

Abstract

Dirhodium(II)-catalyzed reactions of silyl-protected enoldiazoacetates with nitrile oxides exhibit high nitrile oxide substituent dependence in the production rearrangement products via dipolar cycloaddition and either the Neber rearrangement or the Lossen rearrangement.
INTRODUCTION
Catalytically-generated metal carbenes from rhodium-catalyzed reactions of diazo compounds are known mainly for their versatility in reactions that include cyclopropanation and cyclopropenation, ylide formation and rearrangements, and carbon-hydrogen insertion.1,2 However, as we have recently demonstrated,3,4 there are a variety of alternative metal-carbene-dependent processes that offer intriguing possibilities for chemical syntheses. In order to understand metal carbene-dependent extensions in reactivity and selectivity, we have been investigating catalytic reactions of enoldiazoacetates that include 3-(tert-butyldimethylsiloxy)-2-diazo-3-butenoates 1,4 a subset of vinyldiazoacetates that have been introduced and extensively studied by Davies and coworkers.5 We recognized that dinitrogen extrusion by dirhodium(II) changes the polarity of the bound carbon from being electron-rich in the diazo compound to electron-deficient in the metal carbene.6,7,8 Based on this principle, we have developed a novel formal [3+3] cycloaddition7 and an alternative transformation for the functionalized pyrrole synthesis with nitrone.8 Recently, we reported an unexpected catalytic reaction of enoldiazoacetate 1a with nitrile oxides having electron-donating substituents that form 5-arylaminofuran-2(3H)-one-4-carboxylates 3 in high yield (Scheme 1).9 However, a constitutionally isomeric product was detected when nitrile oxides having halide or electron-withdrawing substituents were used. Spectral analyses were insufficient to completely identify the structure of this compound, so crystals of the product from the reaction with o-nitrobenzonitrile-N-oxide (2g) that resulted in the formation of only one isomer were used to obtain its X-ray structure (Figure 1). The constitutionally isomeric product is methyl 3-oxo-1-(o-nitrophenyl)-2-oxa-6-azabicyclo-[3.1.0]hexane-5-carboxylate 4g. We report here the generality of this rearrangement reaction and the mechanism for its formation.
Scheme 1.
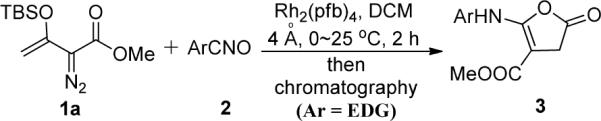
5-Arylaminofuran-2(3H)-one-4-carboxylates from Dirhodium(II) Catalyzed Reactions of 1a with Nitrile Oxides Having Electron-donating Substituents.
Figure 1.

Crystal Structure of the Product 4g from the Dirhodium(II) Catalyzed Reaction Between 1a and o-Nitrobenzonitrile-N-oxide 2g.
RESULTS AND DISCUSSION
Examination of an array of monosubstituted benzonitrile oxides (Table 1) shows that aziridine derivatives, 3-oxo-1-aryl-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylates 4, result from reactions with benzonitrile-N-oxides having electron-withdrawing substituents, whereas 5-arylaminofuran-2(3H)-one-4-carboxylates are the dominant products from reactions with benzonitrile-N-oxides having electron-donating substituents. Both products result from rearrangement reactions that occur from the combination of vinyldiazoacetate 1a with the nitrile oxide in the presence of dirhodium(II) catalysts; in the absence of catalyst or the use of copper catalysts, TBS transfer from 1a to the nitrile oxide occurs.10
Table 1.
Nitrile Oxides Substituent Dependence in the Production Rearrangement Productsa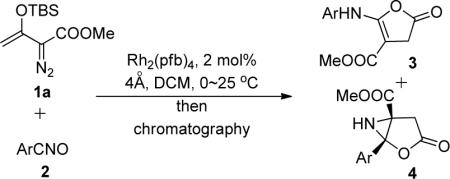
| entry | Ar (2) | product | 3:4b | yield (%)c |
|---|---|---|---|---|
| 1 | 2-MeOC6H4 (2a) | 3a | >95:5 | 92 |
| 2 | 4-MeC6H4 (2b) | 3b, 4b | 90:10 | 84 |
| 3 | 4-FC6H4 (2c) | 3c, 4c | 78:22 | 89 |
| 4 | 4-ClC6H4 (2d) | 3d, 4d | 65:35 | 93 |
| 5 | 4-BrC6H4 (2e) | 3e, 4e | 55:45 | 89 |
| 6 | 4-AcOC6H4 (2f) | 3f, 4f | 39:61 | 81 |
| 7 | 2-NO2C6H4 (2g) | 3g, 4g | <5:95 | 90 |
Reaction conditions: 1a (1.2 mmol) in DCM (3.0 mL) was added over 1 h by syringe pump to mixture of 4 Å molecular sieves (100 mg), Rh2(pfb)4 (2.0 mol %) and nitrile oxide 2 (1.0 mmol) in DCM (5.0 mL) at 0 °C, and stirred for 2 h at room temperature. After solvent removal the product mixture was purified by chromatography on silica gel.
