

Abstract
Metastasis is the major cause of carcinoma-induced death, but mechanisms involved are poorly understood. Metastasis crucially involves epithelial-to-mesenchymal transition (EMT), causing loss of epithelial polarity. Here we identify Annexin A1 (AnxA1), a protein with important functions in intracellular vesicle trafficking, as an efficient suppressor of EMT and metastasis in breast cancer. AnxA1 levels were strongly reduced in EMT of mammary epithelial cells, in metastatic murine and human cell lines and in metastatic mouse and human carcinomas. RNAi-mediated AnxA1 knockdown cooperated with oncogenic Ras to induce TGFβ-independent EMT and metastasis in non-metastatic cells. Strikingly, forced AnxA1 expression in metastatic mouse and human mammary carcinoma cells reversed EMT and abolished metastasis. AnxA1 knockdown stimulated multiple signalling pathways but only Tyk2/Stat3 and Erk1/2 signalling were essential for EMT.
Keywords: Annexin A1, EMT, metastasis suppressor, signalling, Stat3
INTRODUCTION
More than 80% of all cancers are tumours of epithelial origin (carcinomas), which cause >80% of cancer-related deaths due to metastasis concomitant with organ failure (Gupta & Massague, 2006). Thus, a better understanding of cellular and molecular mechanisms leading to metastatic dissemination of carcinoma cells is of utmost importance. A central process in metastasis is epithelial-to-mesenchymal transition (EMT), i.e. the conversion of sessile, polarized epithelial cells to motile fibroblastoid cells expressing mesenchymal markers (reviewed in Huber et al, 2005; Hugo et al, 2007; Kalluri & Weinberg, 2009). Recently, EMT and metastasis were shown to involve deregulation of epithelial polarity, set up and maintained by multiple, complex molecular machines (Humbert et al, 2008; Kalluri & Weinberg, 2009; Tanos & Rodriguez-Boulan, 2008). EMT also emerges as a hallmark of breast cancer initiating/stem cells (Gupta et al, 2009; Mani et al, 2008).
One of many cellular models to study EMT is the EpH4/EpRas cell system. These cells retain full epithelial polarity and allow combined in vitro/in vivo studies, thus being particularly well suited to investigate the relationship between EMT and metastasis (Grunert et al, 2003; Huber et al, 2005). Parental EpH4 cells are spontaneously immortalized, non-tumorigenic mammary epithelial cells (Reichmann et al, 1992), which exhibit normal, physiological responses to relevant cytokines (Oft et al, 1996). EpH4 cells expressing oncogenic RasV12 (EpRas) remain epithelial but are tumorigenic and promote late steps in metastasis. Exposure of EpRas cells to transforming growth factorβ (TGFβ) in culture or in mouse tumours caused them to undergo EMT, stabilized by an autocrine TGFβ1 loop (EpRasXT; Janda et al, 2002a; Oft et al, 1998). These cells invaded collagen gels and induced metastasis after tail vein injection (Janda et al, 2002a).
In addition to Ras plus TGFβ signalling, estradiol-induced activation of a c-FosER fusion protein also caused EMT in EpH4 cells (EpFosER; Eger et al, 2000; Reichmann et al, 1992). Marker analysis also demonstrated EMT to occur in strongly metastatic tumours from MMTV-neu/TGFβ1 double transgenic mice (Jechlinger et al, 2006). Expression profiling in the EpH4/EpRas model identified a number of genes essential for both EMT and metastasis (Jechlinger et al, 2002, 2006; Lahsnig et al, 2009; Waerner et al, 2006). Since a large number of diverse signalling pathways (Etienne-Manneville, 2008; Huber et al, 2005)—including those regulating epithelial polarity (Aranda et al, 2008; Humbert et al, 2008; Tanos & Rodriguez-Boulan, 2008)—induce or contribute to EMT, respective molecular mechanisms are still ill understood.
Recently it has been shown that Annexin A1 (AnxA1) is downregulated in progressed human breast cancer samples as well as in prostate, esophageal and advanced head and neck cancers, but upregulated in other cancers (reviewed in Lim & Pervaiz, 2007; Mussunoor & Murray, 2008). To date, however, metastatic capacity has not been correlated with AnxA1 expression levels. AnxA1 was identified as a mediator of glucocorticoid-dependent anti-inflammatory processes (Lim & Pervaiz, 2007), which did not reveal a clear, causal connection with EMT/metastasis. AnxA1 shows Ca2+-dependent interaction with ceramide-based plasma membrane glycosphingolipids (Babiychuk et al, 2008) and is involved in many aspects of vesicle trafficking, including inward vesiculation of late endosomes into multivesicular bodies (MVB) and enhanced internalization of the epidermal growth factor receptor (EGFR; Futter & White, 2007; Gerke et al, 2005; White et al, 2006). AnxA1 also inhibits phospholipase A2 (PLA2), is tyrosine-phosphorylated by the EGFR and modulates Erk1/2 and p38MAPK signalling (Lim & Pervaiz, 2007; Yang et al, 2006), but functional consequences of these events in epithelial cells remain to be identified.
In this paper, we show that expression of AnxA1 is downregulated in metastatic tumours and further identify AnxA1 as an EMT/metastasis suppressor.
RESULTS
Expression of AnxA1 is downregulated during EMT and metastasis
EpRasXT cells showed strong downregulation of AnxA1 as compared to EpH4 and EpRas cells, as shown by messenger RNA (mRNA) expression profiling (not shown) and qRT-PCR (Fig 1A). Western Blot (WB) analysis showed a corresponding downregulation of AnxA1 protein in EpRasXT cells (Fig 1B). In contrast, five other annexin family members were expressed at similar levels in EpH4, EpRas and EpRasXT cells, showing specific loss of AnxA1 during EMT (Fig S2A). A similar loss of AnxA1 protein was seen in EpFosER cells after EMT induced by estradiol-activated cFosER (Fig 1C; Eger et al, 2000; Reichmann et al, 1992). AnxA1 levels were also downregulated in dedifferentiated and highly metastatic cell lines (HS578T, MDAMB231, SKBR3, ZR751; Fig 1D red) while non-tumorigenic lines (MCF10A, hMEC; Fig 1D green) or epitheloid, tumorigenic cell lines (MCF7, BT474, T47D; Fig 1D blue) showed high or intermediate AnxA1 protein levels, respectively.
Figure 1. AnxA1 is downregulated during EMT and in metastatic mouse and human tumours.

A,B. EpH4, EpRas and EpRasXT cells were analyzed for AnxA1-mRNA (A) by qRT-PCR (normalized to GAPDH) and AnxA1-protein (B) by Western Blot (WB) analysis, respectively.
C. WB analysis of AnxA1 protein levels in EpFosER cells not treated (−) or treated for 7 days with estradiol (+).
D. AnxA1 protein levels analyzed by WB (normalized to actin) in strongly (red) and partially (blue) dedifferentiated human mammary carcinoma cell lines, as compared to normal mammary epithelial cell lines (green).
E. WB analysis for AnxA1 protein in spontaneous mammary gland tumours from MMTV-neu and MMTV-neu x MMTV-TGFβ1 transgenic mice.
F,G. One hundred and forty-one patient samples from two human breast cancer tissue arrays (TA2, TA3) with known case histories were stained for AnxA1 by IHC. (F) Non-metastatic breast carcinomas show brown nuclear and/or cytoplasmic AnxA1 staining which is absent in the metastatic breast carcinomas. (G) AnxA1 staining intensities (scores 0–3+) are depicted in Kaplan–Meier plots for metastasis-free survival (all metastasis types, upper panel) and overall survival (lower panel). For details on in vitro and in vivo models used, see Suppl. Exp. Proc. and Fig S10.
