

Abstract
Apoptosis signal-regulating kinase 1 (ASK1) is an evolutionarily conserved mitogen-activated protein kinase (MAPK) kinase kinase which plays important roles in stress and immune responses. Here, we show that ASK1 deficiency attenuates neuroinflammation in experimental autoimmune encephalomyelitis (EAE), without affecting the proliferation capability of T cells. Moreover, we found that EAE upregulates expression of Toll-like receptors (TLRs) in activated astrocytes and microglia, and that TLRs can synergize with ASK1-p38 MAPK signalling in the release of key chemokines from astrocytes. Consequently, oral treatment with a specific small molecular weight inhibitor of ASK1 suppressed EAE-induced autoimmune inflammation in both spinal cords and optic nerves. These results suggest that the TLR-ASK1-p38 pathway in glial cells may serve as a valid therapeutic target for autoimmune demyelinating disorders including multiple sclerosis.
Keywords: ASK1, chemokines, demyelination, glia, Toll-like receptors
INTRODUCTION
Apoptosis signal-regulating kinase 1 (ASK1) is one of a growing number of mitogen-activated protein kinase (MAPK) kinase kinases identified in the c-Jun N-terminal kinase (JNK) and p38 MAPK pathways (Ichijo et al, 1997). ASK1 is activated by various cytotoxic stressors as well as receptor-mediated inflammatory signals, such as lipopolysaccharide (LPS) and tumour necrosis factor (TNF), and mediates diverse biological signals leading to cell death, differentiation and senescence (Chiang et al, 2006; Saitoh et al, 1998; Tobiume et al, 2001). Recent studies have shown that ASK1 is an evolutionarily conserved signalling intermediate for innate immunity (Kim et al, 2002; Matsuzawa et al, 2005). In mammals, Toll-like receptors (TLRs) activate p38, JNK and NF-κB cascades, leading to the induction of many key cytokine genes (Akira & Takeda, 2004; Beutler, 2004). Among them, ASK1 specifically mediates LPS-induced TLR4 signalling to p38 through a reactive oxygen species (ROS)-dependent pathway in dendritic cells and splenocytes (Matsuzawa et al, 2005). This finding provided a unique link between cellular stress responses and innate immunity. On the other hand, several lines of evidence suggest that ASK1 plays key roles in human diseases that are closely related to dysfunction of cellular responses to oxidative stress and endoplasmic reticulum (ER) stressors (Harada et al, 2006; Kadowaki et al, 2005; Nishitoh et al, 2002, 2008; Takeda et al, 2007). However, these studies focused only on the mechanisms of neural cell death and the detailed function of ASK1 signalling in other cell types in the central nervous system (CNS) is still unknown.
Multiple sclerosis (MS) is an inflammatory disease of the CNS characterized by progressive immune-mediated destruction of the myelin sheath (Sospedra & Martin, 2005). The inflammatory process is thought to be mediated in part by T lymphocytes and microglia/macrophage that are recruited to the CNS in response to chemotactic signals. Recent studies have shown that astrocyte-derived chemokines such as monocyte chemoattractant protein (MCP-1), regulated on activation normal T-cell expressed and secreted proteins (RANTES) and macrophage inflammatory protein-1α (MIP-1α), may play a role in the migration of inflammatory cells into the CNS (Nair et al, 2008; Tanuma et al, 2006; Van Der Voorn et al, 1999). We previously demonstrated that a specific inhibitor of glial cell activation suppressed the release of these key chemokines from astrocytes, thereby ameliorated the severity of experimental autoimmune encephalomyelitis (EAE), an animal model of MS (Guo et al, 2007). Astrocytes are the most abundant cell type in the mammalian CNS and, as well as microglial cells, they are thought to have the potential to affect the immune response by serving as antigen-presenting cells in the target organ (Constantinescu et al, 2005; Girvin et al, 2002). On the other hand, TLR activation in astrocytes may promote an anti-inflammatory and neuroprotective response in human MS (Bsibsi et al, 2006). Several studies have reported that TLR4 and TLR9 may regulate disease severity of EAE, but the detailed functions of TLRs during MS/EAE are still controversial (Kerfoot et al, 2004; Marta et al, 2008; Prinz et al, 2006). In the present study, we attempted to elucidate the potential role of TLRs-ASK1 signalling in glial cells. Our data revealed that ASK1-p38 axis is required for chemokine productions in astrocytes through multiple TLRs. In addition, ASK1 deficiency or inhibition of ASK1 using a pharmacological tool attenuated the severity of EAE, suggesting that TLR-ASK1-p38 pathway in glial cells is a potential therapeutic target for the treatment of MS.
RESULTS
Role of ASK1 on the severity of CNS inflammation, optic neuritis and visual function
In order to elucidate the role of ASK1 and its downstream effector pathway(s) during neuroinflammation, we first examined myelin oligodendrocyte glycoprotein (MOG)-induced EAE susceptibility in ASK1−/− and wild-type (WT) mice. The disease incidence of EAE was not different between WT and ASK1−/− mice, but the severity of the paralytic symptoms was much lower in the ASK1−/− mice than WT mice (Fig 1A). Since EAE is a T-cell-mediated autoimmune disease, we assessed the effect of ASK1 deficiency on T-cell proliferation capability and cytokine profiles. Freshly isolated lymph node cells from WT and ASK1−/− MOG-immunized mice were stimulated with MOG or Concanavalin A (ConA). No significant differences in the T-cell proliferation responses were found between the T cells of WT and ASK1−/− mice (Fig 1B and Fig S1A of Supporting Information). Furthermore, analyses of both in vitro T-cell-derived cytokine release and intracellular cytokine profiles revealed no difference between the two genotypes (Fig S1B–D of Supporting Information), indicating ASK1 deficiency has no effect on the polarization of naive T-cell.