Determined by 1H NMR of the reaction mixture.
Isolated yield of combined 3 and 4 based on limiting reagent 2.
We previously found that under the conditions used for reactions of 1a with nitrile oxides, dinitrogen extrusion produces cyclopropene 5 which undergoes subsequent uncatalyzed [2 + 3]-cycloaddition with nitrile oxides to form 2-oxa-3-azabicyclo[3.1.0]hex-3-ene 6. Subsequent rearrangements of 6 lead to the formation of 3 (Lossen rearrangement)9 and, presumably, 4 (Scheme 2). We anticipated a substituent-dependent correlation between 3 and 4 if the pathways to them are linked through 6, and Figure 2 clearly shows linear correlation between the two processes (log [3]/[4] versus σ). The 6 → 3 conversion occurs through ring opening of 6 (with aryl migration from carbon to nitrogen) to a ketenimine intermediate that has been intercepted and shown to undergo acid-catalyzed ring closure to 3.9 Could a similar ring opening/ring closure process be occurring in the formation of 4 from 6?
Scheme 2.
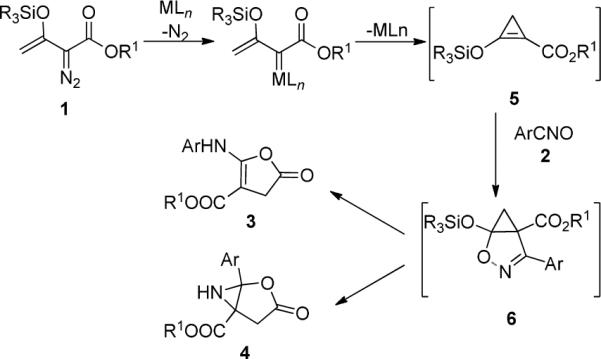
Divergent Outcomes from Catalytic Reactions of Silyl-Protected Enoldiazoacetates with Nitrile Oxides.
Figure 2.
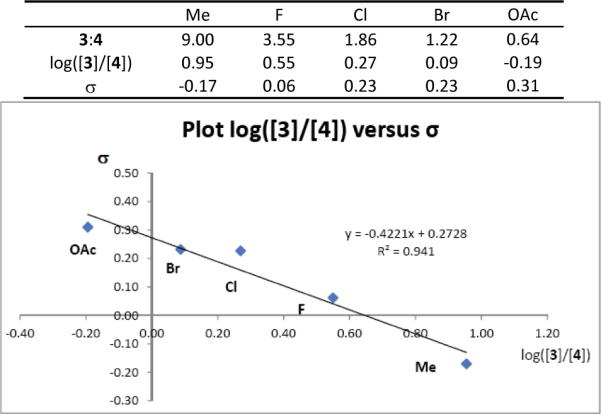
Plot log([3]/[4]) versus σ: r = −0.42 (R2 = 0.94).
Analogous to the formation of 3 from 6, the pathway to 4 is formally a cyclic analog of the Neber reaction.11 In this case ring opening to an azirine intermediate (7 in Scheme 3) would preface formation of the subsequent cyclization product 4. Careful examination of the 1H NMR data for the reaction mixture from Rh2(pfb)4 catalysis of 1a with 2g prior to chromatography10 revealed that the chemical shifts of the CH2 group protons of the product prior to chromatography were different from those of 4g, suggesting that the initially-formed reaction product is transformed into 4g during chromatographic isolation. One possible explanation is that this pre-4g product is the N-TBS-bound aziridine 8 rather than 7. However, the chemical shift of Si is consistent with oxygen attachment in 7 rather than nitrogen attachment in 8.10 Furthermore, catalysis in the formation of 4 occurs either when the product prior to chromatography is treated with saturated aqueous ammonium chloride or with silica gel during chromatography, which is also consistent with 7 as the pre-4 product. Attempts to obtain pure crystalline 7 were not successful.
Scheme 3.