Furthermore, AnxA1 was lost in mammary tumours from MMTV-neu/TGFβ1 double transgenic mice (Fig 1E), which show EMT in vivo as revealed by lack of E-cadherin and high vimentin expression in tumour sections (Jechlinger et al, 2006). Using the Gene Logic database, we also could demonstrate low AnxA1 mRNA levels in 20–80 samples from human prostate, lung and breast carcinomas, while normal breast tissue samples showed high AnxA1 expression (Fig S1A). Likewise, AnxA1 mRNA levels were strongly reduced in 20 invasive human breast carcinomas (Fig S1B; blue), but not in four benign mammary tissue samples (Fig S1B, green). Most importantly, expression analysis of AnxA1 protein by immunohistochemistry (IHC) in two human breast cancer arrays with 16 years documented patients’ histories demonstrated almost exclusive loss of AnxA1 protein expression in 66 primary tumours that progressed to metastasis, while 79 non-metastatic tumours showed detectable to high AnxA1 expression (Fig 1F and G). In conclusion, AnxA1 is downregulated during EMT and metastasis in diverse cell culture and animal models, in human metastatic breast cancer and other carcinoma types pointing to a central role of AnxA1 in tumour progression.
Knockdown of AnxA1 induces EMT
Stable AnxA1 knockdown using shRNAs was performed to address a potential causal function of AnxA1 loss in EMT and metastasis. AnxA1-RNAi induced EMT in EpRas cells (Fig 2A and B), but caused growth arrest/apoptosis in fully polarized murine (EpH4) or human (MCF10A) mammary epithelial cells which are not apoptosis protected by oncogenic ras signalling (not shown). The fibroblastoid EpRas-siAnxA1 cells invaded collagen gels (Fig 2B, top panels), had lost the epithelial marker E-cadherin and strongly expressed the mesenchymal markers vimentin and N-cadherin (Fig 2A and B, bottom panels). This stable AnxA1 knockdown was specific since it did not alter expression of five other annexin family members (Fig S2B).
Figure 2. AnxA1 knockdown induces EMT in EpRas and EpC40 cells.
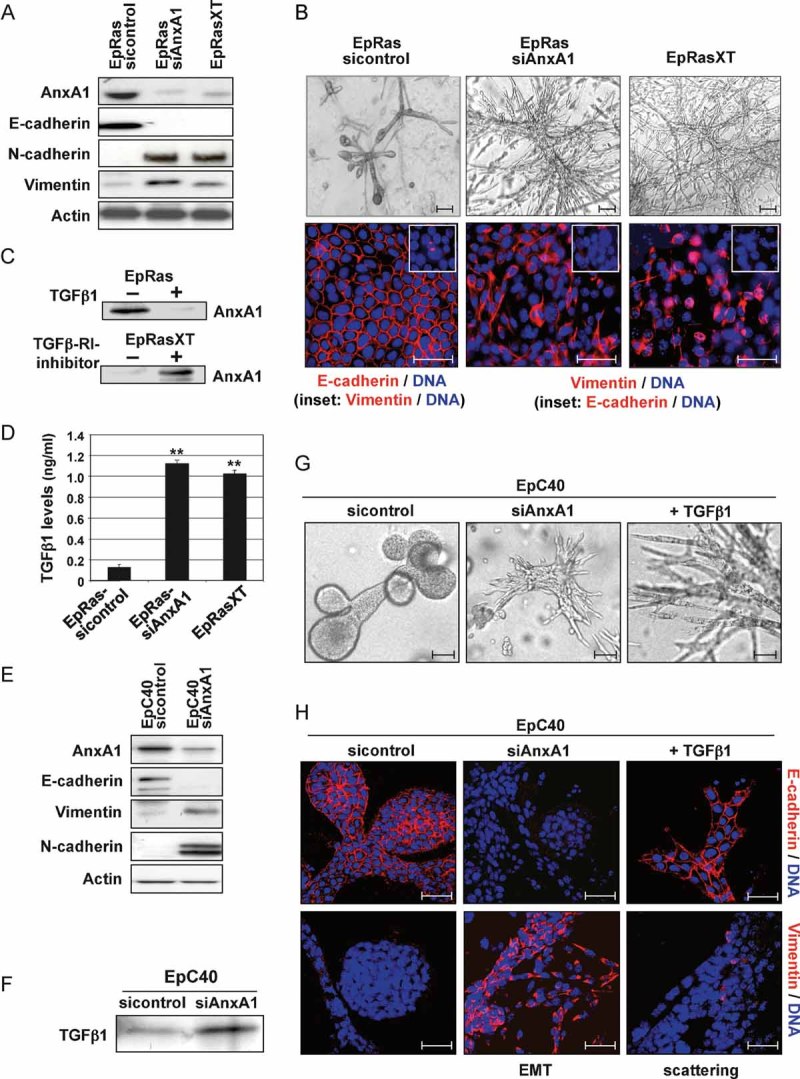
A, E. WB for AnxA1, epithelial (E-cadherin) and mesenchymal (N-cadherin, vimentin) markers in EpRas and EpC40 cells before and after AnxA1 knockdown. Positive control: EpRasXT cells; loading control: β-actin.
B, G, H. The same cell types as in (A) and (E) were cultivated on filters (B, bottom) or in collagen gels (B, top; G, H). Bright field images (B, top; G) and confocal images (B, bottom; H) are shown, the latter stained by immunofluorescence for the EMT markers indicated.
C. EpRas and EpRasXT cells seeded on plastic were treated with 5 ng/ml TGFβ1 (top) and 10 µM TGFβRI inhibitor (bottom), respectively, for 6 days and analyzed for AnxA1 expression by WB.
D, F. Biologically active TGFβ1 or total TGFβ1 were determined by ELISA (D; **p < 0.01) or WB (F), using supernatants or lysates, respectively, from the cell types indicated. For details see Suppl. Exp. Proc.
To determine, if EMT maintained by autocrine TGFβ1 in EpRasXT cells correlates with loss of endogenous AnxA1, we measured AnxA1 protein levels in EpRas and EpRasXT cells treated with TGFβ1 or a potent inhibitor of TGFβRI serine kinase activity (SB 431.542), respectively (see Suppl. Exp. Proc.). TGFβ1 suppressed AnxA1 expression in EpRas cells (Fig 2C, top), while inhibition of TGFβR1 signalling induced strong AnxA1 expression in EpRasXT cells (Fig 2C, bottom). Accordingly, EMT induction in EpRas cells by AnxA1 knockdown caused secretion of TGFβ1 levels comparable to those found in EpRasXT cells (Fig 2D). Respective TGFβ1 levels also cause growth arrest and apoptosis in murine EpH4 and human MCF10A cells, perhaps explaining our inability to stably knock down AnxA1 in these cells in the absence of oncogenic Ras. To determine, whether elevated TGFβ signalling was essential for EMT after AnxA1 knockdown, we employed EpC40 cells, which are apoptosis-protected due to V12RasC40-induced, hyperactive PI3K but not Erk1/2 signalling and do not undergo TGFβ-induced EMT (Janda et al, 2002a). Strikingly, knockdown of AnxA1 in EpC40 cells (Fig 2E) induced complete EMT, yielding fibroblastoid, E-cadherin negative and vimentin/N-cadherin positive cells (Fig 2E, G and H). Exogenous TGFβ1, however, induced only reversible scattering in EpC40 cells, i.e. a fibroblastoid phenotype (Fig 2G, right panels) with plasma membrane E-cadherin but no vimentin expression (Fig 2H, right panels; Grunert et al, 2003; Janda et al, 2002a). Like EpRas-siAnxA1 cells, EpC40-siAnxA1 cells produced enhanced levels of TGFβ1 (Fig 2F). Forced expression of human AnxA1 in EpC40-siAnxA1 cells reversed these mesenchymal cells back to an E-cadherin positive, vimentin negative, epithelial phenotype (Fig S2C and D), confirming that off-target effects played no role in AnxA1-siRNA induced EMT.
Knockdown of AnxA1 confers metastatic potential
Since EMT is a faithful in vitro correlate for late stage tumorigenesis and metastasis in multiple models (Grunert et al, 2003; Hugo et al, 2007; Kalluri & Weinberg, 2009), we addressed potential effects of AnxA1 loss on tumorigenesis and metastasis caused by EpRas- and EpC40 cells in vivo. EpRas-siAnxA1 cells formed larger tumours and more metastases than EpRas cells upon mammary gland fat pad and tail vein injection, respectively (Fig 3A and B; S3A). Strikingly, knockdown of AnxA1 in the non-metastatic EpC40 cells strongly enhanced tumour growth rate (Fig 3C), and induced many, large lung metastases (Fig 3D and E), killing the mice within 27–40 days (Fig 3F). In contrast, EpC40-sicontrol injected mice developed no detectable metastases within 100 days (Fig 3E and F). Cells re-cultivated from the EpC40-siAnxA1 induced lung metastases retained their mesenchymal phenotype and low levels of AnxA1 (Fig S4A and B). In conclusion, knock down of AnxA1 stimulated tumour growth of EpRas and EpC40 cells and most importantly induced metastatic capacity in EpC40 cells, suggesting a key role of AnxA1 in tumour progression.
