted NB TICs in vivo.
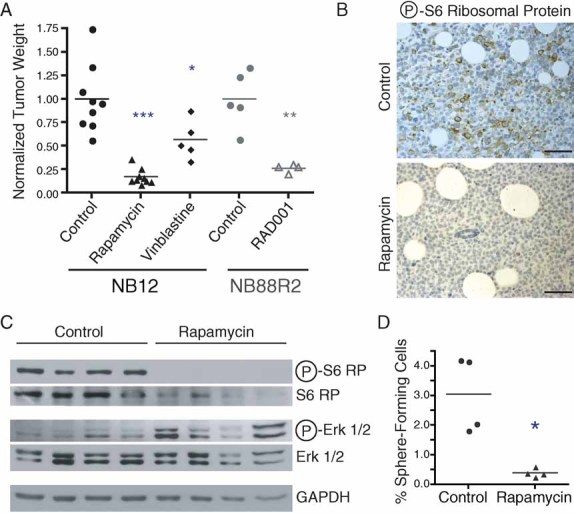
- NB12 or NB88R2 xenograft tumours were established and treated with vehicle, vinblastine or rapamycin as described in Materials and Methods. Three independent experiments were conducted. Points, tumour weight at autopsy. The median for each group is indicated. *, p = 0.0305, ***, p < 0.0001 when compared to NB12 vehicle-treated control group. **, p = 0.0020 when compared to NB88R2 vehicle-treated control group.
- Immunohistochemical staining of control or rapamycin-treated tumours for phosphorylated S6RP (brown staining). Scale bar, 200 µm.
- Tumour lysates from individual control- or rapamycin-treated xenograft tumours were immunoblotted with indicated antibodies.
- Dissociated rapamycin-treated tumours were plated in methylcellulose-containing media and sphere formation analysed from triplicate wells after 10 days. *, p = 0.0066 when compared to control-treated tumour cells.
DISCUSSION
Current cancer therapies that target proliferating tumour cells can dramatically reduce tumour bulk but durable cures may require elimination of TICs that are hypothesized to be responsible for tumour growth, metastases and relapse. In the search for compounds that selectively target TICs, we need to ensure that identified drugs do not affect normal stem cells with which TICs share many properties. This is particularly important for paediatric cancers where normal populations of stem cells are required for proper development. In this paper, we describe the identification of compounds that are selectively toxic to NB TICs while sparing non-transformed paediatric stem cells (SKPs). We chose to screen NB TICs for a number of reasons. First, NB TICs are primary cells that faithfully model high-risk NB tumours. NB TICs express markers of NB and neural crest progenitors and have chromosomal aberrations typical of NB tumours. Second, NB TICs self-renew extensively in vitro as spheres similar to NB bone marrow metastases observed as clusters on bone marrow smears. Screening of spheroid cell cultures may identify a different spectrum of active agents than screening of adherent cell lines that were established and maintained in serum-containing media (Pampaloni et al, 2007). The drug sensitivity of NB TICs appears to be unique when compared to established NB cell lines such as SMS-KCNR, SH-SY5Y and SK-N-AS (this study, Gheeya et al, 2009). Third, NB TICs represent a highly enriched population of TICs with as few as one NB12 cell required to generate a metastatic tumour that phenocopies human NB in immunodeficient mice (Hansford et al, 2007, A. McKee and C. Thiele, unpublished observations). Finally, we were able to screen NB TICs in parallel with non-transformed neural crest-like stem cells (SKPs) under identical in vitro conditions to identify NB TIC-selective compounds.
Using a robust and reproducible cell-based high-throughput assay, we identified compounds that were effective against TICs from multiple NB patients in vitro and were effective against established NB xenograft tumours in vivo. We identified several interesting classes of compounds including several that have not been predicted for use in NB treatment such as antimalarial agents, surfactants and a number of specific protein inhibitors. This parallel screening approach can also be used to evaluate the effect of selected clinical drugs on NB TICs as we have demonstrated for rapamycin.