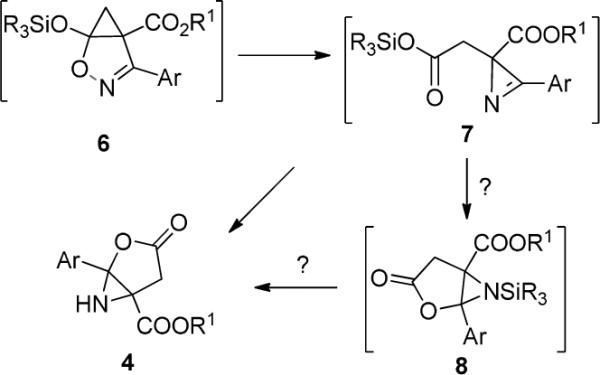
Mechanistic Possibilities for the Formation of 3-Oxo-1-aryl-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylates (4).
Using the combination of rhodium(II) catalyzed formation of 7 followed by ammonium chloride promoted formation of 4, reactions of 1a with a series of nitrile oxides having electron withdrawing groups were examined, and the results of this study are shown in Table 2. These reactions generate aziridine 4 as the sole product in 77–92% isolated yield. Even the tert-butyl ester of the enoldiazoacetate (1b) gives comparable results. The same process and similar reactivity are observed with β-methyl-substituted enoldiazoacetate 1c that produces two diastereoisomers from its reaction with an equal amount of 2g (Scheme 4, 4m, 89% yield). Recognizing that stereocontrol occurs in the nitrile oxide dipolar addition step, we briefly investigated changes in R1 and R2 that could influence this selectivity. By increasing the size of the β-substituent of enoldiazoacetates, very high diastereoselectivity (4o, dr >20:1) is achieved compared to increasing the size of the ester alkyl group (4n, dr 3:1, 83% yield). This rearrangement retains the stereochemical relationship of R2 with the carboxylate group, and retention of configuration occurs in the formation of 4 from 7.
Table 2.
Rhodium(II) Catalyzed Reaction of Silyl-protected Enoldiazoacetates with Nitnle Oxidesa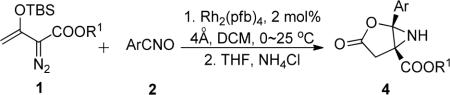
| entry | R1 (1) | Ar (2) | product | yield (%)b |
|---|---|---|---|---|
| 1 | Me (1a) | 2-NO2C6H4 (2g) | 4g | 90 |
| 2 | t-Bu (1b) | 2-NO2C6H4 (2g) | 4h | 92 |
| 3 | Me (1a) | 2,4-(NO2)2C6H4 (2h) | 4i | 81 |
| 4 | t-Bu (1b) | 2,4-(NO2)2C6H4 (2h) | 4j | 88 |
| 5 | Me (1a) | 2-CF3C6H4 (2i) | 4k | 77 |
| 6 | t-Bu (1b) | 2-CF3C6H4 (2i) | 4l | 82 |
Reaction conditions: 1 (1.2 mmol) in DCM (3.0 mL) was added over 1 h via a syringe pump at 0 °C to mixture of 4 Å molecular sieves (100 mg), Rh2(pfb)4 (2.0 mol %) and nitrile oxide 2 (1.0 mmol) in DCM (5.0 mL), and stirred for another 2 h at room temperature. The crude product was hydrolyzed in THF (4.0 mL) with aqueous saturated ammonium chloride solution (2.0 mL) at 60 °C for 1~2 h.
Isolated yield of 4 based on limiting reagent 2.
Scheme 4.

Diastereoselectivity in Reactions of β-Substituted Enoldiazoacetates with o-Nitrobenzonitrile-N-oxide.
Consistent with the proposed mechanism for this transformation, diastereocontrol is established in the intermolecular [3+2]-cycloaddition step that forms cis-6 as the major product. Although the Neber rearrangement is ordinarily associated with the formation of azirines from oximes and their derivatives,12 the formation of azirine 7 from 6 is formally the same process, and the overall scheme represents the first time that the Lossen and Neber rearrangements have been linked through one intermediate (6), especially with the strong correlation that is evident from the data in Figure 2.
In summary, we have discovered an unexpected duality of reactions whose outcome is substrate dependent. Occurring through unstable 2-oxa-3-azabicyclo[3.1.0]hex-3-enes 6 intermediates, electron withdrawing substituents on the aryl group at the 4-position favors the Neber reaction whereas electron donating substituents direct product formation through the Lossen rearrangement. Good to excellent yields have been obtained, and direction to high diastereocontrol has been achieved in the formal Neber process. Evidence is provided that these two processes are interlinked, which has not been previously considered.