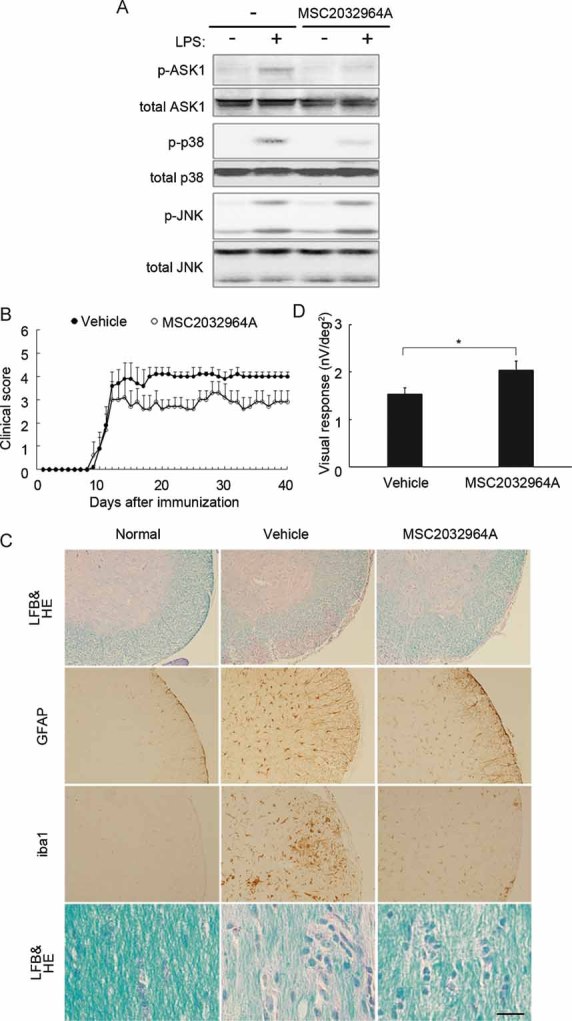
Figure 1. ASK1 deficiency attenuates EAE-induced CNS inflammation, demyelination and glial activation.
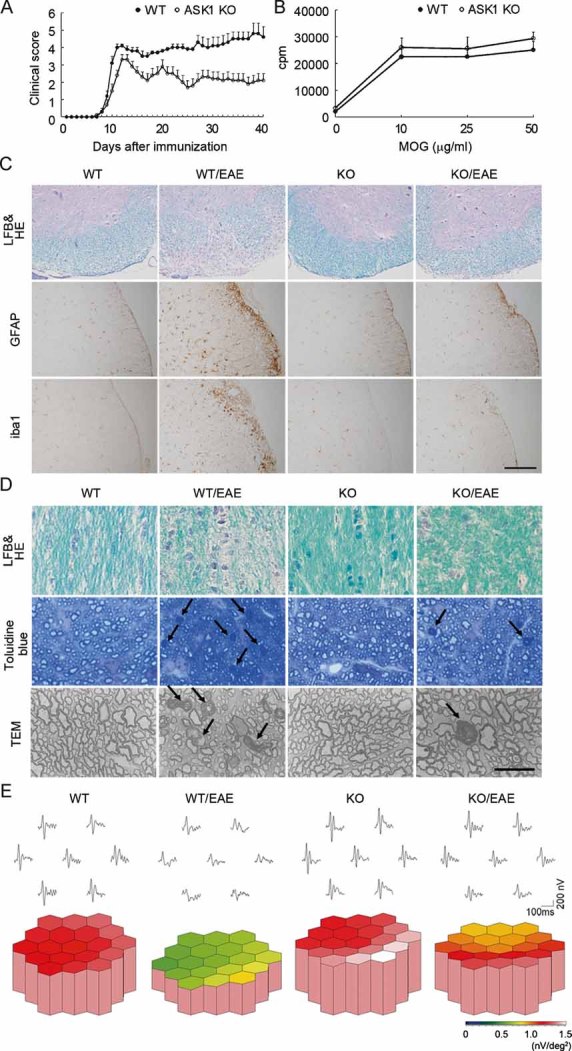
- Clinical evaluation of EAE in wild-type (WT) (n = 13) and ASK1−/− (n = 15) mice during a period of 40 days after MOG immunization.
- Proliferative responses of MOG-specific T cells isolated from WT and ASK1−/− mice (n = 4).
- Representative histology of the spinal cords in EAE mice. Lumbar spinal cords were stained with LFB and HE (upper panels) and either an anti-GFAP (middle panels) or anti-iba1 antibody (lower panels). Scale bar: 40 µm for the upper panel and 220 µm for the middle and lower panels.
- Representative histology of the optic nerves in EAE mice. Optic nerves were stained with LFB and HE (upper panels) or toluidine blue for the semithin transverse sections (middle panels). The arrows point to the degenerating axons, which were observed further with a transmission electron microscope (TEM; lower panels). Scale bar: 100 µm for the upper panel, 50 µm for the middle panel and 15 µm for the lower panel.
- The averaged visual responses from six mice in each group were examined by multifocal electroretinograms. The visual stimulus was applied to seven different areas in the retina. The seven individual traces demonstrate the average responses to the visual stimulus at the corresponding stimulus area (upper panels). Three-dimensional plots show the amplitude variation across the arrays (lower panels). Values are given in nV per square degree (nV/deg2).
Histopathological investigation of the spinal cords of EAE mice revealed that, in ASK1−/− mice, the number of infiltrating cells in the white matter was drastically reduced (Fig S2A of Supporting Information) and the extent of demyelination was milder relative to WT mice (upper panels in Fig 1C). In addition, the increase in the number of glial fibrillary acidic protein (GFAP)-positive astrocytes and iba1-positive microglial cells upon EAE induction was considerably lower in ASK1−/− mice (middle and lower panels in Fig 1C; Fig S2B and C of Supporting Information).
As MS often induces visual disturbance, we next examined the effect of ASK1 deficiency on the severity of optic neuritis. EAE-induced inflammation and demyelination in the optic nerve were milder in ASK1−/− mice than WT mice (upper panels in Fig 1D and Fig S3A of Supporting Information). In addition, the number of degenerating axons was reduced in ASK1−/− EAE mice (middle and lower panels in Fig 1D; Fig S3B of Supporting Information). We next investigated the visual functions of EAE mice using multifocal electroretinograms (mfERG), an established non-invasive method for effectively measuring visual function (Harada et al, 2007). The response topography demonstrated that the visual function of WT EAE mice was impaired in all visual fields, but it was clearly unaffected in ASK1−/− EAE mice (Fig 1E and Fig S4 of Supporting Information). Taken together, these data demonstrate that ASK1 deficiency attenuates both the histological and functional aspects of EAE-induced CNS inflammation and demyelination.