DECA-14 and the related compound dequalinium (DECA-10) were identified as hits in our primary screen. DECA-10 has been shown to inhibit the growth and motility of cancer cell lines in vitro and in vivo (Bodden et al, 1986; Christman et al, 1990; Galeano et al, 2005; Weiss et al, 1987) and its cytotoxicity may be mediated through mechanisms that include impairment of mitochondrial function (Schneider Berlin et al, 1998; Sancho et al, 2007; Weiss et al, 1987) or interactions with the mitochondrial F1-ATPase (Zhuo & Allison, 1988) or protein kinase C (Rotenberg et al, 1990). We found no evidence for an effect of DECA-14 on PKC signalling in NB TICs. Indeed, the IC50 for PKCα activity inhibition by DECA-14 is 2.6 µM (Qin et al, 2000), while the EC50 for NB TIC sphere inhibition by DECA-14 was 0.38 nM. Likewise, growth inhibition of melanoma cancer cell lines was observed at nanomolar concentrations (Sullivan et al, 2000). We examined the effects of several known PKC inhibitors including DECA-10, DECA-14, chelerythrine chloride and Gö6976. Only DECA-14 and chelerythrine chloride were selectively cytotoxic for NB TICs (Fig 2, SI9). Treatment with DECA-14 did not alter phosphorylation of PKCα/β substrates (SI9) or p38MAPK, a downstream target of PKC, nor was expression or phosphorylation of PKCδ, a PKC-isoform implicated in several human cancers, altered over 72 hours of DECA-14 treatment (K. M. S. and D. R. K., unpublished data). Gene expression analysis suggests that DECA-14 treatment of NB TICs induces apoptosis primarily through effects on mitochondria. The most significantly altered transcripts were the mitochondrial NADH dehydrogenase subunits that compose complex I of the mitochondrial electron transport chain. This is consistent with previous reports showing that DECA-10 preferentially accumulates in the mitochondria of tumour cells (Weiss et al, 1987) where it induces selective depletion of mitochondrial DNA (Schneider Berlin et al, 1998). DECA-10 can cause DNA unwinding and may result in loss of mitochondrial DNA through inhibition of DNA synthesis (Schneider Berlin et al, 1998; Wright et al, 1980). We hypothesize that DECA-14 is selectively taken up by the mitochondria, where it alters mitochondrial gene transcription of the NADH dehydrogenase subunit genes ND3, 4, 5 and 6. Inhibition of Complex I function is known to interfere with ATP generation, increase the production of reactive oxygen species and induce apoptosis (Li et al, 2003).
DECA-14 may have use as a therapeutic drug in NB since nanomolar doses of DECA-14 induced rapid cell death of NB TICs in vitro and dramatically reduced tumour weight in vivo. DECA-14 treatment also impaired the ability of NB TICs to form secondary tumours in vivo suggesting that the TIC population was targeted in the primary tumour. Furthermore, the minimal toxicities observed suggest that DECA-14 may be well tolerated and potentially could be combined with other NB agents.
Using this cell based-screening assay, we have also assessed the activity of agents known to be effective against other cancer stem populations. Rapamycin is a macrolide antibiotic originally characterized as an antifungal and immunosuppressive agent (Martel et al, 1977; Sehgal et al, 1975) that inhibits mTOR, a serine/threonine kinase involved in cell growth (reviewed in (Wullschleger et al, 2006)). In addition to antiproliferative properties in a variety of human cancer cell lines (Guertin & Sabatini, 2007), rapamycin has been shown to preferentially target leukaemic stem cells from PTEN-deficient murine leukaemias (Yilmaz et al, 2006). The clinical use of rapamycin has expanded recently with the development of analogues with improved pharmacology and bioavailability.
Rapamycin inhibited the survival and proliferation of NB TICs at nanomolar concentrations that did not affect SKPs or SMS-KCNR cells. Rapamycin has previously been shown to inhibit proliferation of several NB cell lines at micromolar doses but did not significantly increase necrotic or apoptotic cell death (Marimpietri et al, 2007; Misawa et al, 2003). A recent meta-analysis of NB tumour mRNA expression microarrays suggested rapamycin as a potential therapeutic compound (De Preter et al, 2009) and primary NB tumours show expression of activated Akt and mTOR (Johnsen et al, 2008). It was also observed that rapamycin treatment of xenografted NB tumours significantly extended animal survival and decreased tumour volume (Johnsen et al, 2008; Marimpietri et al, 2007). In these studies, it was suggested that rapamycin exerts its antitumour effects by inhibiting both angiogenesis and cell proliferation. Our study suggests that TICs are a clinically relevant target within the bulk xenograft tumour as tumour weight was decreased 82.6% and TIC self-renewal capacity was dramatically decreased in vivo in addition to decreased TIC survival, proliferation and sphere formation in vitro. These results are the first demonstration that rapamycin is effective against NB TICs in vivo and that it can act as a potent and selective tumour cell death inducer, suggesting that rapamycin or a rapamycin analogue may be a valuable chemotherapy adjuvant in a subset of NB patients. Based on preclinical data generated in this study as well as the observation that a combination of rapamycin and vinblastine is effective against NB12 xenograft tumours (L. Z. and S. B., unpublished data), a multicenter North American phase 1 study has opened to evaluate rapamycin in combination with vinblastine for paediatric solid tumours.