EXPERIMENTAL SECTION
General Information
Reactions were performed in oven-dried (140 °C) glassware under an atmosphere of dry N2. Dichloromethane (DCM) was passed through a solvent column prior to use and was kept over 3 Å molecular sieves. Thin layer chromatography (TLC) was carried out using silica gel plates; the developed chromatogram was analyzed by a UV lamp (254 nm). Liquid chromatography was performed using flash chromatography of the indicated system on silica gel (230–400 mesh). 1H NMR and 13C NMR spectra were recorded in CDCl3 on a 400 MHz spectrometer; chemical shifts are reported in ppm with the solvent signals as reference, and coupling constants (J) are given in Hertz. The peak information is described as: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, comp = composite. High-resolution mass spectra (HRMS) were performed on a TOF-CS mass spectrometer using CsI as the standard. Copper hexafluorophosphate and other Lewis acids were purchased and used as received. Dirhodium tetraacetate was purchased. Other dirhodium catalysts1a and silyl-protected enoldiazoacetates 113 were synthesized according to literature procedures. Nitrile oxides 2 were synthesized according to the literature references.14 The general procedure with corresponding data for the synthesis of 5-arylaminofuran-2(3H)-one-4-carboxylates (3) has been reported.9
General Procedure for the Synthesis of Azabicyclo[3.1.0]hexanes (4)
To an oven-dried flask containing a magnetic stirring bar, 4 Å molecular sieves (100 mg), Rh2(pfb)4 (2.0 mol %) and nitrile oxide 2 (1.0 mmol) in dichloromethane (5.0 mL), was added enoldiazoacetate 1 (1.2 mmol) in dichloromethane (3.0 mL) over 1 h via a syringe pump at 0 °C. After addition was complete, the reaction solution was stirred for another 2 h at room temperature. Once the diazo compound was consumed (determined by a 1H NMR spectrum of the crude reaction mixture), then DCM was removed under reduced pressure and THF (2.0 mL) was added; the resulting solution was stirred at 60 °C for 1~2 h with aqueous saturated ammonium chloride solution (2.0 mL). The hydrolyzed product was extracted with ether (5 mL × 3), and the combined organic phase was dried over anhydrous Na2SO4. After evaporating the solvents (if necessary, the reaction mixture was subjected to 1H NMR to determined the diastereoselectivity), the residue was purified by column chromatography on silica gel (eluent: hexanes:EtOAc = 10:1 to 2:1) to give the pure aziridine product 4 in high yield.
Methyl 3-Oxo-1-(p-tolyl)-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylate (4b)
Yellow oil. 21 mg, 8.4% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.38 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 3.70 (s, 3H), 3.57 (d, J = 20.0 Hz, 1H), 3.01 (d, J = 20.0 Hz, 1H), 2.91 (bs, 1H), 2.40 (s, 3H); 13C NMR (100 MHz, CDCl3): 172.67, 166.84, 140.77, 129.77, 127.64, 125.85, 81.54, 53.79, 46.18, 35.90, 21.74; HRMS (ESI) calculated for C13H14NO4 [M+H]+: 248.0917; found: 248.0902.
Methyl 1-(4-Fluorophenyl)-3-oxo-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylate (4c)
White solid, mp 147–150 °C. 49 mg, 19.5% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.50 (m, 2H), 7.11 (m, 2H), 3.68 (s, 3H), 3.57 (d, J = 20.0 Hz, 1H), 3.01 (d, J = 20.0 Hz, 1H), 2.99 (bs, 1); 13C NMR (100 MHz, CDCl3): 172.54, 166.69, 165.25 (d, J = 25.0 Hz),, 129.94 (d, J = 8.0 Hz), 124.96 (d, J = 3.0 Hz), 116.21 (d, J = 22.0 Hz), 80.82, 53.85, 46.23, 35.82; HRMS (ESI) calculated for C12H11FNO4 [M+H]+: 252.0667; found: 252.0671.
Methyl 1-(4-Chlorophenyl)-3-oxo-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylate (4d)
1H Yellow solid, mp 135–136 °C. 87 mg, 32.5% yield. NMR (400 MHz, CDCl3): δ (ppm) 7.44 (q, J = 8.0 Hz, 4H), 3.72 (s, 3H), 3.61 (d, J = 20.0 Hz, 1H), 3.03 (d, J = 20.0 Hz, 1H), 2.91 (bs, 1H); 13C NMR (100 MHz, CDCl3): 172.28, 166.58, 136.75, 129.36, 129.09, 127.52, 80.69, 53.94, 46.46, 35.85; HRMS (ESI) calculated for C12H11ClNO4 [M+H]+: 268.0371; found: 268.0382.