TLR-ASK1 activation in glial cells in EAE
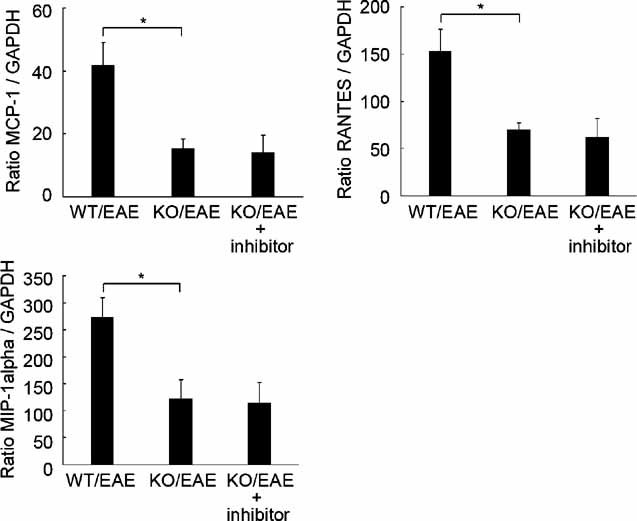
Since the accumulation of activated astrocytes was reduced in the lesion sites of ASK1−/− EAE mice, we next examined the expression levels of MCP-1, RANTES and MIP-1α, which are the key chemokines implicated in the pathogenesis of EAE, in the spinal cord at 12 and 40 days after disease induction (d12 and d40, respectively). The expression of all three chemokines was increased significantly in WT EAE mice, and this chemokine induction was considerably reduced in ASK1−/− mice at d40 (Fig 2A) but not at d12 when ASK1−/− EAE disease reached its peak (Fig S5 of Supporting Information). In addition, activated microglial cells may secrete proinflammatory molecules such as tumour necrosis factor α (TNFα) and nitric oxide, which accelerate the progress of demyelination (Selmaj et al, 1991; Steinman et al, 2002). LPS-induced TNFα release and the production of inducible nitric oxide synthase (iNOS) were significantly reduced in ASK1-deficient cells when compared with WT cells (Fig 2B). We next examined TLR expression levels in EAE mice. Real-time PCR (RT-PCR) analysis of messenger RNA (mRNA) extracted from spinal cord revealed that TLR4 and TLR9 expression levels were significantly upregulated at d40 in WT EAE mice (Fig 2C). Consequently, immunohistochemical analysis demonstrated a significant increase in both TLR4 and TLR9 protein levels in activated astrocytes (Fig 3A and B) and microglia (Fig 3C and D). These results suggest potential roles for TLR-ASK1 signalling in astrocytes and microglia in the pathogenesis and disease progression of EAE.
Figure 2. Activation of TLR-ASK1 signalling in glial cells during neuroinflammation.
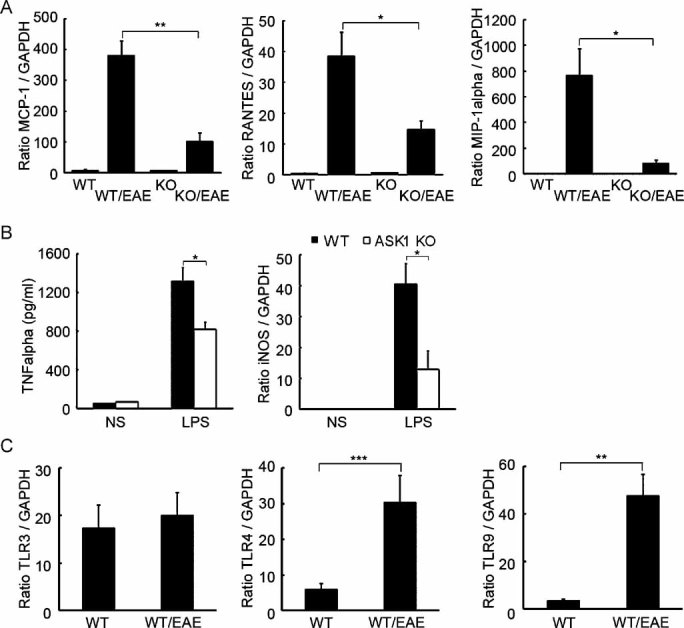
- Impaired chemokine production in the spinal cords of ASK1−/− EAE mice (n = 4). mRNA expression levels of MCP-1, RANTES and MIP-1α at d40 after MOG immunization were determined using quantitative real-time PCR. GAPDH was used as an internal control. **p < 0.01; *p < 0.05.
- Reduced production of TNFα and iNOS in ASK1-deficient microglial cells (n = 4). Cells were stimulated for 16 h with LPS (1 µg/ml) or left unstimulated (NS). TNFα concentrations in the supernatants were determined by ELISA, and iNOS mRNA expression levels were measured by quantitative real-time PCR. *p < 0.05.
- TLR3, TLR4 and TLR9 mRNA expression levels in the spinal cord were determined by quantitative real-time PCR analysis (n = 6). ***p < 0.001; **p < 0.01.
Figure 3. TLR upregulation in glial cells in EAE.
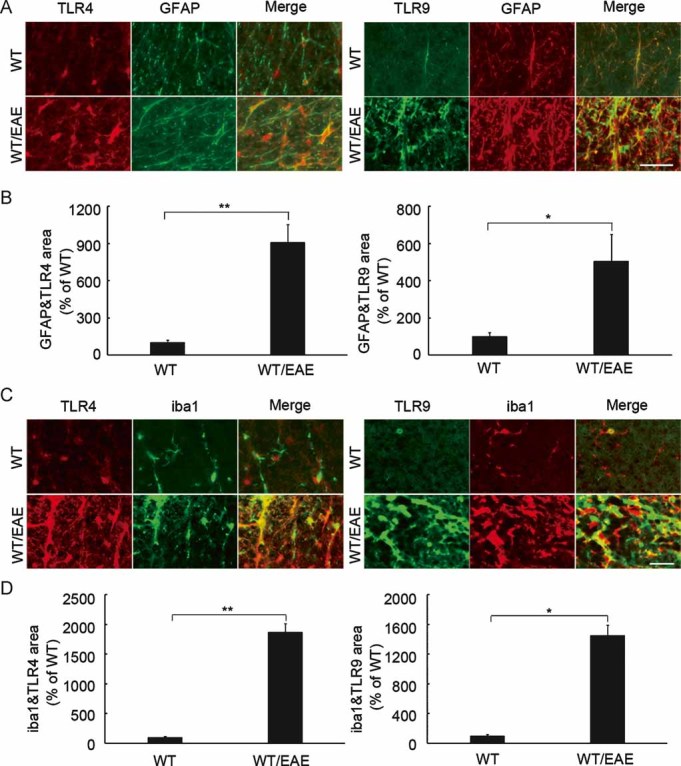
- Representative double-labelling immunohistochemistry for TLR4 or TLR9 with GFAP in the white matter of the spinal cord. Scale bar: 30 µm.
- Quantification of the double-stained areas per unit area (0.048 mm2) in (A). The results are expressed as percentages of the wild-type non-EAE (WT) mice (n = 4). **p < 0.001; *p < 0.01.
- Representative double-labelling immunohistochemistry for TLR4 or TLR9 with iba1 in the white matter of the spinal cord. Scale bar: 30 µm.
- Quantification of the double-stained areas per unit area (0.048 mm2) in (C). The results are expressed as percentages of the wild-type non-EAE (WT) mice (n = 4). **p < 0.001; *p < 0.01.
