

Abstract
Background
Tubulointerstitial fibrosis (fibrosis), a histological feature associated with the failing kidney allograft, is diagnosed using the invasive allograft biopsy procedure. A noninvasive diagnostic test for fibrosis may help improve allograft outcome.
Methods
We obtained 114 urine specimens from 114 renal allograft recipients; 48 from 48 recipients with fibrosis in biopsies and 66 from 66 recipients with normal biopsies. Levels of mRNAs in urinary cells were measured using kinetic, quantitative PCR assays and the levels were related to allograft diagnosis. A discovery set of 76 recipients (32 with allograft fibrosis and 44 with normal biopsies) was used to develop a diagnostic signature and an independent validation set of 38 recipients (16 with allograft fibrosis and 22 with normal biopsies) was used to validate the signature.
Results
In the discovery set, urinary cell levels of the following mRNAs were significantly associated with the presence of allograft fibrosis: vimentin (P<0.0001, logistic regression model), HGF (P<0.0001), α-SMA (P<0.0001), fibronectin 1 (P<0.0001), perforin (P=0.0002), PAI1 (P=0.0002), TGFβ1 (P=0.0004), TIMP1 (P=0.0009), granzyme B (P=0.0009), FSP1 (P=0.006), CD103 (P=0.02), and collagen 1A1 (P=0.04). A 4-gene model comprised of levels of mRNA for vimentin, NKCC2, E-cadherin and 18S rRNA provided the most accurate, parsimonious, diagnostic model of allograft fibrosis with 93.8% sensitivity and 84.1% specificity (P<0.0001). In the independent validation set, this same model predicted the presence of allograft fibrosis with 77.3% sensitivity and 87.5% specificity (P<0.0001).
Conclusions
Measurement of mRNAs in urinary cells may offer a noninvasive means of diagnosing fibrosis in human renal allografts.
INTRODUCTION
Renal allograft fibrosis is currently identified using the invasive allograft biopsy procedure in patients with worsening renal allograft function. However, many challenges exist including early diagnosis of allograft fibrosis (1) and neither serum creatinine nor estimated glomerular filtration rate appears to be an accurate indicator of fibrosis (2). Moreover, the biopsy procedure is costly, complications still occur, sampling errors may bias the diagnosis, and inter-observer variability in grading of biopsies remains a challenge (3–9).
We have reported a method using a quantitative polymerase chain reaction assay to measure mRNA levels of immune products within urinary cells of renal transplant recipients (10–12). In the current study, we investigated the feasibility of developing a noninvasive test for the diagnosis of human renal allograft fibrosis.
The pathogenesis of allograft fibrosis involves immune and non-immune pathways and multiple cell types (1,13–16). We reasoned that measurement in urinary cells of mRNA encoding proteins implicated in fibrogenesis and of mRNA for renal tubule epithelial cell specific proteins would be informative of fibrosis. Because inflammation may co-exist with fibrosis (1,17–20), we measured mRNAs for perforin and granzyme B, previously associated with acute rejection (10). In this report, we describe the discovery and validation of a 4-gene urinary cell mRNA signature for the noninvasive diagnosis of human renal allograft fibrosis.
RESULTS
Study Cohorts for the Discovery Set and Validation Set
We profiled 114 urine samples from 114 renal transplant recipients who had undergone either a clinically indicated renal allograft biopsy or a scheduled (protocol) biopsy. The biopsies were examined for the presence or absence of tubulointerstitial fibrosis as well as classified according to the Banff schema (21) by a pathologist (SVS) blinded to the mRNA results.
Prior to data analysis, the 114 urine samples were assigned, at a 2:1 ratio, to a Discovery set of 76 samples (32 samples from 32 recipients with renal allograft biopsies showing fibrosis and 44 samples from 44 recipients with normal biopsy results) and an independent Validation set of 38 samples (16 samples from 16 recipients with biopsies showing fibrosis and 22 samples from 22 recipients with normal biopsy results) (Figure 1). Neither the recipients’ characteristics nor the transplant or renal allograft related variables differed between those assigned to the Discovery set or the Validation set (Table 1). The risk factors for fibrosis such as acute rejection and deceased donor grafts however were more frequent in the fibrosis biopsy group compared to the normal biopsy group.
Figure 1. Flow chart for the discovery and validation of urinary cell mRNA profiles.