Of those compounds identified as having the greatest selectivity for NB TICs (>10-fold selectivity compared to SKPs), the majority are known chemotherapeutics including teniposide and vinblastine. This was surprising since TICs from other tumours have been shown to be resistant to various chemotherapeutics and the primary screen was conducted with NB TICs derived from a heavily-treated, multiple relapse patient (NB12). These hits were subsequently confirmed with additional patient TICs. One unique characteristic of NB is that advanced stage tumours are sensitive to chemotherapy although clinical progression inevitably occurs (Thiele, 1998). Interestingly, several of the chemotherapeutics found to be cytotoxic against NB12 cells were not cytotoxic to NB67 cells (colchicine, podophyllotoxin, vincristine and vinblastine). NB67 was isolated from the same patient as NB12 but after a period of 2 years during which the patient received additional treatments. Thus, NB67 may be representative of TICs with increased drug resistance. These observations suggest that it may be possible to use TICs to predict individual patient responses to chemotherapy or to study molecular mechanisms of relapse and drug resistance.
This study represents the first chemical screen to compare a primary human TIC and a normal neural crest-like non-transformed counterpart. This type of parallel screen has enormous potential to predict non-tumour-specific cell toxicity at a very early point in the drug discovery process. Furthermore, our results highlight the multiple pathways used by NB cells for survival and proliferation and have identified pathways that may be relevant for NB therapy. Importantly, we have demonstrated that NB TICs from multiple patients show more commonalities with each other than with an established NB cell line. The availability of a parallel screen with patient TICs and normal non-transformed precursor cells (SKPs) also has the potential to identify patient-specific therapies as demonstrated by the patient-specific response to vinblastine. Finally, we have demonstrated that the identified compounds affect TIC renewal both in vitro and in vivo in a solid tumour.
MATERIALS AND METHODS
Cell culture
SKPs (FS89, FS90, FS99, FS105, FS107) were isolated from neonatal foreskin and cultured in serum-free proliferation media (PM) as previously described (Biernaskie et al, 2006; Toma et al, 2005) following ethics approval by The Hospital for Sick Children Research Ethics Board. SKPs were used between passages 1–5. NB TICs (NB12, NB67, NB88R2, NB122R) were isolated as previously described (Hansford et al, 2007). NB TICs were used between passages 3–15. Tumour samples and bone marrow aspirates were obtained from consented patients as approved by The Hospital for Sick Children Research Ethics Board. SMS-KCNR cells were obtained from Dr. Carol Thiele (NIH) and cultured as spheres in PM. SH-SY5Y and SK-N-AS cells were cultured in PM.
Compound libraries and small molecules
The LOPAC1280™ library (Sigma), Prestwick Chemical Library® (Prestwick Chemical, Inc.) and the Spectrum Collection (MicroSource Discovery Systems, Inc.) which include 4383 biologically-active molecules, off-patent drugs and natural products, were provided by the SMART Facility (Samuel Lunenfeld Research Institute, Mt Sinai Hospital, Toronto, Canada). Compound libraries were dissolved in DMSO at 10 mM using the BioMek FX (Beckman Coulter, Inc.) and re-aliquotted as 0.1 mM aqueous solutions prior to dispensing into the assay plates. Additional compounds were purchased from Sigma with the exception of rapamycin (Calbiochem), DECA-14 (Dr. Susan Rotenberg, CUNY-Queens) and parthenolide (BioMol). Dilutions were made in DMSO for secondary assays.