Effect of TLR-ASK1 signalling on chemokine productions in astrocytes
Toll-like receptors activate the p38, JNK and NF-κB cascades, leading to the induction of many key cytokine genes (Akira & Takeda, 2004). Since there was a decrease in the accumulation of activated astrocytes in the lesion sites of ASK1−/− EAE mice, we investigated the effect of ASK1 deficiency on TLR signalling pathways in cultured mouse astrocytes. To this aim, we examined whether TLR ligands activate p38 and JNK using antibodies against phosphorylated proteins. Stimulation with LPS (a ligand for TLR4) or unmethylated cytosine phosphate guanine (CpG) deoxyribonucleic acid (DNA) (a ligand for TLR9) induced strong phosphorylation of p38 in a time-dependent manner and only slight activation of JNK in WT astrocytes (Fig S6A of Supporting Information). Western blotting analysis demonstrated that TLR-induced p38 activity was significantly reduced in ASK1-deficient astrocytes (Fig 4A), while the phosphorylation of JNK was unaffected (Fig S6B of Supporting Information). In contrast, the TLR-induced NF-κB activation pattern in ASK1-deficient astrocytes was indistinguishable from WT astrocytes (Fig S6B of Supporting Information). To explore the biological functions of TLR-ASK1-p38 signalling, we next measured the release of key chemokines from WT and ASK1-deficient astrocytes using an enzyme-linked immunosorbent assay (ELISA). TLR-induced secretion of MCP-1, RANTES and MIP-1α in ASK1-deficient astrocytes was significantly lower than in WT astrocytes (Fig 4B). In order to confirm that TLR-ASK1-p38 pathway is responsible for the changes in chemokine production, we have pre-treated the WT astrocytes with a p38 inhibitor. The production of the aforementioned chemokines was significantly inhibited by the p38 inhibitor (Fig S7 of Supporting Information), demonstrating that in astrocytes, the ASK1-p38 pathway contributes to TLR-mediated chemokine production.
Figure 4. ASK1 is required for TLR ligand-induced p38 activation and chemokine production in astrocytes.
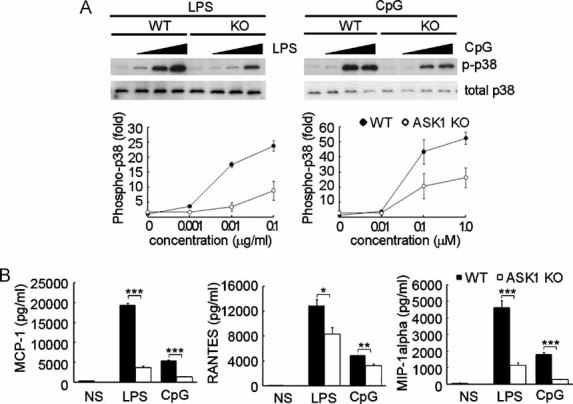
- Effect of ASK1 on TLR ligand-induced p38 activation. Astrocytes derived from WT or ASK1−/− (KO) mice were stimulated with the indicated concentration of the specific TLR ligand for 30 min, followed by immunoblot analysis of total and phosphorylated p38 in cell lysates.
- Impaired chemokine production in ASK1-deficient astrocytes (n = 4). Cells were stimulated for 16 h with LPS (10 µg/ml), unmethylated CpG DNA (1 µM) or left unstimulated (NS). Concentrations of MCP-1, RANTES and MIP-1α in culture medium were measured by ELISA. ***p < 0.001; **p < 0.01; *p < 0.05.
ASK1-specific inhibitor ameliorates the severity of EAE and optic neuritis
The previous experiments using ASK1−/− mice suggest that the pharmacological inhibition of ASK1 activity might be beneficial for the treatment of EAE and optic neuritis. To this aim, we have performed a high-throughput screening with purified ASK1 and have identified one small molecule inhibitor Hit Series. The most potent compound from the Series, MSC1946002A, showed a moderate inhibition of the enzymatic activity of ASK1 (IC50 of 3000 ± 320 nM). Medicinal chemistry optimization efforts to improve the potency and the in vitro absorption, distribution, metabolism and excretion (ADME) properties have led to the identification of the lead compound MSC2032964A (Fig 5A). This compound showed not only a high potency on ASK1 (IC50 of 93 ± 16 nM), but also an excellent selectivity profile. MSC2032964A was tested against a panel of 210 kinases (Table S1 of Supporting Information). The kinases were initially screened at a single concentration of 10 µM MSC2032964A, and any kinases whose activity was inhibited by over 70% were selected for determination of IC50 values. An IC50 value of less than 10 µM was only found for two kinases: ASK1 at 93 nM and CK1δ at 4800 nM (Fig 5B). In addition, the profiling of MSC2032964A revealed an excellent overall in vitro ADME profile. In particular, it showed a very good metabolic stability (<4 ml/min/mg, as measured by intrinsic human and rat microsomal clearance), a good apparent permeability (Papp: 51 × 10−6 cm/s, measured in Caco-2 cells) and a moderate tendency to be bound to plasma protein (5–7% unbound fraction for both human and rat serum). In vivo rat pharmacokinetics of MSC2032964A confirmed these findings and showed an excellent oral bioavailability of 82% (5 mg/kg, administered as a suspension), a moderate clearance (1.1 L/kg/h), a long half-life (5.2 h) and a high volume of distribution (Vss: 1.0 L/kg), indicating widespread distribution above total body water and accumulation in some peripheral compartments (Fig 6 and Table S2–4 of Supporting Information). Interestingly, MSC2032964A was exposed in brain (ratio of the exposure in brain over the exposure in plasma is 0.55, after dosing 0.6 mg/kg, iv). Pharmacokinetics in mice showed very similar results (data not shown).
Figure 5. Chemistry optimization and IC50s of ASK1 inhibitor.
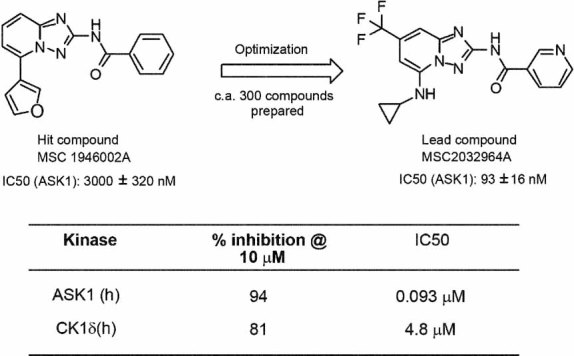
- Medicinal chemistry optimization led to the identification of the compound MSC2032964A.
- MSC2032964A showed IC50 values below 10 µM for only two kinases: ASK1 and CK1δ, at 93 and 4800 nM, respectively.
Figure 6.