The 114 renal allograft recipients (48 with biopsies showing fibrosis and 66 with normal biopsy results) were rank ordered within group (Fibrosis group or Normal Biopsy group) by the copy number of 18S rRNA and partitioned into triplets. Within each triplet, the first and third patients were assigned to the Discovery set and the second patient was assigned to the Validation set, resulting in the two sets being exactly matched on fibrosis status and very closely matched on 18S rRNA copy number. Twice as many patients were assigned to the Discovery set in order to enhance statistical power for the exploratory analyses which included a procedure to protect against the risk of a Type I error.
Table 1.
Characteristics of the Renal Allograft Recipients
| Variable | Discovery Set (N=76) | Validation Set (N=38) | Pa Discovery Set vs. Validation Set | ||||||
|---|---|---|---|---|---|---|---|---|---|
| All Patients (N=76) | Fibrosis Biopsy Group (N=32) | Normal Biopsy Group (N=44) | Pa Fibrosis vs. Normal | All Patients (N=38) | Fibrosis Biopsy Group (N=16) | Normal Biopsy Group (N=22) | Pa Fibrosis vs. Normal | ||
| Recipient characteristics | |||||||||
| Age (mean±SD, years) | 46±13 | 46±14 | 46±12 | 0.88 | 44±10 | 44±9 | 44±12 | 0.80 | 0.40 |
| Gender (Male/female) | 37/39 | 17/15 | 20/24 | 0.50 | 21/17 | 8/8 | 13/9 | 0.57 | 0.50 |
| Ethnicity (White/Black/Other) | 26/19/31 | 12/7/13 | 14/12/18 | 0.82 | 13/11/14 | 4/9/3 | 9/2/11 | 0.006 | 0.88 |
| Cause of End Stage Renal Disease, N (%) | 0.18 | 0.29 | 0.20 | ||||||
| Glomerulonephritis | 18 (24) | 10 (31) | 8 (18) | 7 (18) | 4 (25) | 3 (14) | |||
| Diabetes | 21 (28) | 9 (28) | 12 (27) | 6 (16) | 1 (6) | 5 (23) | |||
| Cystic/hereditary/congenital | 11 (15) | 4 (13) | 7 (16) | 10 (26) | 4 (25) | 6 (27) | |||
| Secondary glomerulonephritis | 4 (5) | 1 (3) | 3 (7) | 4 (11) | 3 (19) | 1 (5) | |||
| Hypertension | 6 (8) | 1 (3) | 5 (11) | 6 (16) | 3 (19) | 3 (14) | |||
| Interstitial nephritis | 4 (5) | 3 (9) | 1 (2) | 0 (0) | 0 (0) | 0 (0) | |||
| Miscellaneous conditions | 2 (3) | 2 (6) | 0 (0) | 1 (3) | 1 (6) | 0 (0) | |||
| Neoplasm | 0 (0) | 0 (0) | 0 (0) | 1 (3) | 0 (0) | 1 (5) | |||
| Etiology uncertain | 10 (13) | 2 (6) | 8 (18) | 3 (8) | 0 (0) | 3 (14) | |||
| Peak pre-transplant HLA class I or II PRA (mean±SD, %)b | 17.6±23.7 | 18.8±25.3 | 17.0±23.0 | 0.77 | 26.4±28.3 | 34.8±33.1 | 20.3±23.1 | 0.12 | 0.09 |
| Peak pre-transplant HLA class I or II PRA ≥50%, N (%) | 6 (9) | 2 (9) | 4 (9 | 0.95 | 6 (16) | 4 (25) | 2 (9) | 0.18 | 0.29 |
| Transplant variables | |||||||||
| Deceased donor, N (%) | 26 (34) | 19 (59) | 7 (16) | <0.0001 | 18 (47) | 12 (75) | 6 (27) | 0.004 | 0.17 |
| Donor age (mean±SD, years) | 44±11 | 48±11 | 42±10 | 0.03 | 45±14 | 49±13 | 42±14 | 0.12 | 0.65 |
| HLA mismatches (mean±SD) | 3.5±2.0 | 3.8±2.2 | 3.4±1.8 | 0.41 | 3.3±1.8 | 4.6±1.4 | 2.4±1.5 | <0.0001 | 0.57 |