Primary screening assay
The Beckman BioMek FX and the Samuel Lunenfeld Research Institute High-Throughput Screening Robotics platform were used for cell seeding, treatment and viability assessment. NB TICs (NB12, NB88R2, NB122R), SKPs (FS90) and NB cell lines (SMS-KCNR, SK-N-AS, SH-SY5Y) were cultured to ∼85% confluency, dissociated and resuspended in PM + 30% SKPs conditioned media (CM). Three-thousand cells were seeded in 100 µl per well in non-tissue culture-treated 96-well plates (Corning Life Sciences). Compounds were immediately added to a final concentration of 5 µM (LOPAC1280) or 1 µM (Prestwick, Spectrum). These concentrations were chosen to obtain hit rates in the 1–2% range, as estimated via pilot experiments. Cells treated with 0.1% DMSO vehicle alone (eight wells/plate) were used as the negative controls, while media only (eight wells/plate) was used to determine the assay noise. Ten microlitre of alamarBlue® was added to each well after 30 h and incubated an additional 24 h. Fluorescence intensity was measured using PHERAstar SpectraMax Plus384 microplate reader (BMG LABTECH) with an excitation filter of 535 nm and an emission filter of 590 nm.
Assay quality and hit selection
Under these conditions, the alamarBlue® signal displayed a linear response with time, background was minimal, and the dynamic range satisfactory (i.e. >10). For both NB TICs and SKPs, the variability of alamarBlue® signals was low, with coefficient of variation values ranging between 3.5 and 4.5% across the plates, and the dimensionless, statistical parameters Z′ and Z factors greater than 0.5 (Zhang et al, 1999), suggesting an excellent assay quality. The results were normalized to negative controls using the formula 100*(signal-noise/negative control-noise) to obtain cell survival percentage. The results were subjected to statistical analysis and corrected for systematic errors using the B-score method (Brideau et al, 2003). Compounds with a normalized signal ≥3 standard deviations away from the mean B score of the sample population were considered hits.
To confirm hits, compounds were cherry-picked from stock library plates and diluted in water to generate a 3-point, two-fold dilution series. Cells were seeded, treated with compound and assayed for alamarBlue® reduction as above. IC50 curves were then generated for confirmed hits from a 10-point, two-fold dilution dose curve of each compound in triplicate.
Sphere-formation assay
Cells were seeded in triplicate in 96-well non-tissue culture-treated plates at 3000 cells/well (2000 cells/well for NB12) in 50 µl PM + 30% SKPs CM. Compounds were diluted in PM + CM to the indicated concentrations and immediately added in a volume of 50 µl. Cells were retreated 3 days post-plating and fixed with 4% paraformaldehyle (Electron Microscopy Sciences) at day 6. Sphere number was determined manually. Percentage of control sphere number was calculated as (mean sphere number for treated wells/mean sphere number of 0.05% DMSO-treated wells)*100. EC50 curves were generated using GraphPad Prism software (GraphPad Software, Inc.).
Cell viability
10,000 cells were seeded in triplicate in 24-well non-tissue culture-treated plates in 1 ml PM + CM containing compounds at indicated concentrations or 0.05% DMSO. Spheres were collected, dissociated in trypan blue and viable cells counted. For knockdown experiments, NB TICs were infected with either mock treatment or lentivirus encoding shGFP_437, shFRAP1_193, shRaptor_273, shRaptor_275 and shRictor_333 at an MOI of 10. Seventy-two hours post-infection, the virus was removed and cells were seeded in triplicate at a density of 10,000 per ml in 24-well plates. The remaining cells were used for RNA isolation to determine the efficiency of knockdown by qRT-PCR. Viable cell numbers were determined as above.
Nuclear morphology
40,000 NB12 cells were seeded on 5% lysine/2% laminin coated glass chamber slides (Nunc Lab-Tek). Cells were treated in duplicate with DMSO, 100 nM DECA-14 or 50 nM rapamycin 24 h after plating. At indicated time points, cells were fixed with 4% paraformaldehyde and 0.5 µg/ml Hoechst 33342 (Sigma), washed with PBS− and mounted with Geltol (Thermo Shandon). Nuclei from 15 random fields were quantified using a 40× objective.
Propidium iodide staining for DNA content
200,000 NB TICs were seeded in 25 cm2 flasks and treated with DMSO, 100 nM DECA-14 or 50 nM rapamycin after 48 h. At indicated times, cells were collected, washed in PBS− and fixed in 70% ethanol. Fixed cells were treated with RNaseA, stained with propidium iodide at 37 °C for 1 h and the cellular DNA content analysed with a FACScan (Becton Dickinson) and FlowJo software (Tree Star, Inc.)