Figure 3. Knockdown of AnxA1 stimulates tumour growth and metastasis.
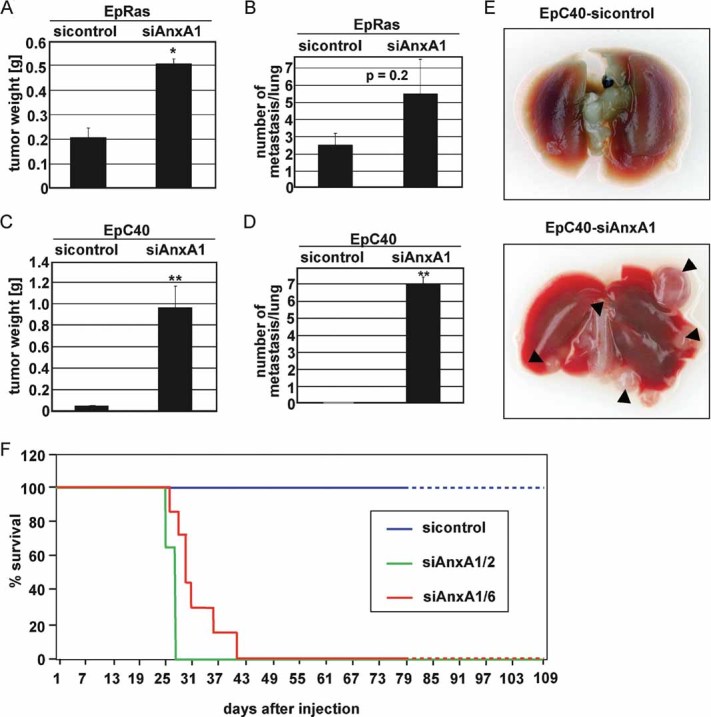
A,D. The indicated cell types were injected into mammary gland fat pads (A, C) or tail veins (B, D) of ≥6 nude mice. Tumour weight (A, C) was determined after 2–3 weeks, while numbers of metastases on lung surfaces were counted after sacrificing the mice after 15–20 (B) and 25–40 days (D; **p = 0.01).
E. Photographs of lungs 35 days after tail vein injection of EpC40-sicontrol and EpC40-siAnxA1 cells. Arrowheads: metastases.
F. Kaplan–Meier plots after tail vein injection of EpC40 cell clones expressing sicontrol- or two different shRNAs targeting AnxA1 (AnxA1/2; −1/6; 7 mice per cell type).
Forced re-expression of AnxA1 reverts EMT and interferes with metastasis
Next, we tested whether AnxA1 re-expression would reverse the mesenchymal phenotype of EpRasXT cells. Inhibition of TGFβR1 caused these cells to form compact, unordered structures of cells still expressing vimentin but re-expressing plasma membrane E-cadherin (Fig S6A; Jechlinger et al, 2002). Indeed, AnxA1 overexpressing EpRasXT (Fig S2E) cells became epithelial on plastic and on filters (Fig S2F), expressing plasma membrane E-cadherin and very little/no vimentin (Fig S2E and F). Thus, AnxA1 re-expression but not blockade of TGFβ-signalling completely reversed EMT of EpRasXT cells.
Consequently we addressed whether forced AnxA1 expression would reverse EMT and suppress metastasis in human MDA-MB-231 mammary carcinoma cells. These cells essentially lack AnxA1 (Figs 1D and 4A), exhibit a stable EMT phenotype, i.e. fibroblastoid morphology, high vimentin, no E-cadherin and cytoplasmic or stressfibre localized β-actin (Fig 4B and C, left panels; Eger et al, 2005) and show high metastatic capacity in xenografts (Fig 4F). This phenotype was maintained even after exposure to multiple pharmacological inhibitors that reverse EMT in the EpRas model (Fig S4C; Janda et al, 2002a).
Figure 4. Forced expression of AnxA1 in MDA-MB-231 cells reverses EMT and suppresses metastasis formations.

A. MDA-MB-231 cells after forced expression of AnxA1 or empty vector were analyzed for AnxA1 and the markers E- and N-cadherin by WB. β-actin: loading control.
B,C. The same cell types as in (A) were cultivated on plastic (B, top), on filters (B, centre, bottom) or in collagen gels (C). Shown are bright field (B, C, top) and confocal images of cells co-stained for the markers indicated (B, C, centre, bottom).
D. Tumour weights 2–3 weeks after fat pad injection of the cell types indicated.
E. Kaplan–Meier plots for survival of three mice injected with the above cell types.
F. Photographs of lungs from mice sacrificed 5 (MDA-MB-231 + empty vector) and 8 weeks (MDA-MB-231 + AnxA1) after tail vein injection.
Strikingly, forced expression of human AnxA1 in MDA-MB-231 cells completely reversed their EMT phenotype into an epithelial one (Fig 4A). MDA-MB-231 + AnxA1 cells formed cobblestone-like epithelial monolayers (Fig 4B), and compact collagen gel structures (Fig 4C), showing plasma-membrane-localized E-cadherin, cortical β-actin and no or little vimentin (Fig 4B and C; right panels). MDA-MB-231 + AnxA1 cells were still tumorigenic (Fig 4D), but no metastases were detectable in tail vein injected mice within 60 days (Fig 4E and F). MDA-MB-231 control cells, however, killed all mice after 37 days through many, large lung metastases (Fig 4E and F). Taken together, forced AnxA1 expression in cells with a constitutive, inhibitor-resistant EMT phenotype induces epithelial polarity and suppresses metastasis, indicating, that AnxA1 is a key player in maintenance of the epithelial phenotype.
TGFβ1-Smad signalling is not essential for AnxA1-siRNA induced EMT
Does AnxA1-RNAi induced EMT require activation of TGFβR-Smad signalling, as suggested by the observed induction of TGFβ1 secretion (Fig 2D and F)? Both EpRas-siAnxA1 and EpC40-siAnxA1 cells showed elevated levels of phospho-Smad2 in the absence of exogenous TGFβ1 (Fig 5A and B), as compared to EpRas- and EpC40-sicontrol cells. To test, if this was a direct effect of AnxA1 knockdown or a consequence of EMT, EpRas cells were co-transfected with siAnxA1-knockdown plasmids and Smad2- or Smad3-dependent reporter gene constructs. In the absence of exogenous TGFβ1, EpRas-siAnxA1 cells showed strong Smad2 reporter gene activation as compared to control cells, hardly enhanced by TGFβ1 (Fig 5C). In contrast, the Smad3 reporter gene was unresponsive to AnxA1-RNAi in the absence of TGFβ1, but clearly stronger activated by TGFβ1 than in control cells (Fig 5C). These results were obtained already 36 h post-transfection, while completion of EMT requires more than 5 days (Janda et al, 2002a; Oft et al, 1996). Thus, the TGFβR-Smad pathway was directly activated by AnxA1 knockdown and not only as a consequence of EMT. Respective co-transfections of EpC40 cells yielded similar results (not shown).
Figure 5. Knockdown of AnxA1 activates TGFβRI/Smad2 signalling.