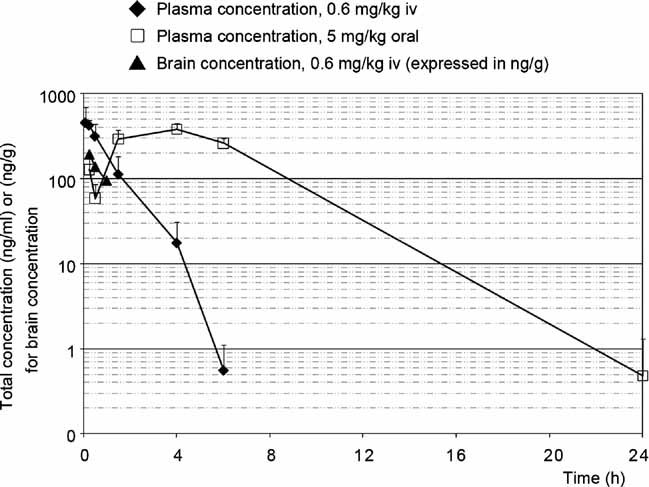
In vivo pharmacokinetics study of MSC2032964A in rats. Concentrations in plasma and brain were measured at different time points. The average for each group was expressed as mean ± SEM (n = 3 for each group).
Since MSC2032964A showed outstanding overall in vitro and in vivo profiles, and has a very good exposure in brain, we proceeded for further validation of this compound as a therapeutically effective ASK1 inhibitor. First, we tested MSC2032964A in in vivo inflammation models and found that it blocked LPS-induced ASK1 and p38 phosphorylation in cultured mouse astrocytes (Fig 7A). Next, we examined the effect of pharmacological inhibition of ASK1 enzymatic activity during EAE using MSC2032964A. The disease incidence of EAE was not different between vehicle- and 30 mg/kg MSC2032964A-treated groups, but MSC2032964A induced a significantly attenuated disease course after d18, nearly reproducing the phenotype observed in ASK1−/− mice (Fig 7B). Consistent with these results, histopathological analysis of the spinal cord demonstrated reduced demyelination (upper panels in Fig 7C) and decreased astrocyte and microglia activation (middle panels in Fig 7C) in the MSC2032964A-treated EAE group. In addition, MSC2032964A partially prevented optic nerve demyelination (lower panels in Fig 7C). Consequently, the average visual responses were significantly preserved in the MSC2032964A-treated EAE group (Fig 7D), further suggesting the strong potential of ASK1 inhibition as a treatment of MS and optic neuritis.
Figure 7. Effect of MSC2032964A on LPS-induced ASK1 activation in vitro and the severity of EAE in vivo.
- Effect of MSC2032964A on LPS-induced ASK1 activation, as assessed by the phosphorylation of ASK1, p38 and JNK in cultured mouse astrocytes. Confluent astrocytes seeded in 12-well plates were pre-treated with MSC2032964A (10 µM) for 90 min and then treated with LPS (10 µg/ml) for 30 min. Cell lysates were subjected to immunoblotting with the indicated antibodies.
- Clinical evaluation of EAE mice treated with vehicle (n = 7) or MSC2032964A (n = 7) for a period of 40 days after MOG immunization.
- Representative histology of the spinal cords and optic nerves of EAE mice treated with vehicle or MSC2032964A. Lumbar spinal cords were stained with LFB and HE (upper panels) and either an anti-GFAP or anti-iba1 antibody (middle panels), and optic nerves were stained with LFB and HE (lower panels). Scale bar: 150 µm for the upper three panels and 20 µm for the lower panel.
- Averaged visual response amplitudes from each mouse group (n = 7) were examined by multifocal electroretinograms. *p < 0.05. Values are given in nV per square degree (nV/deg2).
To elucidate whether MSC2032964A has targets other than ASK1, we also investigated the effect of MSC2032964A on the chemokine production in ASK1−/− EAE mice. Spinal cords from WT EAE, ASK1−/− EAE treated with MSC2032964A or vehicle were sampled on d20 after MOG immunization. Quantitative RT-PCR analysis revealed significantly reduced chemokine production in ASK1−/− EAE mice compared with WT EAE mice. However, no difference was found between ASK1−/− EAE mice treated with MSC2032964A or vehicle (Fig 8). These results demonstrate a high selectivity of MSC2032964A for ASK1 during EAE, and thus introducing this compound to mice that lack ASK1 has no additional effects on the suppression of cytokine production.
Figure 8.
Selective inhibition of ASK1 by MSC2032964A during EAE. ASK1−/− EAE mice were administrated with vehicle or MSC2032964A (30 mg/kg), and mRNA expression levels of MCP-1, RANTES and MIP-1α at day 20 after MOG immunization were detected using quantitative real-time PCR. GAPDH was used as an internal control. *p < 0.05.
DISCUSSION
Our present results suggest that TLR-ASK1-p38 signalling in astrocytes regulates chemokine production and recruitment of activated microglia into the lesion site. In addition, the same signalling pathway in microglial cells seems to modulate the progress of demyelination by altering the release of proinflammatory components such as TNFα and iNOS. With dramatically increased glial TLR4 and TLR9 expression in EAE mice, our findings are consistent with previous reports showing that TLR4- or TLR9-mediated innate immunity contributes to the pathogenesis of EAE (Kerfoot et al, 2004; Prinz et al, 2006). In addition, mice deficient in the TLR adaptor protein Myeloid differentiation factor 88 (MyD88) were completely resistant to MOG-induced EAE (Prinz et al, 2006). Since TRAF6-ASK1-p38 axis is a part of MyD88-dependent pathway (Akira & Takeda, 2004; Beutler, 2004) and ASK1 had no effect on T-cell proliferation capability (Fig 1B), it seems to be reasonable that all ASK1−/− mice developed EAE with decreased severity.