Immunoblotting analyses
200,000 NB TICs were seeded in 25 cm2 flasks. Cells were treated with DMSO, 100 nM DECA-14 or 50 nM rapamycin 48 h post-plating. At indicated times, cells were collected, washed in cold PBS− and lysates were prepared in NP-40 lysis buffer. Equal amounts of protein were resolved on polyacrylamide gels and subjected to immunoblotting with the following antibodies: anti-cleaved human PARP, anti-phospho-p70S6K (T389), anti-p70S6K, anti-phospho-S6 ribosomal protein (S235/236), anti-S6RP, anti-phospho-p42/p44 MAPK (T202/Y204 ERK1/2), anti-ERK1 (Santa Cruz), anti-phospho-p38, anti-p38, anti-PKC α/β Substrate, anti-phospho-Akt (S473), anti-Akt, anti-γ-tubulin (Sigma) and anti-GAPDH (Ambion). Unless otherwise noted, primary antibodies were supplied by Cell Signaling Technologies. HRP-conjugated goat anti-mouse IgG and goat anti-rabbit IgG secondary antibodies were used.
Microarray and bioinformatics
NB12, NB88R2 and NB122R cells were treated with 100 nM DECA-14 or DMSO for 24 h. Cells were collected and lysed in Trizol and RNA was purified using RNeasy mini kit (Qiagen). RNA samples were analysed on Affymetrix GeneChip Human Gene 1.0 ST Arrays. Microarray data are available in the ArrayExpress database (accession number E-MEXP-2835). The data were background corrected and normalized using the Robust Multichip Average (RMA) procedure implemented in the Affymetrix Expression Console software. Gene-level expression summaries were computed based on all core probes. Differential gene expression was assessed between the DECA-treated and untreated cells using the LIMMA Bioconductor package (Smyth, 2004). The F-statistic with Benjamini–Hochberg (BH) multiple testing correction implemented in the eBayes function was used to assess the significance of differential expression. Those genes with BH-corrected p-value <0.05 were considered statistically significant. Genes that showed evidence for differential expression based on both uncorrected p-value <0.01 and BH-corrected p-value <0.05 were analysed with Ingenuity Pathway Analysis software (http://www.ingenuity.com) for functional enrichment. Microarray results were confirmed by multiplex semi-quantitative RT-PCR. cDNA was prepared using the Superscript First Strand Synthesis System (Invitrogen) with 2.5 µg total RNA and 50 ng random hexamer primer. 0.5 µl of cDNA was amplified using primers of the specific gene and that of an internal control (GAPDH or β2-microglobulin) as detailed in SI7.
The paper explained
PROBLEM
Fewer than 40% of patients older than 1 year with high-risk neuroblastoma (NB) survive due to disease relapse and extensive metastatic disease. For those patients who survive their disease, there is significant morbidity due to treatment-related toxicities. We hypothesize that new therapies are needed that target cancer stem cell-like tumour initiating cells (TICs) to provide durable cures for NB. These new therapies must not affect normal stem cells in these young patients.
RESULTS
Using a high-throughput cell-based screening assay, we identified compounds that selectively targeted NB TICs while having little effect on normal paediatric stem cells. We further characterized two compounds and demonstrate that dequalinium analogue, C-14 linker (DECA-14) and rapamycin prevent TIC self-renewal in vitro and in vivo.
IMPACT
Both DECA-14 and rapamycin may have potential use in the treatment of NB. On the basis of this study, a multicenter North American phase 1 study has opened to evaluate rapamycin in combination with vinblastine for paediatric solid tumours.
In vivo assays of tumourigenicity
Four- to 5-week-old NOD/SCID mice (Charles River Laboratories) were housed in a facility certified by the Canadian Council of Animal Care and used for this study according to a protocol approved by The Hospital for Sick Children Animal Care Committee. 50,000 NB TICs (NB12 or NB88R2) were resuspended in 50 µl PM, mixed 1:1 with Matrigel (BD Biosciences) and immediately injected subcutaneously into NOD/SCID mice. For SK-N-AS xenograft tumours, 200,000 SK-N-AS cells cultured in PM were resuspended in 20 µl PBS, mixed 1:3 with Matrigel (Trevigen) and injected as above. Tumour growth was monitored weekly and treatment was begun when tumour size reached 0.5 cm2. For experiments with DECA-14, mice were injected intraperitoneally (IP) every other day with DECA-14 (2.5 mg/kg, 14 days or 4.5 mg/kg, 10 days) or vehicle (0.36% citrate). Animals treated with 2.5 mg/kg DECA-14 received daily subcutaneous injections of lactated Ringer's solution. For rapamycin treatment, mice were injected IP daily for 14 days with rapamycin (3 mg/kg, prepared every other day), vinblastine (0.25 mg/kg) or vehicle (0.2% carboxymethylcellulose). All mice underwent complete necropsy examination. A portion of the tumour was lysed by mechanical homogenization in Loading Buffer. Equal protein amounts were separated on a 10% polyacrylamide gel and immunoblotted as above. Tumour and organs were fixed in 10% formalin for 24 h before paraffin embedding and haematoxylin-and-eosin (H&E) staining. Tumour sections were prepared for immunohistochemical analysis by standard protocols and stained for phospho-S6RP (1:100, Cell Signaling Technology) as described by the manufacturer.