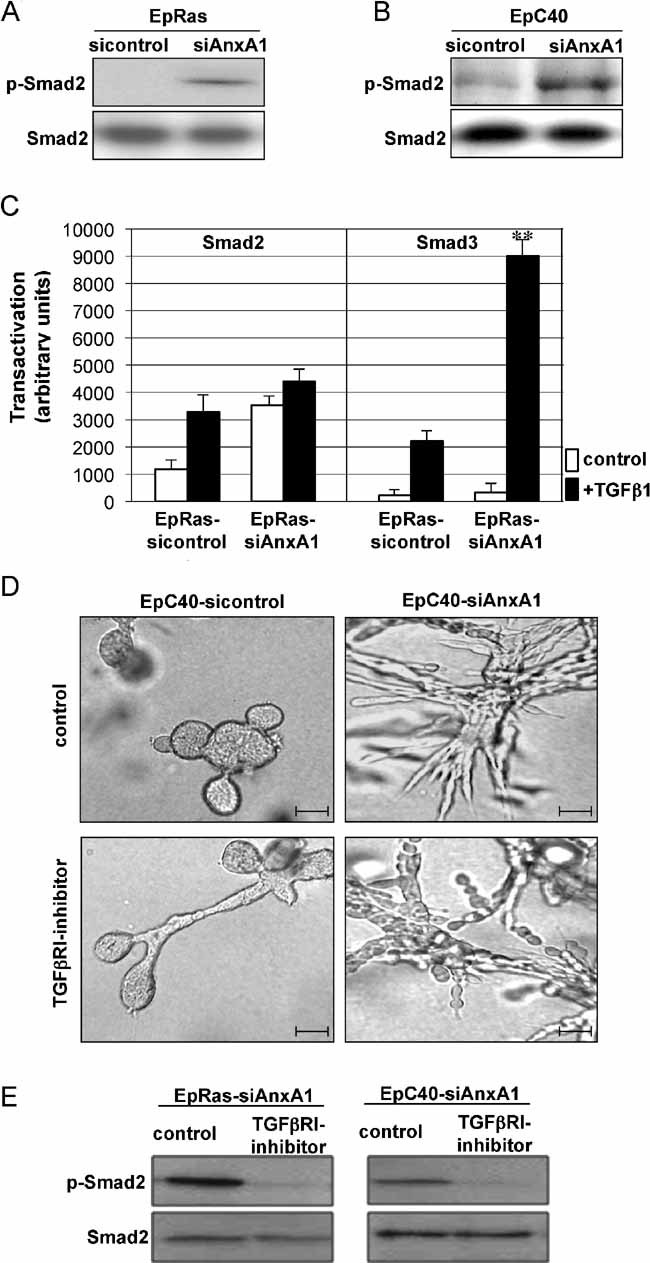
A,B. WB analysis of EpRas (A) and EpC40 cells (B) before and after AnxA1 knockdown for phospho-Smad2 (p-Smad2); loading control: total Smad2.
C. Basal and TGFβ1-induced reporter gene activity of Smad2- or Smad3-specific reporter constructs in EpRas cells co-transfected with control and AnxA1 knockdown vectors (**p = 0.01).
D. Shown are bright field images of EpC40-sicontrol and EpC40-siAnxA1 cells not treated or treated with TGFβRI inhibitor.
E. WB analysis of p-Smad2 before and after treatment with TGFβRI inhibitor. Loading control: total Smad2.
To address, whether or not TGFβ signalling was essential for maintenance of EMT in EpC40-siAnxA1 cells, we exposed these cells to the TGFβRI inhibitor SB 431.542, using EpRasXT cells as controls. Treatment of EpRas-siAnxA1 and EpC40-siAnxA1 cells with the TGFβR1 inhibitor completely abolished phosphorylation of Smad2 (Fig 5E) but treated EpC40-siAnxA1 cells still formed unordered strands of slightly more rounded, vimentin-positive but E-cadherin low/negative cells, similar to untreated EpC40-siAnxA1 cells (Figs 5D and S5B). EpRasXT cells, however, showed the expected reversion of EMT described above (Fig S6A). Thus, TGFβRI/Smad2 signalling is not essential for EMT caused by knockdown of AnxA1.
Jak/Stat3 signalling is essential for EMT induction by loss of AnxA1
Recent findings suggesting an important role for Jak/Stat signalling in tumour progression (Zhao et al, 2009), prompting us to analyze the expression of Stat3 in EpRas and EpC40 cells, before and after AnxA1-RNAi. AnxA1-RNAi enhanced both total and pY-Stat3 protein expression (Fig 6A), but did not affect Stat1 and Stat5 (data not shown). Autocrine IL6 did not contribute to Stat3-induction, (i) since AnxA1 knockdown did not significantly elevate IL6 secretion in EpC40 cells (Fig 6B) and (ii) since neither a neutralizing IL6 antibody nor a pharmacological inhibitor of the p130 IL6 receptor subunit affected AnxA1-RNAi induced EMT (not shown). Since Jaks are essential for Stat signalling (Yu & Jove, 2004), we employed a pan Jak-inhibitor (Jak-inhibitor I, see Suppl. Exp. Proc.) to test, whether elevated Jak/Stat3 signalling was necessary for loss of AnxA1-induced EMT. Indeed, pan Jak-inhibitor had no effect on EpRas and EpC40 control cells (Fig 6C and D; left panels), but completely reversed the EMT of EpRas-siAnxA1 and EpC40-siAnxA1 cells, forming compact hollow structures expressing plasma membrane E-cadherin, cortical β-actin but no vimentin, indistinguishable of control cells (Fig 6C and D; right panels).
Figure 6. Tyk2/Stat 3 signalling is required for EMT induced by knockdown of AnxA1.

A. WB analysis for phospho (pY-) Stat3 and total Stat3 protein in EpRas and EpC40 cells before and after AnxA1 knockdown.
B. IL6 secretion in control and AnxA1 knockdown cells measured by ELISA (**p = 0.01).
C,D. The same cells as in (A) were cultivated in collagen gels and treated with pan-Jak-inhibitor I. Shown are bright field (C) or confocal immunofluorescence images (D) after staining for the markers indicated (insets: 300% magnification of representative parts of main images).
E,F. EpC40-sicontrol and EpC40-siAnxA1 cells in collagen gels were treated or not treated with the Tyk2-inhibitor AG1 for 5 days (E) and stained for the markers indicated (F).
To identify the Jak family member(s) important in AnxA1-RNAi induced EMT, we tried to abolish Jak1, Jak2 and Tyk2 expression and activity by RNAi and specific pharmacological inhibitors. While lentiviral RNAi-knockdown did not yield viable clones with significant downregulation of these Jaks, the Tyk2 inhibitor AG1 reversed the EMT of EpC40-siAnxA1 cells in a similar fashion as the pan Jak-inhibitor (Fig 6E and F). The Jak2 inhibitor AG490, however, did not affect EMT on plastic and inhibited cell proliferation in collagen gels (not shown). Interestingly, elevated Jak/Stat signalling may also play a role in Ras/TGFβ induced EMT, since total and pY-Stat3 were elevated in EpRasXT cells (not shown), in which the Tyk2 inhibitor AG1 partially reversed EMT (compact structures, cytoplasmic E-cadherin, reduced vimentin expression; Fig S6B).
In conclusion, elevated Jak/Stat3 signalling is necessary for AnxA1-RNAi induced EMT, perhaps involving unconventional mechanisms of Stat3 action such as modulation of epithelial polarity by Stat3 (Guo et al, 2006; Ng et al, 2006). Besides the TGFβRI/Smad pathway, AnxA1-RNAi activated two other signalling pathways active in Ras/TGFβ induced EMT, i.e. Erk1/2 and p38MAPK signalling, while PI3K signalling via AKT showed AnxA1-independent hyperactivation through V12RasC40 (Figs S5A). The p38MAPK pathway however, was not essential for AnxA1-RNAi induced EMT, as shown by respective pharmacological inhibitors. Inhibition of MEK/Erk1/2 signalling caused EMT reversal in EpC40-siAnxA1 cells concomitant with upregulation of E-cadherin and loss of vimentin (Fig S5B). Importantly, inhibition of Ras and PI3K signalling by respective pharmacological inhibitors clearly induced cell death in collagen gels (Fig S5C) supporting the findings in normal mammary epithelial cells that knockdown of AnxA1 is highly dependent on apoptosis protection by Ras and PI3K signalling, respectively.