| Cold ischemia time (deceased donor grafts, mean±SD, hours) | 20.8±7.6 | 20.5±8.1 | 21.3±7.1 | 0.83 | 22.2±5.0 | 22.9±5.5 | 20.7±3.6 | 0.39 | 0.51 |
| Delayed graft functionc, (deceased donor grafts, N (%)) | 8 (39) | 6 (32) | 2 (29) | 0.88 | 6 (33) | 5 (42) | 1 (17%) | 0.28 | 0.85 |
| History of acute rejection before biopsy, N (%) | 9 (12) | 7 (22) | 2 (5) | 0.02 | 9 (24) | 8 (50) | 1 (5) | 0.001 | 0.10 |
| History of BK virus nephropathy before biopsy, N (%) | 2 (3) | 2 (6) | 0 (0) | 0.09 | 2 (5) | 2 (13) | 0 (0) | 0.09 | 0.47 |
| Graft failure within 12 months after biopsy, N (%) | 13 (17) | 13 (41) | 0 (0) | <0.0001 | 8 (21) | 7 (44) | 1 (5) | 0.003 | 0.61 |
| Renal allograft variables | |||||||||
| Time of biopsy (mean±SD, months since transplant) | 25.4±53.1 | 54.4±72.7 | 4.3±4.4 | <0.0001 | 14.1±23.3 | 29.2±29.9 | 3.0±3.8 | 0.0002 | 0.21 |
| Serum creatinine at biopsy (mean±SD, mg/dL) | 2.1±1.5 | 3.2±1.9 | 1.3±0.4 | <0.0001 | 2.3±1.5 | 3.3±1.8 | 1.5±0.4 | <0.0001 | 0.63 |
| eGFR at biopsy (mean±SD, mL/min/1.73m2) | 45.1±20.6 | 26.2±13.2 | 58.5±13.1 | <0.0001 | 42.8±19.5 | 29.1±19.0 | 52.7±12.9 | <0.0001 | 0.56 |
| Urinary protein:creatinine ratiod at biopsy | 1.4±2.9 | 3.6±4.3 | 0.3±0.3 | <0.0001 | 0.5±0.9 | 1.2±1.4 | 0.2±0.1 | 0.004 | 0.11 |
| Allograft fibrosis grade | |||||||||
| No fibrosis, N (%) | 44 (58) | 0 (0) | 44 (100) | 22 (58) | 0 (0) | 22 (100) | |||
| Grade I (<25% of cortical area), N (%) | 7 (9) | 7 (22) | 0 (0) | 2 (5) | 2 (13) | 0 (0) | |||
| Grade II (26–50% of cortical area), N (%) | 9 (12) | 9 (28) | 0 (0) | 7 (18) | 7 (44) | 0 (0) | |||
| Grade III (>50% of cortical area), N (%) | 16 (21) | 16 (50) | 0 (0) | 7 (18) | 7 (44) | 0 (0) | |||
| Mean±SD fibrosis grade | 1.0±1.2 | 2.3±0.8 | 0.0±0.0 | 1.0±1.2 | 2.3±0.7 | 0.0±0.0 | |||
| Banff Classification categoriese | |||||||||
| Normal | 39 (51) | 0 (0) | 39(89) | 19 (50) | 0 (0) | 19 (86) | |||
| Chronic active antibody-mediated rejection | 4 (5) | 4 (13) | 0 (0) | 2 (5) | 2 (13) | 0 (0) | |||
| Chronic active T-cell mediated rejection | 3 (4) | 3 (9) | 0 (0) | 3 (8) | 3 (19) | 0 (0) | |||
| IF/TA, no evidence of any specific etiology | 22 (29) | 22 (69) | 0 (0) | 8 (21) | 8 (50) | 0 (0) | |||
| Other | 8 (11) | 3 (9)f | 5 (11)g | 6 (16) | 3 (19)f | 3 (19)g | |||
P-values determined by Chi-square or Fisher’s exact tests for categorical variables or independent samples T-test for continuous variables.
Panel reactive antibodies (PRA) directed to the HLA class I or II antibodies were identified using the complement dependent cytotoxicity assay, and PRA value was available in 67 of 76 patients in the Discovery set (23 of 32 patients in the fibrosis biopsy group and 44 of 44 patients in the normal biopsy group) and 38 of 38 patients in the Validation set.
Defined by the need for hemodialysis in the first week post-transplantation
Urinary protein:creatinine ratio is the urinary protein concentration (mg/dL) divided by the urinary creatinine concentration (mg/dL) in a random urine specimen.