Re-isolation of TICs from treated xenograft tumours
Compound- or vehicle-treated tumours were cut into 2 mm2 pieces and enzymatically dissociated with collagenase type XI (1 mg/ml in H2O, Sigma) for 15–45 min at 37 °C. Tumour cells were triturated and filtered through a 40 µm cell strainer. To determine the sphere formation capacity of treated tumours, dissociated cells were plated in 24-well non-tissue culture-treated plates in PM + CM containing 0.8% methylcellulose (Sigma) and sphere formation was scored after 10 days. To examine the ability of treated tumour to generate secondary tumour in vivo, 50,000 dissociated tumours cells were resuspended in 50 µl PM, mixed 1:1 with Matrigel (BD Biosciences) and injected subcutaneously into 5-week-old NOD/SCID mice. Tumour growth was monitored twice weekly. Animals were sacrificed when tumour diameter reached approximately 1 cm. Tumour volume upon autopsy was calculated as 4/3πabc where a, b and c are tumour radii (Sorensen et al, 2001).
Statistical analyses
Statistical analyses were performed with GraphPad Prism 4.0 using an unpaired, two-tailed t-test with p < 0.05 as the significance cutoff. To analyse the secondary tumour formation in Fig 4C, the SAS Procedure PROC LIFETEST was used (SAS v8.02). The time to tumour formation was determined as the shortest time for tumour formation among animals injected with the same tumour cells. The Wilcoxon and the Log-Rank statistics were used to determine whether a statistical difference exists between the times to tumour occurrence in these two groups (DECA-14, Control).
Acknowledgments
We thank Frederick Vizeacoumar at the SMART Facility for assistance with robotics and data analysis as well as members of the Kaplan and Miller laboratories especially Jean-François Lavoie for statistical advice. This work was supported by the Canadian Institutes of Health Research, the National Cancer Institute of Canada, The James Birrell Fund for Neuroblastoma Research, Lilah's Fund, McLaughlin Centre for Molecular Medicine, Solving Kid's Cancer, Ontario institute for Cancer Research, Terry Fox Research Institute and the Canadian Stem Cell Network. F. D. M. is an HHMI International Research Scholar and F. D. M. and D. R. K. hold Canada Research Chairs.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author Contributions
KMS, AD, and DRK conceived and designed the experiments. KMS, AD, MF, NG, LZ and KMB performed the experiments. KMS, AD, and OM analysed the data. SAR, LMH, FDM, HY, MSI, JM, MAM, SB, JLW contributed reagents or analysis tools. KMS and DRK wrote the paper.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Biernaskie JA, McKenzie IA, Toma JG, Miller FD. Isolation of skin-derived precursors (SKPs) and differentiation and enrichment of their Schwann cell progeny. Nat Protoc. 2006;1:2803–2812. doi: 10.1038/nprot.2006.422. [DOI] [PubMed] [Google Scholar]
- Biernaskie J, Paris M, Morozova O, Fagan BM, Marra M, Pevny L, Miller FD. SKPs Derive from Hair Follicle Precursors and Exhibit Properties of Adult Dermal Stem Cells. Cell Stem Cell. 2009;5:610–623. doi: 10.1016/j.stem.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodden WL, Palayoor ST, Hait WN. Selective antimitochondrial agents inhibit calmodulin. Biochem Biophys Res Commun. 1986;135:574–582. doi: 10.1016/0006-291x(86)90032-x. [DOI] [PubMed] [Google Scholar]
- Brideau C, Gunter B, Pikounis B, Liaw A. Improved statistical methods for hit selection in high-throughput screening. J Biomol Screen. 2003;8:634–647. doi: 10.1177/1087057103258285. [DOI] [PubMed] [Google Scholar]
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman JE, Miller DS, Coward P, Smith LH, Teng NN. Study of the selective cytotoxic properties of cationic, lipophilic mitochondrial-specific compounds in gynecologic malignancies. Gynecol Oncol. 1990;39:72–79. doi: 10.1016/0090-8258(90)90402-7. [DOI] [PubMed] [Google Scholar]
- Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- De Preter K, De Brouwer S, Van Maerken T, Pattyn F, Schramm A, Eggert A, Vandesompele J, Speleman F. Meta-mining of neuroblastoma and neuroblast gene expression profiles reveals candidate therapeutic compounds. Clin Cancer Res. 2009;15:3690–3696. doi: 10.1158/1078-0432.CCR-08-2699. [DOI] [PubMed] [Google Scholar]
- Fernandes KJL, McKenzie IA, Mill P, Smith KM, Akhavan M, Barnabe-Heider F, Biernaskie J, Junek A, Kobayashi NR, Toma JG, et al. A dermal niche for multipotent adult skin-derived precursor cells. Nat Cell Biol. 2004;6:1082–1093. doi: 10.1038/ncb1181. [DOI] [PubMed] [Google Scholar]
- Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010;120:41–50. doi: 10.1172/JCI41004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeano E, Nieto E, Garcia-Perez AI, Delgado MD, Pinilla M, Sancho P. Effects of the antitumoural dequalinium on NB4 and K562 human leukemia cell lines. Mitochondrial implication in cell death. Leuk Res. 2005;29:1201–1211. doi: 10.1016/j.leukres.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Gheeya JS, Chen QR, Benjamin CD, Cheuk AT, Tsang P, Chung JY, Metaferia BB, Badgett TC, Johansson P, Wei JS, et al. Screening a panel of drugs with diverse mechanisms of action yields potential therapeutic agents against neuroblastoma. Cancer Biol Ther. 2009;8:2386–2395. doi: 10.4161/cbt.8.24.10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, Holyoake TL. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99:319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Hansford LM, McKee AE, Zhang L, George RE, Gerstle JT, Thorner PS, Smith KM, Look AT, Yeger H, Miller FD, et al. Neuroblastoma cells isolated from bone marrow metastases contain a naturally enriched tumor-initiating cell. Cancer Res. 2007;67:11234–11243. doi: 10.1158/0008-5472.CAN-07-0718. [DOI] [PubMed] [Google Scholar]
- Johnsen JI, Segerstrom L, Orrego A, Elfman L, Henriksson M, Kagedal B, Eksborg S, Sveinbjornsson B, Kogner P. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene. 2008;27:2910–2922. doi: 10.1038/sj.onc.1210938. [DOI] [PubMed] [Google Scholar]
- Konig H, Holtz M, Modi H, Manley P, Holyoake TL, Forman SJ, Bhatia R. Enhanced BCR-ABL kinase inhibition does not result in increased inhibition of downstream signaling pathways or increased growth suppression in CML progenitors. Leukemia. 2008;22:748–755. doi: 10.1038/sj.leu.2405086. [DOI] [PubMed] [Google Scholar]
- Laverdiere C, Liu Q, Yasui Y, Nathan PC, Gurney JG, Stovall M, Diller LR, Cheung N-K, Wolden S, Robison LL, et al. Long-term Outcomes in Survivors of Neuroblastoma: A Report From the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2009;101:1131–1140. doi: 10.1093/jnci/djp230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Marimpietri D, Brignole C, Nico B, Pastorino F, Pezzolo A, Piccardi F, Cilli M, Di Paolo D, Pagnan G, Longo L, et al. Combined therapeutic effects of vinblastine and rapamycin on human neuroblastoma growth, apoptosis, and angiogenesis. Clin Cancer Res. 2007;13:3977–3988. doi: 10.1158/1078-0432.CCR-06-2757. [DOI] [PubMed] [Google Scholar]
- Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- Martel RR, Klicius J, Galet S. Inhibition of the immune response by rapamycin, a new antifungal antibiotic. Can J Physiol Pharmacol. 1977;55:48–51. doi: 10.1139/y77-007. [DOI] [PubMed] [Google Scholar]
- Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. N Engl J Med. 1999;341:1165–1173. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- Misawa A, Hosoi H, Tsuchiya K, Sugimoto T. Rapamycin inhibits proliferation of human neuroblastoma cells without suppression of MycN. Int J Cancer. 2003;104:233–237. doi: 10.1002/ijc.10914. [DOI] [PubMed] [Google Scholar]