In ASK1-deficient astrocytes, LPS-induced activation of p38 was attenuated, but neither JNK activation nor the signalling pathway leading to the activation of NF-κB was affected (Fig S4 of Supporting Information). Although the reason for the lack of impairment of JNK and NF-κB activation in ASK1-deficient cells is unclear, activation of ASK1-p38 axis might be more tightly regulated than pathways involving other MAP3Ks such as TAK1 and MEKK3 (Akira & Takeda, 2004; Beutler, 2004; Wang et al, 2001). ASK1 is required for ROS-dependent p38 activation through TLR4 in dendritic cells, but either LPS or unmethylated CpG DNA could activate this pathway in astrocytes. One plausible explanation for this discrepancy is that expression level and the upregulation pattern of TLRs vary between species, cell types or culture conditions, in response to different stimuli (Bsibsi et al, 2006; Carpentier et al, 2005; Jack et al, 2005; McKimmie & Fazakerley, 2005). Moreover, a recent study showed that ROS is also produced downstream of TLR9 signalling in macrophages (Lee et al, 2008). TLR9 is known to recruit multiple Toll-interleukin 1 receptor domain-containing adaptors such as MyD88, TRAF6, TIRAP/Mal and the kinases RICK/RIP2 and IRAK, which eventually activates p38 (Akira & Takeda, 2004; Beutler, 2004). Thus, it seems to be reasonable that both TLR4- and TLR9-dependent signalling pathways could activate the ASK1 pathway in astrocytes. In addition, LPS-induced TNFα production was partially suppressed in ASK1-deficient microglial cells, suggesting a possible role of ASK1 deficiency in TNFα signalling (Fig 2B). Although our data strongly indicate that TLRs-ASK1-p38 pathways play important roles in the severity of EAE (Figs 4 and 7 and Fig S6 of Supporting Information), we cannot exclude the possibility that a deficient TNF-ASK1-p38 MAPK pathway might partly account for the alleviated severity of EAE in ASK1−/− mice. Another important point is that glial TLRs may induce the production of a variety of neurotrophic factors, which lead to the protection of surrounding CNS cells (Bsibsi et al, 2006). Since glia–glia and glia–neuron networks utilizing neurotrophic factors play important roles in the CNS (Harada et al, 2000, 2002), more detailed functions of the TLR-ASK1 pathway in resident glial cells, especially its beneficial and adverse roles during CNS inflammation, should be elucidated.
A recent study demonstrated that ASK2, a new ASK1 family member, formed a heteromeric complex with ASK1, and ASK2 in this complex exhibited sufficient basal activity toward the JNK and p38 pathways as a MAP3K (Takeda et al, 2007). Since ASK2 also activates ASK1 by direct phosphorylation, endogenous ASK2 expression level may modulate the capacity and sensitivity of downstream signalling pathways including p38. Thus, detailed ASK2 gene expression profiles in various cell types and functional analysis using ASK2−/− and ASK1−/−/ASK2−/− mice will be required in future investigations. In our study, the phosphorylation of ASK1, which is required for ASK1 activation, was inhibited by MSC2032964A in cultured astrocytes (Fig 7A). This finding might be explained by two mechanisms: the compound inhibited an upstream component required for the activation of ASK1, or the compound inhibited ASK1 phosphorylation by directly binding to it. Thus, it is possible that ASK2, which acts upstream of ASK1, could also be a target of MSC2032964A. However, ASK2 contributes little in ROS-induced p38 activation (Iriyama et al, 2009). Combined with the findings that MSC2032964A demonstrated almost no inhibition on other upstream kinases such as GCK (Table S1), it seems to be more likely for MSC2032964A to act directly on ASK1.
Optic neuritis is an acute inflammatory demyelinating syndrome of the CNS that often occurs in MS, and even a past history of idiopathic optic neuritis is a risk factor for developing MS (Optic Neuritis Study Group, 2008). Here, we assessed the effect of ASK1 deficiency on the severity of optic neuritis, and demonstrated reduced optic nerve inflammation and axonal loss in ASK1−/− mice. In addition, we measured visual function of EAE mice in vivo, and showed mild visual disturbance in ASK1−/− mice. These results suggest a possibility that ASK1 is a potential therapeutic target for optic neuritis that may occur independent of, or as a complication of MS. Although the detailed etiology of irreversible optic neuritis is yet unknown, several studies reported that glutamate neurotoxicity may play a role in the pathogenesis of CNS inflammation (Bal-Price & Brown, 2001; Maier et al, 2007). Interestingly, stimulation of astrocytes with TLR ligands inhibits expression of GLAST, a major glial glutamate transporter, and reduces glutamate uptake activity (Scumpia et al, 2005). We recently found that GLAST-deficient mice show glaucomatous optic nerve degeneration due to increased glutamate neurotoxicity and oxidative stress (Harada et al, 2007). These observations suggest that TLR-mediated innate immunity may be involved in the pathogenesis of various neurodegenerative diseases (Boivin et al, 2007; Kilic et al, 2008) including glaucoma (Shibuya et al, 2008). We are currently investigating the severity of optic nerve degeneration in GLAST−/− EAE mice as well as GLAST−/−/ASK1−/− mice (Harada et al, 2010).
The paper explained
PROBLEM
Multiple sclerosis is an autoimmune and inflammatory disease of the CNS that causes neurological disability in young adults. To date, there is no cure for this debilitating disease. We previously demonstrated that a specific inhibitor of glial cell activation suppressed the release of some key chemokines from astrocytes, thereby ameliorating the severity of EAE, an animal model of MS. In the present study, we attempted to elucidate a potential role of TLRs–ASK1 signalling in glial cells during MS.
RESULTS
By using ASK1 knockout mice, we demonstrated that ASK1 deficiency attenuates neuroinflammation in EAE, without affecting the proliferation capability of T cells. Moreover, we found that EAE upregulates expression of TLRs in activated astrocytes and microglia, and that TLRs can synergize with ASK1-p38 MAPK signalling in the release of key chemokines from astrocytes. Furthermore, oral treatment with a specific small molecular weight inhibitor of ASK1 suppressed EAE-induced autoimmune inflammation in both spinal cords and optic nerves.
IMPACT
These findings suggest that the TLR-ASK1-p38 pathway in glial cells may serve as a valid therapeutic target for autoimmune demyelinating disorders including MS.
In conclusion, we demonstrated that ASK1-p38 pathway is required for multiple TLRs-mediated innate immunity in resident glial cells, such as the production of chemokines in astrocytes and of toxic factors in microglia. Consistently, ASK1−/− EAE mice showed attenuated neurological symptoms in both spinal cord and optic nerve lesions. Thus, with careful attention to the potential side effects, ASK1-selective inhibitors, such as MSC2032964A, may be useful in future treatments of neuroinflammatory diseases including MS.
MATERIALS AND METHODS
Mice
Female C57BL/6J and ASK1−/− mice (Harada et al, 2006) were 6–8 weeks of age at the time of immunization. Animal treatments were performed in accordance with the Tokyo Metropolitan Institute for Neuroscience Guidelines for the Care and Use of Animals.
EAE induction, ASK1 inhibitor administration and clinical scoring
EAE was induced with MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) as previously reported (Mendel et al, 1995). To evaluate the effect of MSC2032964A, MOG-immunized mice were treated with either MSC2032964A (30 mg/kg) or vehicle (0.5% carboxymethylcellulose/0.25% Tween 20 in distilled water) once daily by oral gavage throughout the whole experimental period. Clinical signs were scored daily as follows: 0, no clinical signs; 1, loss of tail tonicity; 2, flaccid tail; 3, impairment of righting reflex; 4, partial hind limb paralysis; 5, complete hind limb paralysis; 6, partial body paralysis; 7, partial forelimb paralysis; 8, complete forelimb paralysis or moribund; 9, death.