Methyl 1-(4-Bromophenyl)-3-oxo-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylate (4e)
Yellow solid, mp 148–150 °C. 138 mg, 44.4% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.58 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 8.0 Hz, 2H), 3.72 (s, 3H), 3.58 (d, J = 20.0 Hz, 1H), 3.03 (d, J = 20.0 Hz, 1H), 2.90 (bs, 1H); 13C NMR (100 MHz, CDCl3): 172.26, 166.56, 132.32, 129.30, 128.04, 125.00, 80.74, 53.95, 46.46, 35.85; HRMS (ESI) calculated for C12H11BrNO4 [M+H]+: 311.9866; found: 311.9874.
Methyl 1-(4-acetoxyphenyl)-3-oxo-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylate (4f)
Yellow oil. 144 mg, 49.5% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.54 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 8.0 Hz, 2H), 3.73 (s, 3H), 3.58 (d, J = 20.0 Hz, 1H), 3.04 (d, J = 20.0 Hz, 1H), 2.89 (bs, 1H), 2.33 (s, 3H); 13C NMR (100 MHz, CDCl3): 171.97, 169.01, 166.22, 151.94, 128.55, 126.07, 121.93, 80.48, 53.52, 46.11, 35.53, 21.16; HRMS (ESI) calculated for C14H14NO6 [M+H]+: 292.0816; found: 292.0833.
Methyl 1-(2-Nitrophenyl)-3-oxo-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylate (4g)
Colourless crystal (after crystallization in DCM, EtOAc and Hexanes), mp 155–156 °C. 250 mg, 90% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.17 (d, J = 8.0 Hz, 1H), 7.82–7.69 (comp, 3H), 3.74 (d, J = 16.0 Hz, 1H), 3.72 (s, 3H), 3.03 (d, J = 16.0 Hz, 1H), 2.80 (bs, 1H); 13C NMR (100 MHz, CDCl3): 172.45, 167.54, 147.73, 134.48, 132.16, 131.91, 126.02, 124.91, 78.89, 54.07, 46.53, 34.99; HRMS (ESI) calculated for C12H11N2O6 [M+H]+: 279.0612; found: 279.0611.
tert-Butyl 1-(2-Nitrophenyl)-3-oxo-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylate (4h)
Brown oil. 294 mg, 92% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.19 (d, J = 8.0 Hz, 1H), 7.80–7.67 (comp, 3H), 3.70 (d, J = 20.0 Hz, 1H), 2.97 (d, J = 20.0 Hz, 1H), 2.74 (bs, 1H), 1.28 (s, 9H); 13C NMR (100 MHz, CDCl3): 172.60, 165.23, 147.11, 134.10, 131.64, 131.60, 125.49, 125.06, 84.15, 78.33, 46.83, 34.72, 27.53; HRMS (ESI) calculated for C15H17N2O6 [M+H]+: 321.1081; found: 321.1077.
Methyl 1-(2,4-Dinitrophenyl)-3-oxo-2-oxa-6-azabicyclo[3.1.0]hexane-5- carboxylate (4i)
Yellow solid, mp 168–170 °C. 261 mg, 81% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.95–8.09 (comp, 3H), 3.78 (d, J = 20.0 Hz, 1H), 3.72 (s, 3H), 3.07 (d, J = 16.0 Hz, 1H), 2.84 (bs, 1H); 13C NMR (100 MHz, CDCl3): 171.90, 167.11, 149.48, 148.16, 133.76, 131.02, 128.36, 121.33, 54.26, 47.01, 34.98; HRMS (ESI) calculated for C12H10N3O8 [M+H]+: 324.0462; found: 324.0469.
tert-Butyl 1-(2,4-Dinitrophenyl)-3-oxo-2-oxa-6-azabicyclo[3.1.0]hexane-5- carboxylate (4j)
Dark brown oil. 321 mg, 88% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.20–7.67 (comp, 3H), 3.70 (d, J = 20.0 Hz, 1H), 2.97 (d, J = 20.0 Hz, 1H), 2.74 (bs, 1H), 1.28 (s, 9H); 13C NMR (100 MHz, CDCl3): 172.20, 165.49, 149.41, 147.89, 133.83, 131.59, 128.37, 121.22, 85.25, 47.67, 35.06, 28.02; HRMS (ESI) calculated for C15H16N3O8 [M+H]+: 366.0932; found: 366.09312.