AnxA1 function in EMT: mechanistic approaches
One unifying and therefore mechanistically relevant process during EMT in multiple systems is the upregulation of E-cadherin repressors (Eger et al, 2005; Moreno-Bueno et al, 2008; Peinado et al, 2007), some of which also regulate EMT-associated miRNAs (Wellner et al, 2009). It was thus particularly interesting to determine, if these transcriptional regulators were also upregulated in AnxA1-RNAi induced EMT, which differed from Ras/TGFβ induced EMT by (i) its independence of TGFβRI-signalling and (ii) its response to TGFβRI- and Tyk2 inhibitors (compare Figs 5D, S5B and S6). mRNA levels of snail, slug, deltaEF1, Sip1, Twist2 and Fra-1 were determined by qRT-PCR (Fig 7). Snail, slug and deltaEF1 mRNAs were 5.5–97-fold upregulated in EpC40-siAnxA1 cells, while snail and Fra-1 mRNAs were 7.4 and 4.3-fold elevated in EpRas-siAnxA1 cells, as compared to the respective control cells. The observed upregulation of E-cadherin repressors (exept twist2) supports our conclusion that AnxA1 knockdown indeed induces EMT, but is not directly caused by loss of AnxA1 since these repressors were not upregulated by AnxA1 knockdown in a transient transfection approach (48 h) analyzed via chip analysis (data not shown). This suggests that upregulation of E-cadherin repressors is the consequence of AnxA1-RNAi-induced EMT rather than a direct consequence of AnxA1 knockdown. Most interestingly, reversal of EMT and metastasis suppression in MDA-MB-231 + AnxA1 cells (Fig 4) led to 11–166-fold downregulation of mRNAs for all factors except Sip1. In conclusion, E-cadherin repressor upregulation was much more evident in EpC40-siAnxA1 cells than in EpRas-siAnxA1 cells, while dramatic downregulation of almost all E-cadherin repressors accompanied EMT reversal in human MDA-MB-231 + AnxA1 cells (Fig 7).
Figure 7. Up- or downregulation of E-cadherin repressors by loss or gain of AnxA1 expression.
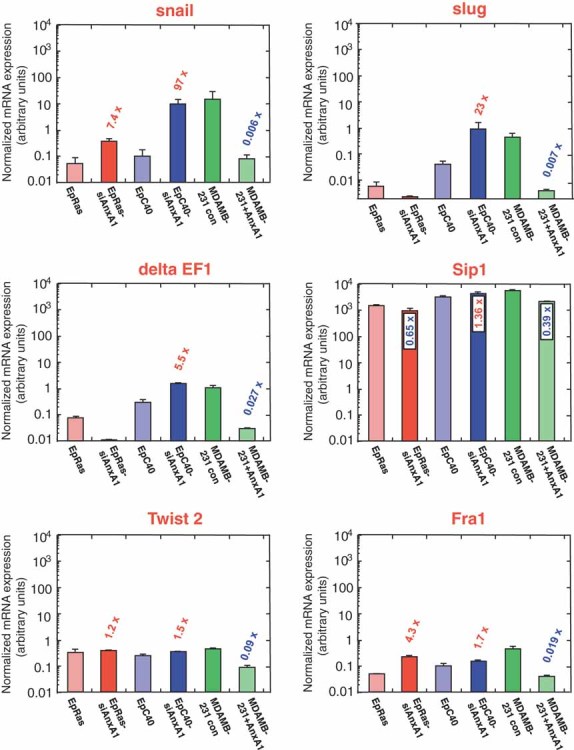
EpRas- and EpC40 cells before and after AnxA1-RNAi-induced EMT, and MDA-MB-231 human mammary carcinoma cells before and after EMT-reversal by forced AnxA1expression were processed for quantitative, real-time RT-PCR analysis to determine mRNA-expression levels for five transcription factors repressing E-cadherin, and Fra-1 (presumed to act upstream of these factors; I. Custic, unpublished). Results were normalized to GAPDH expression. For details, see Suppl. Exp. Proc. Note: bars in all confocal fluorescence photographs 50 µm.
Another possible function of AnxA1 relevant to EMT could be its interaction with cytoplasmic EGFR signalling (Taub et al, 2007). The EGF-activated EGFR phosphorylates AnxA1 on tyrosine, while AnxA1−/− fibroblasts are defective for internalization of plasma-membrane-bound EGFR (Gerke et al, 2005). Indeed, AnxA1 knockdown strongly elevated plasma membrane levels of EGFR without affecting EGFR biosynthesis (Fig S7A and B). Further results, however, were not compatible with a role of EGFR signalling in AnxA1-RNAi induced EMT. Firstly, the EGFR-tyrosine kinase inhibitor H14 completely abolished Erk1/2 signalling in EpC40-sicontrol cells (Fig S7C), also leading to inhibition of growth in collagen gels (Fig S7D), but did not affect Erk1/2 signalling and growth in EpRas-sicontrol, EpRas-siAnxA1 and EpC40-siAnxA1 cells (Fig S7C and not shown), suggesting that AnxA1 may activate Erk1/2 signalling via EGFR-independent pathways. Secondly, EGFR-kinase inhibition completely failed to reverse EMT in EpC40-siAnxA1 and almost completely in EpRasXT cells (Fig S7D and E).
Recently, EMT was identified as an important property of breast carcinoma stem cells (Mani et al, 2008), which could be selectively targeted by Salinomycin in murine breast cancer models, probably acting through EMT reversal (Beug, 2009; Gupta et al, 2009). We therefore studied the possible effects of salinomycin on Ras/AnxA1-RNAi- versus Ras/TGFβ-induced EMT, the latter analyzed in MDCK-DDRafER cells since they are the model of choice for mechanistic studies in epithelial polarity (Lehmann et al, 2000; Tanos & Rodriguez-Boulan, 2008). After exposure to estradiol (E2) plus TGFβ1, MDCK-DDRafER cells showed a particularly striking, complete EMT phenotype (Fig S9, see Suppl. Exp. Proc.), which is completely prevented by Salinomycin (Fig S8B). In striking contrast, Salinomycin induced expression of—mostly cytoplasmic—E-cadherin and low ZO-1 levels at the plasma membrane in EpC40-siAnxA1 cells, while mesenchymal markers (vimentin, fibronectin) were not suppressed at all (Fig S8A). In conclusion, Ras/AnxA1-RNAi-induced EMT in EpC40-siAnxA1 cells is largely resistant to treatment with Salinomycin, which completely reverses Ras/TGFβ induced EMT, strengthening the difference between both types of EMT (see Discussion Section).
DISCUSSION
In this paper, we identified the multifunctional protein AnxA1 (Futter & White, 2007; Gerke et al, 2005) as a suppressor of both EMT and metastasis in murine and human cells and tumours. Loss of AnxA1 induced by RNAi actively promoted EMT and metastasis in mouse mammary epithelial cells, requiring deregulation of Tyk2/Stat3- and Erk1/2-signalling. Forced AnxA1 expression, however, abolished the metastatic capacity of a fibroblastoid, highly metastatic human breast cancer cell line through reversal to an epithelial phenotype. Loss of AnxA1 also predicted metastasis and impaired survival in a breast cancer tissue arrays from 141 patients, suggesting AnxA1 as an excellent biomarker for human metastatic breast cancer. Since AnxA1 abolishes metastatic capacity by re-inducing epithelial properties in dedifferentiated tumour cells, it might also be a candidate for gene therapy in metastatic cancer, although AnxA1 overexpression slightly increased cytotoxic drug resistance in certain tumour cell lines (Mussunoor & Murray, 2008).
How could AnxA1 function as a metastasis suppressor in murine and human cancer?
AnxA1 expression is lost or strongly reduced in many different human tumour types, predominantly representing later stages of tumour progression, but upregulated in other tumours, which might predominantly represent early tumour stages such as adenomas (Lim & Pervaiz, 2007; Mussunoor & Murray, 2008). Our own expression analyses of AnxA1 in breast cancer tissue arrays from 141 patients—and in multiple murine and human tumour cell lines and tumour types—clearly establish that AnxA1 is downregulated during late stage of tumour progression and metastasis (Figs 1D–G; S1A and B).