Histopathology and immunohistochemistry
Optic nerves and spinal cords were examined by hematoxylin and eosin (HE) and luxol fast blue (LFB) staining as previously reported (Guo et al, 2007). Immunohistochemistry was performed using the following primary antibodies: rabbit anti-iba1 (1.0 µg/ml) (Harada et al, 2002), mouse anti-GFAP (50 µg/ml; Progen), rabbit anti-GFAP (1:1; Abcam), goat anti-TLR4 (2.0 µg/ml; Santa Cruz Biotechnology) and mouse anti-TLR9 (1.0 µg/ml; Santa Cruz Biotechnology). Quantitative analysis of the immunopositive cell number or stained region was carried out using NIH Image (ImageJ 1.38).
Cell culture
Primary astrocytes and microglial cells were obtained as previously reported (Guo et al, 2007). Astrocytes were treated with LPS or unmethylated CpG DNA at various concentrations and for different amounts of time. To investigate the effects of a p38 inhibitor on chemokine productions, astrocytes were pre-treated with SB203580 (2 µM) for 30 min followed by LPS stimulation (0.1 µg/ml) for 16 h. Microglial cells were treated for 16 h with LPS (1 µg/ml). Concentrations of chemokines (MCP-1, RANTES, MIP-1α) and TNFα in the cell culture media were determined by ELISA (Guo et al, 2007).
Immunoblot analysis
Immunoblotting was performed as previously reported (Namekata et al, 2008, 2010). Membranes were incubated with an antibody against ASK1, phospho-ASK1, p38 (1:1000; BD Biosciences), phospho-p38 (1:1000; BD Biosciences), JNK (1:1000; Santa Cruz Biotechnology), phospho-JNK (1:1000; Santa Cruz Biotechnology) or IκB (1:1000; BD Biosciences).
Quantitative real-time PCR
Quantitative RT-PCR was performed using the ABI 7500 fast RT-PCR system (Applied Biosystems) with SYBR Green PCR Master Mix (Applied Biosystems) as previously reported (Guo et al, 2007). Primer probe pairs used are listed in Table S5 of Supporting Information.
Proliferation assay and T-cell-derived cytokine analysis
Proliferative responses of lymph node cells from WT and ASK1−/− mice 9 days after MOG immunization were assayed in microtiter wells by the uptake of [3H]thymidine as previously reported (Guo et al, 2007). T-cell culture media were collected and concentrations of cytokines in the media were determined by ELISA.
Multifocal electroretinograms (mfERGs)
The second-order kernel of the mfERGs was measured using a VERIS 6.0 system (Electro-Diagnostic Imaging) as previously reported (Harada et al, 2007).
Selectivity profile
The selectivity profile of the compound MSC2032964A over the 210 kinases was determined at Millipore (KinaseProfiler). The inhibition of the kinases by the compound was tested at concentrations of Km for ATP and at 10 µM for the compound. The protocols for testing can be found at www.millipore.com.
In vivo pharmacokinetics study in rats
Eight-week-old male Sprague–Dawley rats received the test compound (MSC2032964A) by oral (5 mg/kg by gavage) or by intravenous (0.6 mg/kg into the tail vein) routes. Serial blood samples were collected from the sublingual vein up to 24 h after dosing. Plasma samples were analyzed by LC/MS/MS (Sciex API 4000). Pharmacokinetic parameters were determined using non-compartmental analysis.
Statistics
Data are presented as means ± SEM. When statistical analyses were performed, the Student's t-test was used to estimate the significance of the results.
Acknowledgments
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the Japan Society for the Promotion of Science; the Ministry of Health, Labour and Welfare of Japan; and the Japan Medical Association.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
XG: Study design, acquisition of data, analyses and interpretation of data, manuscript preparation. CH: acquisition of data, analyses and interpretation of data. KN: acquisition of data, analyses and interpretation of data. AM: acquisition of data, analyses and interpretation of data. MC: screened and characterized the compound, manuscript preparation. HJ: screened and characterized the compound. DS: screened and characterized the compound, manuscript preparation. CJ-L: screened and characterized the compound. MM: screened and characterized the compound. P-AV: screened and characterized the compound. TR: screened and characterized the compound. AK: analyses and interpretation of data, manuscript preparation. KK: acquisition of data, analyses and interpretation of data. YM: contributed important reagent, analyses and interpretation of data. HI: contributed important reagent, analyses and interpretation of data. TH: study design, manuscript preparation and managed the whole project.
For more information
National multiple sclerosis society
http://www.nationalmssociety.org
Multiple sclerosis association of America (MSAA)
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21:6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430:257–263. doi: 10.1038/nature02761. [DOI] [PubMed] [Google Scholar]
- Boivin A, Pineau I, Barrette B, Filali M, Vallières N, Rivest S, Lacroix S. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J Neurosci. 2007;27:12565–12576. doi: 10.1523/JNEUROSCI.3027-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia. 2006;53:688–695. doi: 10.1002/glia.20328. [DOI] [PubMed] [Google Scholar]
- Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- Chiang E, Dang O, Anderson K, Matsuzawa A, Ichijo H, David M. Cutting edge: apoptosis-regulating signal kinase 1 is required for reactive oxygen species-mediated activation of IFN regulatory factor 3 by lipopolysaccharide. J Immunol. 2006;176:5720–5724. doi: 10.4049/jimmunol.176.10.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescu CS, Tani M, Ransohoff RM, Wysocka M, Hilliard B, Fujioka T, Murphy S, Tighe PJ, Sarma JD, Trinchieri G, et al. Astrocytes as antigen-presenting cells: expression of IL-12/IL-23. J Neurochem. 2005;95:331–340. doi: 10.1111/j.1471-4159.2005.03368.x. [DOI] [PubMed] [Google Scholar]
- Girvin AM, Gordon KB, Welsh CJ, Clipstone NA, Miller SD. Differential abilities of central nervous system resident endothelial cells and astrocytes to serve as inducible antigen-presenting cells. Blood. 2002;99:3692–3701. doi: 10.1182/blood-2001-12-0229. [DOI] [PubMed] [Google Scholar]
- Guo X, Nakamura K, Kohyama K, Harada C, Behanna HA, Watterson DM, Matsumoto Y, Harada T. Inhibition of glial cell activation ameliorates the severity of experimental autoimmune encephalomyelitis. Neurosci Res. 2007;59:457–466. doi: 10.1016/j.neures.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Harada T, Harada C, Nakayama N, Okuyama S, Yoshida K, Kohsaka S, Matsuda H, Wada K. Modification of glial-neuronal cell interactions prevents photoreceptor apoptosis during light-induced retinal degeneration. Neuron. 2000;26:533–541. doi: 10.1016/s0896-6273(00)81185-x. [DOI] [PubMed] [Google Scholar]