Methyl 3-Oxo-1-(2-(trifluoromethyl)phenyl)-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylate (4k)
Brown oil. 231 mg, 77% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.78–7.62 (comp, 4H), 3.71 (s, 3H), 3.56 (d, J = 20.0 Hz, 1H), 3.01 (d, J = 20.0 Hz, 1H), 2.85 (bs, 1H); 13C NMR (100 MHz, CDCl3): 172.20, 167.23, 132.80, 131.56 (d, J = 5.0 Hz), 129.45 (q, J = 32.0 Hz), 127.81 (q, J = 5.0 Hz), 127.06, 125.38, 122.66, 79.67, 53.91, 46.02, 34.89; HRMS (ESI) calculated for C13H11F3NO4 [M+H]+: 302.0635; found: 302.0627.
tert-Butyl 3-Oxo-1-(2-(trifluoromethyl)phenyl)-2-oxa-6-azabicyclo[3.1.0]hexane-5-carboxylate (4l)
Brown oil. 281 mg, 82% yield. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.19 (d, J = 8.0 Hz, 1H), 7.80–7.67 (comp, 3H), 3.70 (d, J = 20.0 Hz, 1H), 2.97 (d, J = 20.0 Hz, 1H), 2.74 (bs, 1H), 1.28 (s, 9H); 13C NMR (100 MHz, CDCl3): 172.68, 165.23, 132.58, 131.33 (d, J = 3.3 Hz), 129.74 (q, J = 32.0 Hz), 127.72 (q, J = 5.2 Hz), 127.4, 125.36, 122.64, 84.57, 79. 34, 46.70, 34.93, 27.93; HRMS (ESI) calculated for C16H17F3NO4 [M+H]+: 344.1104; found: 344.1107.
Methyl 4-Methyl-1-(2-nitrophenyl)-3-oxo-2-oxa-6-azabicyclo[3.1.0]hexane-5- carboxylate (4m)
cis + trans: 260 mg, 89% yield. trans:cis = 1:1 (trans and cis relative chemistry was determined by 1D-NoE experiment).10trans-4m: Yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.14 (d, J = 8.0 Hz, 1H), 7.80–7.67 (comp, 3H), 3.85 (q, J = 8.0 Hz, 1H), 3.72 (s, 3H), 2.80 (bs, 1H), 1.48 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): 175.42, 167.65, 147.73, 134.46, 132.12, 131.91, 125.95, 124.94, 77.30, 54.05, 50.30, 39.82, 11.86; cis-4m: Yellow solid, mp 117–119 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.14 (d, J = 8.0 Hz, 1H), 7.80–7.67 (comp, 3H), 3.85 (q, J = 8.0 Hz, 1H), 3.72 (s, 3H), 2.80 (bs, 1H), 1.48 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): 176.78, 167.65, 147.75, 134.36, 132.21, 131.99, 125.78, 125.37, 79.46, 53.66, 49.05, 43.35, 14.21; HRMS (ESI) calculated for C13H13N2O6 [M+H]+: 293.0768; found: 293.0771.
Benzyl 4-Methyl-1-(2-nitrophenyl)-3-oxo-2-oxa-6-azabicyclo[3.1.0]hexane-5- carboxylate (4n)
cis + trans: 305 mg, 83% yield. trans:cis = 3:1 (trans and cis relative chemistry was determined by comparison the analogue 1H NMR specrta with 4m). trans-4n: Yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.08 (d, J = 8.0 Hz, 1H), 7.75–7.14 (comp, 8H), 5.16 (d, J = 12.0 Hz, 1H), 5.01 (d, J = 12.0 Hz, 1H), 3.91 (q, J = 8.0 Hz, 1H), 2.65 (bs, 1H), 1.47 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): 175.35, 166.89, 147.57, 134.72, 134.32, 131.97, 131.79, 129.11, 129.04, 128.73, 125.98, 124.98, 77.25, 68.81, 50.41, 39.83, 11.90; cis-4n: Yellow oil. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.06 (d, J = 8.0 Hz, 1H), 7.69–7.08 (comp, 8H), 5.09 (d, J = 12.0 Hz, 1H), 4.97 (d, J = 12.0 Hz, 1H), 3.18 (q, J = 8.0 Hz, 1H), 2.80 (bs, 1H), 1.82 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): 176.83, 166.69, 147.55, 134.53, 134.62, 132.09, 131.83, 129.09, 129.01, 128.86, 125.97, 125.80, 79.45, 68.61, 49.01, 43.47, 14.18; HRMS (ESI) calculated for C19H17N2O6 [M+H]+: 369.1081; found: 369.1062.
Methyl 1-(2-Nitrophenyl)-3-oxo-4-phenyl-2-oxa-6-azabicyclo[3.1.0]hexane-5- carboxylate (4o)
White solid, mp 189–191 °C. 202 mg, 57% yield. trans:cis > 20:1 (trans and cis relative chemistry was determined by 1D-NoE experiment).10 1H NMR (400 MHz, CDCl3): δ (ppm) 8.19 (d, J = 8.0 Hz, 1H), 7.75–7.38 (comp, 8H), 5.14 (s, 1H), 3.72 (s, 3H), 2.80 (bs, 1H); 13C NMR (100 MHz, CDCl3): 172.84, 167.39, 147.72, 134.53, 133.25, 132.24, 131.97, 129.69, 129.18, 128.71, 126.06, 124.79, 54.20, 50.78, 50.19; HRMS (ESI) calculated for C18H15N2O6 [M+H]+: 355.0925; found: 355.0905.