Annexins are a gene family of >10 isoforms, some members of which function both extra- and intracellularly. Annexins regulate interactions of membranous organelles with each other, the plasma membrane and the actin cytoskeleton (Bandorowicz-Pikula & Pikula, 1998; Gerke et al, 2005). This includes inhibition of EGF-induced inward vesiculation of late endosomes into MVB in cells from AnxA1−/− mice (Futter & White, 2007). Current research on AnxA1 function, however, predominantly focuses on extracellular events, namely the interaction of secreted AnxA1 and its N-terminal peptide with formyl-peptide receptor (FRP)-family G protein-coupled receptor (GPCR)-receptors, or direct inhibition of PLA2 (Lim & Pervaiz, 2007). In our experiments, supernatants from EpRas, EpC40 and EpRasXT cells did not contain detectable AnxA1, and extracellularly added AnxA1-N-terminal peptide did not affect the phenotypes of these cells, suggesting that these extracellular mechanisms are not relevant for AnxA1-RNAi induced EMT. Thus we favour an intracellular mechanism of AnxA1 action in EMT, perhaps by modulating vesicle trafficking through endocytic and/or secretory pathways. This is in line with enhanced secretion of IL6, TGFβ1 and multiple chemokines, and with activation of the Erk1/2, p38MAPK, Stat3 and TGFβ signalling pathways. The latter pathways all involve signalling from both the plasma membrane and from intracellular organelles such as late endosomes (Bokel et al, 2006; Schenck et al, 2008; Shah et al, 2006; Taub et al, 2007). For instance, manipulation of the subcellular location of late endosomes strongly altered strength and duration of EGFR-activated Erk1/2 and p38MAPK signalling (Taub et al, 2007). AnxA1-RNAi might thus deregulate signal strength from this ‘organelle signalling’ via effects on molecular machines involved in vesicle trafficking and polarity complexes, possibly explaining the pleiotropic effects of AnxA1 loss on cytokine secretion, receptor internalization and multiple signalling pathways. Importantly, however, only enhanced Stat3 activation by Tyk2—and to a certain extent hyperactive Erk1/2 signalling—were essential for EMT induced by loss of AnxA1 (Figs 6 and S5B).
Studies to unravel possible mechanisms, how loss of AnxA1 disturbs epithelial polarity with the outcome of EMT, are hindered by the enormous complexity of these processes (Tanos & Rodriguez-Boulan, 2008). While AnxA1-RNAi correlates with upregulation of several transcription factors acting as E-cadherin repressors (Fig 7) these changes appear to be a consequence rather than a cause for the EMT phenotype. Stimulation of TGFβR signalling (Fig 5) and altered EGFR internalization and signalling (Fig S7) by AnxA1-RNAi did not affect the respective EMT. We also obtained evidence suggesting that induction of EMT through loss of AnxA1 plus Ras may involve mechanisms distinct from those operating in Ras/TGFβ-induced EMT (see above). Strikingly, reversal of EMT by the antibiotic Salinomycin, which eradicates breast cancer stem cells by reversal of their EMT (Beug, 2009; Gupta et al, 2009) was complete in Ras/TGFβ-induced EMT, but only partial in Ras/AnxA1-RNAi-dependent EMT (Fig S8).
EMT caused by AnxA1-RNAi: why is cooperation with oncogenic Ras required?
Strikingly, knockdown of AnxA1 in normal mammary epithelial EpH4 and MCF10A cells caused proliferation arrest and apoptosis rather than EMT. Additionally, treatment of AnxA1 knockdown cells with Ras and PI3K inhibitors induced apoptosis, strengthening the idea that hyperactive Ras and PI3K signalling is required for EMT induced by loss of AnxA1. Possible consequences of AnxA1-RNAi like altered vesicular trafficking, receptor internalization and cytoplasmic signalling may be incompatible with normal cell function, thus eliciting apoptotic responses that are prevented by oncogenic Ras. In line with this, the Ras effector mutant V12RasC40—selectively hyperactivating the anti-apoptotic PI3K pathway—causes apoptosis protection and tumour cell hyperproliferation (Janda et al, 2002b) enabling EMT induction rather than apoptosis by other pathways such as Erk1/2 signalling (Janda et al, 2002a), interleukin like EMT inducer (ILEI) and cellular repressor of E1A-stimulated genes (CREG) overexpression (Waerner et al, 2006; Alacakaptan et al, unpublished) and loss of AnxA1 (this paper) or basolateral polarity complex proteins such as scribble (Humbert et al, 2008, HB unpublished). It remains unclear, however, whether or not apoptosis protection (Janda et al, 2002a,b) is sufficient to allow EMT induction by AnxA1-RNAi, or whether hyperactive, cytoplasmic PI3K signalling (Schenck et al, 2008) elicited by V12RasC40 may also be required.
Does lung metastasis caused by loss of AnxA1 in mice also require cooperation with oncogenic Ras or respective downstream signal transducers? To address this, we crossbred MMTV-neu (very late metastasis) and MMTV-neu × MMTV-TGFβ1 mice (early metastasis) onto an AnxA1−/− background (see Supplementary Material). Loss of AnxA1 did not enhance lung metastasis in these mice, suggesting that HER2 cannot replace oncogenic Ras in metastasis formation by AnxA1-RNAi (see Supplementary Material). Likewise, expression of activated HER2* in EpH4 cells did not substitute for oncogenic Ras in TGFβ-induced EMT, causing partial and reversible rather than complete and metastable EMT (HB unpublished). Since AnxA2, AnxA4 and AnxA5 are upregulated in AnxA1−/− mice (Hannon et al, 2003) in vivo compensation by other annexins might also be involved. This effect, however, does not occur in the RNAi approach (Fig S2B).
Finally, tumour formation by AnxA1 knockdown in mice also required oncogenic Ras. This was shown using hTERT-immortalized human mammary epithelial cells (hMEC) which lacked one of the three defined oncogenes required for carcinoma formation (Zhao et al, 2003). After knockdown of AnxA1, the resulting cells required oncogenic Ras, but not Polyoma small t for anchorage independent growth (not shown) and tumour formation by loss of AnxA1 (see Fig S3B and Supplementary Material).
MATERIALS AND METHODS
Cells and cell culture
The origin, cultivation and properties of EpH4-derived cell types has been described previously (Janda et al, 2002a; Waerner et al, 2006), for details see Suppl. Exp. Proc. and Fig S10. Human mammary carcinoma cell lines—before and after AnxA1 overexpression—and derivatives of MDCK-cells are also described in Suppl. Exp. Proc. and Fig S10. HMEC-hTERT and their derivatives (see Suppl. Exp. Proc. and Fig S5), a generous gift from W. C. Hahn, were cultivated as described (Zhao et al, 2003). Human mammary epithelial MCF10A cells were grown in mammary epithelial growth medium (MEGM) (Promocell).
Generation of retroviral vectors
The different siRNA-expression vectors containing targeting sequences for mouse AnxA1 were constructed using the Gateway-compatible pMSCV-puromycin vector according to the Gateway protocol's instructions (Invitrogen, for details see Suppl. Exp. Proc.). Vectors containing different target sequences for human AnxA1 were generated as described above for the mouse constructs. Expression vectors for full length human AnxA1 complementary DNA (cDNA) were generated according to the Gateway protocol (Invitrogen, see Suppl. Exp. Proc.) in pMSCV-pgk-puromycin (MDA-MB-231) and pMSCV-GFP target vectors (EpC40-siAnxA1). All vectors were confirmed by sequencing of the target inserts.
Transfection and generation of cell lines stably expressing genes of interest
Retrovirus producer cell lines (NIH-3T3gp + 86 cells) were transfected with the above retroviral constructs using Lipofectamine2000 reagent (Invitrogen) as described earlier (Oft et al, 1998; Waerner et al, 2006). Virus supernatants were used to infect the various target cell lines (for details see Suppl. Exp. Proc.). Stable knockdown of Jak1, Jak2 and Tyk2 was performed using MISSION Lentiviral Transduction particles (Sigma–Aldrich) according to the manufacturer's instructions (for details see Suppl. Exp. Proc.).
Reagents and antibodies
Antibodies to EGFR, AnxA1, AnxA5, AnxA6, AnxA7 were from Santa Cruz, to AnxA2 and AnxA4 from BD Transduction Laboratories and to N-cadherin from Zymed. Antibodies to vimentin, β-actin, pErk1/2 and total Erk1/2 were from Sigma, to total and phosphorylated Stat3, Smad2, AKT and p38MAPK from cell signalling, to E-cadherin from BD and to TGFβ1 from Promega. Tyrosine phosphorylated EGFR was detected using 4G10 anti-P-Tyr-antibody (Millipore), after immunoprecipitation with an EGFR antibody (Santa Cruz). Pharmacological inhibitors for Jaks, MEK1, p38MAPK and PI3K were from Calbiochem and Sigma, inhibitors for Ha-Ras, EGFR and TGFβRI were kind gifts of BI Vienna. Estradiol was from Sigma–Aldrich, TGFβ1 from Peprotech and Salinomycin from Sigma.