- Harada T, Harada C, Kohsaka S, Wada E, Yoshida K, Ohno S, Mamada H, Tanaka K, Parada LF, Wada K. Microglia-Müller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J Neurosci. 2002;22:9228–9236. doi: 10.1523/JNEUROSCI.22-21-09228.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada C, Nakamura K, Namekata K, Okumura A, Mitamura Y, Iizuka Y, Kashiwagi K, Yoshida K, Ohno S, Matsuzawa A, et al. Role of apoptosis signal-regulating kinase 1 in stress-induced neural cell apoptosis in vivo. Am J Pathol. 2006;168:261–269. doi: 10.2353/ajpath.2006.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T, Harada C, Nakamura K, Quah HM, Okumura A, Namekata K, Saeki T, Aihara M, Yoshida H, Mitani A, et al. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J Clin Invest. 2007;117:1763–1770. doi: 10.1172/JCI30178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada C, Namekata K, Guo X, Yoshida H, Mitamura Y, Matsumoto Y, Tanaka K, Ichijo H, Harada T. ASK1 deficiency attenuates neural cell death in GLAST-deficient mice, a model of normal tension glaucoma. Cell Death Differ. 2010;17:1751–1759. doi: 10.1038/cdd.2010.62. [DOI] [PubMed] [Google Scholar]
- Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Iriyama T, Takeda K, Nakamura H, Morimoto Y, Kuroiwa T, Mizukami J, Umeda T, Noguchi T, Naguro I, Nishitoh H, et al. ASK1 and ASK2 differentially regulate the counteracting roles of apoptosis and inflammation in tumorigenesis. EMBO J. 2009;28:843–853. doi: 10.1038/emboj.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K, Masutani H, Yodoi J, Urano Y, Nagano T, et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005;12:19–24. doi: 10.1038/sj.cdd.4401528. [DOI] [PubMed] [Google Scholar]
- Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, Zanardo RC, Bonder C, James WG, Robbins SM, Kubes P. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J Immunol. 2004;173:7070–7077. doi: 10.4049/jimmunol.173.11.7070. [DOI] [PubMed] [Google Scholar]
- Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM. TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiol Dis. 2008;31:33–40. doi: 10.1016/j.nbd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, et al. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science. 2002;297:623–626. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- Lee JG, Lee SH, Park DW, Lee SH, Yoon HS, Chin BR, Kim JH, Kim JR, Baek SH. Toll-like receptor 9-stimulated monocyte chemoattractant protein-1 is mediated via JNK-cytosolic phospholipase A2-ROS signaling. Cell Signal. 2008;20:105–111. doi: 10.1016/j.cellsig.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Maier K, Merkler D, Gerber J, Taheri N, Kuhnert AV, Williams SK, Neusch C, Bähr M, Diem R. Multiple neuroprotective mechanisms of minocycline in autoimmune CNS inflammation. Neurobiol Dis. 2007;25:514–525. doi: 10.1016/j.nbd.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Marta M, Andersson A, Isaksson M, Kämpe O, Lobell A. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur J Immunol. 2008;38:565–575. doi: 10.1002/eji.200737187. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–592. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- McKimmie CS, Fazakerley JK. In response to pathogens, glial cells dynamically and differentially regulate Toll-like receptor gene expression. J Neuroimmunol. 2005;169:116–125. doi: 10.1016/j.jneuroim.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T-cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 1995;25:1951–1957. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- Nair A, Frederick TJ, Miller SD. Astrocytes in multiple sclerosis: a product of their environment. Cell Mol Life Sci. 2008;65:2702–2720. doi: 10.1007/s00018-008-8059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekata K, Harada C, Kohyama K, Matsumoto Y, Harada T. Interleukin-1 stimulates glutamate uptake in glial cells by accelerating membrane trafficking of Na+/K+-ATPase via actin depolymerization. Mol Cell Biol. 2008;28:3273–3280. doi: 10.1128/MCB.02159-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekata K, Harada C, Taya C, Guo X, Kimura H, Parada LF, Harada T. Dock3 induces axonal outgrowth by stimulating membrane recruitment of the WAVE complex. Proc Natl Acad Sci USA. 2010;107:7586–7591. doi: 10.1073/pnas.0914514107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22:1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol. 2008;65:727–732. doi: 10.1001/archneur.65.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, Piesche M, Schroers R, Weiss E, Kirschning CJ, et al. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. 2006;116:456–464. doi: 10.1172/JCI26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scumpia PO, Kelly KM, Reeves WH, Stevens BR. Double-stranded RNA signals antiviral and inflammatory programs and dysfunctional glutamate transport in TLR3-expressing astrocytes. Glia. 2005;52:153–162. doi: 10.1002/glia.20234. [DOI] [PubMed] [Google Scholar]
- Selmaj KW, Raine CS, Cross AH. Anti-tumor necrosis factor therapy abrogates autoimmune demyelination. Ann Neurol. 1991;30:694–700. doi: 10.1002/ana.410300510. [DOI] [PubMed] [Google Scholar]
- Shibuya E, Meguro A, Ota M, Kashiwagi K, Mabuchi F, Iijima H, Kawase K, Yamamoto T, Nakamura M, Negi A, et al. Association of Toll-like receptor 4 gene polymorphisms with normal tension glaucoma. Invest Ophthalmol Vis Sci. 2008;49:4453–4457. doi: 10.1167/iovs.07-1575. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Steinman L, Martin R, Bernard C, Conlon P, Oksenberg JR. Multiple sclerosis: deeper understanding of its pathogenesis reveals new targets for therapy. Annu Rev Neurosci. 2002;25:491–505. doi: 10.1146/annurev.neuro.25.112701.142913. [DOI] [PubMed] [Google Scholar]
- Takeda K, Shimozono R, Noguchi T, Umeda T, Morimoto Y, Naguro I, Tobiume K, Saitoh M, Matsuzawa A, Ichijo H. Apoptosis signal-regulating kinase (ASK) 2 functions as a mitogen-activated protein kinase kinase kinase in a heteromeric complex with ASK1. J Biol Chem. 2007;282:7522–7531. doi: 10.1074/jbc.M607177200. [DOI] [PubMed] [Google Scholar]
- Tanuma N, Sakuma H, Sasaki A, Matsumoto Y. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 2006;112:195–204. doi: 10.1007/s00401-006-0083-7. [DOI] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Voorn P, Tekstra J, Beelen RH, Tensen CP, Van Der Valk P, De Groot CJ. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am J Pathol. 1999;154:45–51. doi: 10.1016/S0002-9440(10)65249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.