- Pampaloni F, Reynaud EG, Stelzer EHK. The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol. 2007;8:839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- Qin D, Sullivan R, Berkowitz WF, Bittman R, Rotenberg SA. Inhibition of protein kinase C(alpha) by dequalinium analogues: dependence on linker length and geometry. J Med Chem. 2000;43:1413–1417. doi: 10.1021/jm990340z. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Rietze RL. Neural stem cells and neurospheres-re-evaluating the relationship. Nat Methods. 2005;2:333–336. doi: 10.1038/nmeth758. [DOI] [PubMed] [Google Scholar]
- Reynolds CP, Biedler JL, Spengler BA, Reynolds DA, Ross RA, Frenkel EP, Smith RG. Characterization of human neuroblastoma cell lines established before and after therapy. J Natl Cancer Inst. 1986;76:375–387. [PubMed] [Google Scholar]
- Rotenberg SA, Smiley S, Ueffing M, Krauss RS, Chen LB, Weinstein IB. Inhibition of rodent protein kinase C by the anticarcinoma agent dequalinium. Cancer Res. 1990;50:677–685. [PubMed] [Google Scholar]
- Sancho P, Galeano E, Nieto E, Delgado MD, Garcia-Perez AI. Dequalinium induces cell death in human leukemia cells by early mitochondrial alterations which enhance ROS production. Leuk Res. 2007;31:969–978. doi: 10.1016/j.leukres.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Schneider Berlin KR, Ammini CV, Rowe TC. Dequalinium induces a selective depletion of mitochondrial DNA from HeLa human cervical carcinoma cells. Exp Cell Res. 1998;245:137–145. doi: 10.1006/excr.1998.4236. [DOI] [PubMed] [Google Scholar]
- Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo) 1975;28:727–732. doi: 10.7164/antibiotics.28.727. [DOI] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- Sorensen AG, Patel S, Harmath C, Bridges S, Synnott J, Sievers A, Yoon Y-H, Lee EJ, Yang MC, Lewis RF, et al. Comparison of diameter and perimeter methods for tumor volume calculation. J Clin Oncol. 2001;19:551–557. doi: 10.1200/JCO.2001.19.2.551. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Stone M, Marshall JF, Uberall F, Rotenberg SA. Photo-induced inactivation of protein kinase calpha by dequalinium inhibits motility of murine melanoma cells. Mol Pharmacol. 2000;58:729–737. doi: 10.1124/mol.58.4.729. [DOI] [PubMed] [Google Scholar]
- Thiele CJ. Neuroblastoma. In: Masters JRW, Palsson B, editors. Human Cell Culture. Lancaster, UK: Kluwer Academic Publishers; 1998. pp. 21–53. [Google Scholar]
- Toma J, Akhavan M, Fernandes K, Barnabe-Heider F, Sadikot A, Kaplan D, Miller F. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol. 2001;3:78–784. doi: 10.1038/ncb0901-778. [DOI] [PubMed] [Google Scholar]
- Toma JG, McKenzie IA, Bagli D, Miller FD. Isolation and characterization of multipotent skin-derived precursors from human skin. Stem Cells. 2005;23:727–737. doi: 10.1634/stemcells.2004-0134. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Dirks PB. Cancer stem cells: at the headwaters of tumor development. Annu Rev Pathol. 2007;2:175–189. doi: 10.1146/annurev.pathol.2.010506.091847. [DOI] [PubMed] [Google Scholar]
- Weiss MJ, Wong JR, Ha CS, Bleday R, Salem RR, Steele GD, Chen LB. Dequalinium, a topical antimicrobial agent, displays anticarcinoma activity based on selective mitochondrial accumulation. Proc Natl Acad Sci. 1987;84:5444–5448. doi: 10.1073/pnas.84.15.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright RGM, Wakelin LPG, Fieldes A, Acheson RM, Waring MJ. Effects of ring substituents and linker chains on the bifunctional intercalation of diacridines into deoxyribonucleic acid. Biochemistry. 1980;19:5825–5836. doi: 10.1021/bi00566a026. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- Zhang J-H, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhuo S, Allison WS. Inhibition and photoinactivation of the bovine heart mitochondrial F1-ATPase by the cytotoxic agent, dequalinium. Biochem Biophys Res Commun. 1988;152:968–972. doi: 10.1016/s0006-291x(88)80378-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.