General Procedure for Rearrangement Reactions of TIPS-Protected Enoldiazoacetate 1 with Nitrile Oxides 2g
To an oven-dried flask containing a magnetic stirring bar, 4 Å molecular sieves (100 mg), Rh2(pfb)4 (2.0 mol %) and nitrile oxide 2g (1.0 mmol) in dichloromethane (5.0 mL), was added enoldiazoacetate 1 (1.2 mmol) in dichloromethane (3.0 mL) over 1 h via a syringe pump at 0 °C. After addition was complete, the reaction solution was stirred for another 2 h at room temperature, then DCM was removed under reduced pressure and the crude reaction mixture was subjected to NMR analysis. 100% Conversion to 7. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.23–7.78 (comp, 4H), 3.73 (s, 3H), 3.43 (d, J = 16.0 Hz, 1H), 2.97 (d, J = 16.0 Hz, 1H), 1.24 (m, 3H), 1.02 (m, 18H); 13C NMR (100 MHz, CDCl3): δ (ppm) 171.87, 170.80, 163.72, 147.98, 134.21, 133.97, 132.49, 125.50, 119.50, 53.19, 40.59, 38.72, 18.04, 18.03, 12.25; 29Si NMR (99 MHz, CDCl3): δ (ppm) 22.73.
Control Reaction of Enoldiazoacetate 1a with 4-Chlorobenzonitrile Oxide (2d) in the Absence of Catalyst

4-Chlorobenzaldehyde O-(tert-Butyldimethylsilyl) oxime (10)
To an oven-dried flask containing a magnetic stirring bar, 4 Å molecular sieves (100 mg), and nitrile oxide 2d (1.0 mmol) in dichloromethane (5.0 mL), was added diazo compound 1a (1.2 mmol) in dichloromethane (3.0 mL) over 1 h via a syringe pump at 0 °C. After addition was complete, the reaction solution was stirred overnight at room temperature. The crude product was purified by column chromatography on silica gel (eluent: hexanes: EtOAc = 30:1 to 10:1) to give the hydrolyzed diazo compound 9 and TBS protected oxime 10 (Colorless oil, 193 mg, 72% yield of 10). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.18 (s, 1H), 7.55 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 1.00 (s, 9H), 0.25 (s, 6H); 13C NMR (100 MHz, CDCl3): δ (ppm) 152.47, 136.00, 131.62, 129.32, 128.59, 26.49, 18.68; HRMS (ESI) calculated for C13H21ClNOSi [M+H]+: 270.1075; found: 270.1093.
Supplementary Material
ACKNOWLEDGMENT
The support of the National Institutes of Health (GM 46503) for this research is gratefully acknowledged.
Footnotes
Supporting information. Product characterization (1H and 13C spectra) and X-ray diffraction analysis data of 4g. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Doyle MP, McKervey MA, Ye T. Modern CatalyticMethods for Organic Synthesis with Diazo Compounds. John Wiley & Sins; New York: 1998. [Google Scholar]; (b) Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem. Rev. 2010;110:704. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]; (c) Davies HML, Morton D. Chem. Soc. Rev. 2011;1857;40 doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]; (d) Davies HML, Beckwith REJ. Chem. Rev. 2003;103:2861. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; (e) Padwa A, Weingarten MD. Chem. Rev. 1996;96:223. doi: 10.1021/cr950022h. [DOI] [PubMed] [Google Scholar]; For review:
- 2.(a) Selander JN, Fokin VV. J. Am. Chem. Soc. 2012;134:2477. doi: 10.1021/ja210180q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nadeau E, Ventura DL, Brekan JA, Davies HML. J. Org. Chem. 2010;75:1927. doi: 10.1021/jo902644f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Qian Y, Xu X, Jiang L, Prajapati D, Hu W. J. Org. Chem. 2010;75:7483. doi: 10.1021/jo101559p. [DOI] [PubMed] [Google Scholar]; (d) Cui X, Xu X, Lu H, Zhu S, Wojtas L, Zhang XP. J. Am. Chem. Soc. 2011;3304;133 doi: 10.1021/ja111334j. [DOI] [PubMed] [Google Scholar]; (e) Zhang Z, Liu Y, Ling L, Li Y, Dong Y, Gong M, Zhao X, Zhang Y, Wang J. J. Am. Chem. Soc. 2011;133:4330. doi: 10.1021/ja107351d. [DOI] [PubMed] [Google Scholar]; (f) Takeda K, Oohara T, Shimada N, Nambu H, Hashimoto S. Chem. Eur. J. 2011;17:13992. doi: 10.1002/chem.201102733. [DOI] [PubMed] [Google Scholar]; (g) Weathers TM, Jr., Wang Y, Doyle MP. J. Org. Chem. 2006;71:8183. doi: 10.1021/jo0614902. [DOI] [PMC free article] [PubMed] [Google Scholar]; Selected recent examples:
- 3.Jaber DM, Burgin RN, Hepler M, Zavalij P, Doyle MP. Chem. Commun. 2011;47:7623. doi: 10.1039/c1cc12443a. [DOI] [PubMed] [Google Scholar]
- 4.(a) Doyle MP, Ratnikov M, Liu Y. Org. Biomol. Chem. 2011;9:4007. doi: 10.1039/c0ob00698j. [DOI] [PubMed] [Google Scholar]; (b) Xu X, Hu W, Doyle MP. Angew. Chem. Int. Ed. 2011;50:6392. doi: 10.1002/anie.201102405. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Y, Bakshi K, Zavalij PY, Doyle MP. Org. Lett. 2010;12:4304. doi: 10.1021/ol101744h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xu X, Hu W, Zavalij PY, Doyle MP. Angew. Chem. Int. Ed. 2011;50:11152. doi: 10.1002/anie.201105557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(b) Qin C, Boyarskikh V, Hansen JH, Hardcastle KI, Musaev DG, Davies HML, Lian Y, Davies HML. J. Am. Chem. Soc. J. Am. Chem. Soc. 2011;2011;133133:11940. doi: 10.1021/ja2074104. [DOI] [PubMed] [Google Scholar]; (c) Lian Y, Hardcastle KI, Davies HML. Angew. Chem., Int. Ed. 2011;50:9370. doi: 10.1002/anie.201103568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Davies HML, Houser JH, Thornley C. J. Org. Chem. 1995;60:7529. [Google Scholar]; (b) Davies HML, Ahmed D, Churchill MR. J. Am. Chem. Soc. 1996;118:10774. [Google Scholar]
- 7.Wang X, Xu X, Zavalij PJ, Doyle MP. J. Am. Chem. Soc. 2011;133:16402. doi: 10.1021/ja207664r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu X, Ratnikov MO, Zavalij PY, Doyle MP. Org. Lett. 2011;13:6122. doi: 10.1021/ol2026125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu X, Shabashov D, Zavalij PY, Doyle MP. Org. Lett. 2012;14:800. doi: 10.1021/ol203331r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.See Supportting Information.
- 11.(a) Ueda S, Naruto S, Yoshida T, Sawayama T, Uno H. J. Chem. Soc. Chem. Comm. 1985;4:218. [Google Scholar]; (b) Qi X, Jiang Y, Park C. Chem.Commun. 2011;47:7848. doi: 10.1039/c1cc11683e. [DOI] [PubMed] [Google Scholar]; (c) Sakamoto S, Inokuma T, Takemoto Y. Org. Lett. 2011;13:6374. doi: 10.1021/ol2026747. [DOI] [PubMed] [Google Scholar]; (d) Ooi T, Takahashi M, Doda K, Maruoka K. J. Am. Chem. Soc. 2002;124:7640. doi: 10.1021/ja0118791. [DOI] [PubMed] [Google Scholar]
- 12.Pereira MMA, Santos PP. In: The Chemistry of Hydroxylamines, Oximes, and Hydroxamic Acids. Rappoport Z, Liebman JF, editors. Chapter 9. Wiley-Interscience; Chichester, England: 2009. [Google Scholar]
- 13.(a) Davies HML, Peng Z-Q, Houser JH. Tetrahedron Lett. 1994;35:8939. [Google Scholar]; (b) Ueda Y, Roberge G, Vinet V. Can. J. Chem. 1984;62:2936. [Google Scholar]; (c) Davies HML, Ahmed G, Churchill MR. J. Am. Chem. Soc. 1996;118:10774. [Google Scholar]; (d) Schwartz BD, Denton JR, Lian Y, Davies HML, Williams CM. J. Am. Chem. Soc. 2009;131:8329. doi: 10.1021/ja9019484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Liu KC, Shelton BR, Howe RK. J. Org. Chem. 1980;45:3916. [Google Scholar]; (b) Larsen KE, Torssel KBG. Tetrahedron. 1984;40:2985. [Google Scholar]; (c) Minakata S, Okumura S, Nagamachi T, Takeda Y. Org. Lett. 2011;13:2966. doi: 10.1021/ol2010616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.