Western Blot analysis
Western blot analysis was carried out as described previously (Maschler et al, 2004), WB data for Fig 1D were quantified by densitometry (ImageJ). For details and small modifications see Suppl. Exp. Proc.
Immunoprecipitation of surface labelled and metabolically labelled proteins
Cell surface biotinylation was performed as described previously (Maschler et al, 2005), with small modifications described in Suppl. Exp. Proc. Equal cell numbers were metabolically labelled with [35S]-l-methionine/l-cysteine, lysates immunoprecipitated with EGFR-antibody and processed as detailed in Suppl. Exp. Proc.
Collagen gel cell culture
Serum-free, three-dimensional cultures in Collagen I gels were performed as described previously (Janda et al, 2002a; Maschler et al, 2004; Waerner et al, 2006). For details and small modifications see Suppl. Exp. Proc.
Immunofluorescence analysis of cells grown on filters or in 3D collagen gels
Fluorescent antibody staining of cells cultivated on porous supports or in collagen gels was carried out exactly as described earlier (Janda et al, 2002a; Maschler et al, 2004). Confocal analysis was performed using a Leica-TCS-NT confocal microscope.
Immunohistochemical staining of breast cancer tumour tissue arrays
Two breast cancer tissue arrays from primary tumour tissue of 141 breast cancer patients with recorded disease history since maximally 16 years were processed for IHC staining of AnxA1 as described in Suppl. Exp. Proc. and (Waerner et al, 2006). Analysis was performed according to protocols approved by the institutional bioethics review board of the Medical University Vienna.
The paper explained
PROBLEM:
Metastasis is the major cause of cancer related death but the mechanisms involved are not completely understood due to the complexity of the process. EMT is considered a prerequisite for cancer cells to invade adjacent tissue and form metastases at distinct sites. AnxA1 is a protein with diverse functions in secretion and signalling and we investigated its role in EMT and metastasis.
RESULTS:
Annexin A1 expression is highly reduced during tumour progression and metastasis in breast cancer and AnxA1 expression correlates with reduced patient's survival. We demonstrate that knock down of AnxA1 stimulates EMT in the EpH4/EpRas cell system and metastasis formation in vivo. In addition, forced overexpression of AnxA1 in highly metastatic human breast cancer cells reverts EMT and inhibits metastasis. JAK/Stat and MAPK/ERK signalling is essential for EMT induced by loss of AnxA1.
IMPACT:
The data highlight the importance of AnxA1 in EMT and metastasis and we propose that AnxA1 might also be used as a marker for tumour progression in clinical analysis.
Reporter assay
1–3 × 105 EpRas or EpC40 cells were cultivated in 2 ml of medium in 6-well plates and transiently transfected (Lipofectamin 2000) with siAnxA1- or siFFL-vectors plus Smad2- or Smad3 reporter constructs (Germain et al, 2000), and a β-Gal construct for normalization. Twenty-four hours after transfection, the 60% confluent cells were treated with 10 ng/ml TGFβ1 for 8 h, lysed and reporter gene activation was assayed using the Invitrogen Dual Luciferase System according to manufacturer's instructions.
ELISA
The amount of biologically active, secreted TGFβ1 and the levels of secreted IL6 were measured in serum-free cell supernatants from test cells (3 × 104 cells/12 well, 1.5 ml medium after 24 h incubation in serum free medium), using the TGFβ1 ELISA kit (Promega) and the Quantikine IL6 ELISA kit (R&D systems), respectively, according to the manufacturer's instructions.
Non-permeant cellular ELISA
Cell surface EGFR was determined using a non-permeant cellular ELISA assay as described in (Lee et al, 2002) with minor modifications (for details and references, see Suppl. Exp. Proc.).
RNA Isolation and cDNA synthesis
Total ribonucleic acid (RNA) was isolated using the TRIzol reagent (Invitrogen) according to the manufacturer's instruction. cDNA was synthesized from 2 µg of total RNA using the ‘Ready to go you prime First Strand’ beads kit (Amersham Biosciences) and random oligonucleotides according to the manufacturer's instructions.
Determination of gene expression levels in cell lines and tumour sample by qRT-PCR
qRT-PCR of the expression levels of AnxA1, Jak1, Jak2, Tyk2, snail, slug, deltaEF1, Sip1, twist2, GAPDH and Fra-1 in murine and humans breast cancer cells and human tumour samples was performed using the SYBR green 2x PCR kit (Applied Biosciences) and a primer mix from Qiagen. For details see Suppl. Exp. Proc.
Tumour formation and metastasis induction
Tumour formation ability in nude mice was determined by mammary gland fat pad- or subcutanous injection as described earlier (Janda et al, 2002a; Waerner et al, 2006; Zhao et al, 2003). Metastasis formation was measured after cell injection into the tail vein of nude mice (Janda et al, 2002a; Waerner et al, 2006; Zhao et al, 2003). For minor modifications, details and analysis of tumours from transgenic MMTV-neu and MMTV neu × MMTV-TGFβ1 mice see Suppl. Exp. Proc. Experiments were performed according to IMP-held animal experiment approvals from the Austrian Federal Ministry of Science and Research.
Evaluation of the gene logic database for AnxA1 expression
These methods, performed for us by Boehringer Ingelheim International (BI Austria, Vienna) are described in Suppl. Exp. Proc.
Acknowledgments
We thank Dr. R. J. Flower, London, UK for his kind gift of AnxA1−/− mice, Dr. W. Hahn for the hMEC cell series, Dr. K. Zatloukal, Graz, Austria for mammary carcinoma- and normal mammary tissue samples, Boehringer Ingelheim (BI) Vienna for pharmacological inhibitors of EGFR and active help with the Gene Logics screen, Gabi Litos for expert technical assistance and Drs. Lukas Huber, Kai Simons and Norbert Kraut for critically reading the manuscript. This work was supported by grants from the Austrian Science Foundation (FWF SFB028; FWF P17699-B12) and the Austrian FFG (project no. 814.184).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
SM and HB planned the experiments and devised the concept. CAG generated EpRas-siAnxA1 cells and EpRasXT + AnxA1 cells and performed qPCR analysis of AnxA1 expression in EpH4/EpRas cell system. E-MW performed immunofluorescence analysis. MA processed human tumour samples for IHC. MS provided human tumour tissue array and the statistics of the survival data. IC helped with qRTPCR for E-Cadherin repressors. SM and HB wrote and edited the manuscript.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Aranda V, Nolan ME, Muthuswamy SK. Par complex in cancer: a regulator of normal cell polarity joins the dark side. Oncogene. 2008;27:6878–6887. doi: 10.1038/onc.2008.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiychuk EB, Monastyrskaya K, Draeger A. Fluorescent Annexin A1 reveals dynamics of ceramide platforms in living cells. Traffic. 2008;9:1757–1775. doi: 10.1111/j.1600-0854.2008.00800.x. [DOI] [PubMed] [Google Scholar]
- Bandorowicz-Pikula J, Pikula S. Annexins and ATP in membrane traffic: a comparison with membrane fusion machinery. Acta Biochim Pol. 1998;45:721–733. [PubMed] [Google Scholar]
- Beug H. Breast cancer stem cells: eradication by differentiation therapy. Cell. 2009;138:623–625. doi: 10.1016/j.cell.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Bokel C, Schwabedissen A, Entchev E, Renaud O, Gonzalez-Gaitan M. Sara endosomes and the maintenance of Dpp signaling levels across mitosis. Science. 2006;314:1135–1139. doi: 10.1126/science.1132524. [DOI] [PubMed] [Google Scholar]
- Eger A, Stockinger A, Schaffhauser B, Beug H, Foisner R. Epithelial mesenchymal transition by c-Fos estrogen receptor activation involves nuclear translocation of beta-catenin and upregulation of beta-catenin/lymphoid enhancer binding factor-1 transcriptional activity. J Cell Biol. 2000;148:173–188. doi: 10.1083/jcb.148.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24:2375–2385. doi: 10.1038/sj.onc.1208429. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S. Polarity proteins in migration and invasion. Oncogene. 2008;27:6970–6980. doi: 10.1038/onc.2008.347. [DOI] [PubMed] [Google Scholar]
- Futter CE, White IJ. Annexins and endocytosis. Traffic. 2007;8:951–958. doi: 10.1111/j.1600-0854.2007.00590.x. [DOI] [PubMed] [Google Scholar]
- Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- Germain S, Howell M, Esslemont GM, Hill CS. Homeodomain and winged-helix transcription factors recruit activated Smads to distinct promoter elements via a common Smad interaction motif. Genes Dev. 2000;14:435–451. [PMC free article] [PubMed] [Google Scholar]
- Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- Guo W, Pylayeva Y, Pepe A, Yoshioka T, Muller WJ, Inghirami G, Giancotti FG. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell. 2006;126:489–502. doi: 10.1016/j.cell.2006.05.047. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon R, Croxtall JD, Getting SJ, Roviezzo F, Yona S, Paul-Clark MJ, Gavins FN, Perretti M, Morris JF, Buckingham JC, et al. Aberrant inflammation and resistance to glucocorticoids in annexin 1−/− mouse. FASEB J. 2003;17:253–255. doi: 10.1096/fj.02-0239fje. [DOI] [PubMed] [Google Scholar]
- Huber MA, Kraut N, Beug H. Molecular requirements for epithelial–mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- Humbert PO, Grzeschik NA, Brumby AM, Galea R, Elsum I, Richardson HE. Control of tumourigenesis by the Scribble/Dlg/Lgl polarity module. Oncogene. 2008;27:6888–6907. doi: 10.1038/onc.2008.341. [DOI] [PubMed] [Google Scholar]
- Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, Grunert S. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002a;156:299–313. doi: 10.1083/jcb.200109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda E, Litos G, Grunert S, Downward J, Beug H. Oncogenic Ras/Her-2 mediate hyperproliferation of polarized epithelial cells in 3D cultures and rapid tumor growth via the PI3K pathway. Oncogene. 2002b;21:5148–5159. doi: 10.1038/sj.onc.1205661. [DOI] [PubMed] [Google Scholar]
- Jechlinger M, Grunert S, Beug H. Mechanisms in epithelial plasticity and metastasis: insights from 3D cultures and expression profiling. J Mammary Gland Biol Neoplasia. 2002;7:415–432. doi: 10.1023/a:1024090116451. [DOI] [PubMed] [Google Scholar]
- Jechlinger M, Sommer A, Moriggl R, Seither P, Kraut N, Capodiecci P, Donovan M, Cordon-Cardo C, Beug H, Grunert S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J Clin Invest. 2006;116:1561–1570. doi: 10.1172/JCI24652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahsnig C, Mikula M, Petz M, Zulehner G, Schneller D, van Zijl F, Huber H, Csiszar A, Beug H, Mikulits W. ILEI requires oncogenic Ras for the epithelial to mesenchymal transition of hepatocytes and liver carcinoma progression. Oncogene. 2009;28:638–650. doi: 10.1038/onc.2008.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FJ, Xue S, Pei L, Vukusic B, Chery N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F. Dual regulation of NMDA receptor functions by direct protein–protein interactions with the dopamine D1 receptor. Cell. 2002;111:219–230. doi: 10.1016/s0092-8674(02)00962-5. [DOI] [PubMed] [Google Scholar]
- Lehmann K, Janda E, Pierreux CE, Rytomaa M, Schulze A, McMahon M, Hill CS, Beug H, Downward J. Raf induces TGFbeta production while blocking its apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells. Genes Dev. 2000;14:2610–2622. doi: 10.1101/gad.181700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LH, Pervaiz S. Annexin 1: the new face of an old molecule. FASEB J. 2007;21:968–975. doi: 10.1096/fj.06-7464rev. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou A, Brooks F, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschler S, Grunert S, Danielopol A, Beug H, Wirl G. Enhanced tenascin-C expression and matrix deposition during Ras/TGF-beta-induced progression of mammary tumor cells. Oncogene. 2004;23:3622–3633. doi: 10.1038/sj.onc.1207403. [DOI] [PubMed] [Google Scholar]
- Maschler S, Wirl G, Spring H, Bredow DV, Sordat I, Beug H, Reichmann E. Tumor cell invasiveness correlates with changes in integrin expression and localization. Oncogene. 2005;24:2032–2041. doi: 10.1038/sj.onc.1208423. [DOI] [PubMed] [Google Scholar]
- Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27:6958–6969. doi: 10.1038/onc.2008.346. [DOI] [PubMed] [Google Scholar]
- Mussunoor S, Murray GI. The role of annexins in tumour development and progression. J Pathol. 2008;216:131–140. doi: 10.1002/path.2400. [DOI] [PubMed] [Google Scholar]
- Ng DC, Lin BH, Lim CP, Huang G, Zhang T, Poli V, Cao X. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J Cell Biol. 2006;172:245–257. doi: 10.1083/jcb.200503021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF-beta1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996;10:2462–2477. doi: 10.1101/gad.10.19.2462. [DOI] [PubMed] [Google Scholar]
- Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype. Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- Reichmann E, Schwarz H, Deiner EM, Leitner I, Eilers M, Berger J, Busslinger M, Beug H. Activation of an inducible c-FosER fusion protein causes loss of epithelial polarity and triggers epithelial-fibroblastoid cell conversion. Cell. 1992;71:1103–1116. doi: 10.1016/s0092-8674(05)80060-1. [DOI] [PubMed] [Google Scholar]
- Schenck A, Goto-Silva L, Collinet C, Rhinn M, Giner A, Habermann B, Brand M, Zerial M. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell. 2008;133:486–497. doi: 10.1016/j.cell.2008.02.044. [DOI] [PubMed] [Google Scholar]
- Shah M, Patel K, Mukhopadhyay S, Xu F, Guo G, Sehgal PB. Membrane-associated STAT3 and PY-STAT3 in the cytoplasm. J Biol Chem. 2006;281:7302–7308. doi: 10.1074/jbc.M508527200. [DOI] [PubMed] [Google Scholar]
- Tanos B, Rodriguez-Boulan E. The epithelial polarity program: machineries involved and their hijacking by cancer. Oncogene. 2008;27:6939–6957. doi: 10.1038/onc.2008.345. [DOI] [PubMed] [Google Scholar]
- Taub N, Teis D, Ebner HL, Hess MW, Huber LA. Late endosomal traffic of the epidermal growth factor receptor ensures spatial and temporal fidelity of mitogen-activated protein kinase signaling. Mol Biol Cell. 2007;18:4698–4710. doi: 10.1091/mbc.E07-02-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waerner T, Alacakaptan M, Tamir I, Oberauer R, Gal A, Brabletz T, Schreiber M, Jechlinger M, Beug H. ILEI: a cytokine essential for EMT, tumor formation, and late events in metastasis in epithelial cells. Cancer Cell. 2006;10:227–239. doi: 10.1016/j.ccr.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, Hausen AZ, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- White IJ, Bailey LM, Aghakhani MR, Moss SE, Futter CE. EGF stimulates annexin 1-dependent inward vesiculation in a multivesicular endosome subpopulation. EMBO J. 2006;25:1–12. doi: 10.1038/sj.emboj.7600759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YH, Toh ML, Clyne CD, Leech M, Aeberli D, Xue J, Dacumos A, Sharma L, Morand EF. Annexin 1 negatively regulates IL-6 expression via effects on p38 MAPK and MAPK phosphatase-1. J Immunol. 2006;177:8148–8153. doi: 10.4049/jimmunol.177.11.8148. [DOI] [PubMed] [Google Scholar]
- Yu H, Jove R. The STATS of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4:655–665. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, Hahn WC. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell. 2003;3:483–495. doi: 10.1016/s1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]
- Zhao E, Xu J, Yin X, Sun Y, Shi J, Li X. Detection of deregulated pathways to lymphatic metastasis in oral squamous cell carcinoma. Pathol Oncol Res. 2009;15:217–223. doi: 10.1007/s12253-008-9102-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.