In addition to screening the allograft biopsies for the presence or absence of tubulointerstial fibrosis and grading the extent of fibrosis, presence or absence of inflammation, the allograft biopsies were also classified using the Banff 07 updated version of the Banff 97 diagnostic categories (21). All 6 biopsies classified as chronic antibody mediated rejection were positive for C4d deposition; cryostat or paraffin sections of the for-cause biopsies were examined for C4d deposition with the use of anti-human C4d antibody. All other biopsies in the fibrosis group were negative for C4d deposition.
6 biopsies in the Other diagnosis category; 3 in the Discovery set and 3 in the Validation set include diabetic nephropathy (N=4), and glomerulonephritis recurrence (N=2).
8 biopsies in the Other diagnosis category; 5 in the Discovery set and 3 in the Validation set includes vascular changes but no interstitial fibrosis.
Diagnostic Value of Individual mRNA Levels in the Discovery Set
We used our pre-amplification enhanced kinetic quantitative PCR assay (11) for the absolute quantification of mRNAs in the urine of renal allograft recipients. This assay enables measurement of a large number of mRNAs using a very small quantity of cDNA and the sequence and location of the gene specific oligonucleotide primers and TaqMan probes we designed for quantifying the mRNAs in the PCR assays are listed in supplemental digital content (SDC) Table 1.
We used LOESS (locally weighted scatterplot smoothing) methods in the discovery phase of the analysis to initially examine the bivariate relationship of each mRNA measure to diagnosis in the Discovery set comprised of 32 renal transplant recipients with biopsy-confirmed fibrosis and 44 recipients with normal allograft biopsy results, controlling for the quadratic relationship of 18S rRNA. Logistic regression analysis was then used to parsimoniously model each relationship as a piece-wise linear model.
Figure 2 illustrates that the levels of twelve of the twenty-two mRNAs measured are significantly associated with the diagnosis of fibrosis after using the Holm modified (22) Bonferoni procedure to control the risk of a Type I error. The lack of association between the remaining 10 mRNAs and allograft diagnosis is shown in SDC Figure 1.
Figure 2. Predicted probability of fibrosis as a function of urinary cell mRNA copy number in the Discovery set, for LOESS model and piece-wise linear logistic regression model, after controlling for 18S rRNA copy number.
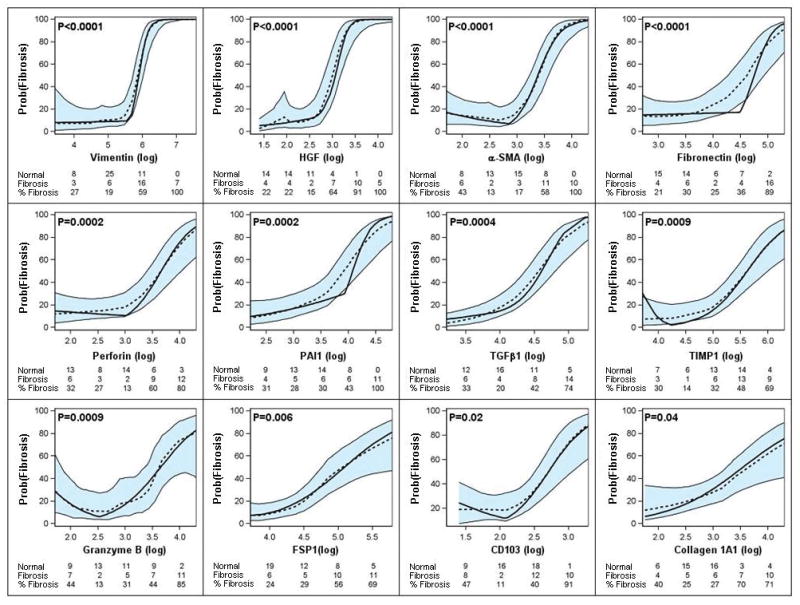
Urine samples were collected from 32 renal transplant recipients with graft dysfunction and biopsy-confirmed fibrosis and 44 recipients with stable allograft function and normal allograft biopsy, and levels of mRNA in urinary cells were measured with the use of pre-amplification enhanced kinetic quantitative PCR assays. Figure shows the predicted probability of fibrosis (Y-axis), controlling for 18S rRNA, as a function of individual log10-transformed mRNA copy numbers (X-axis). Each plot shows the LOESS model’s predicted probabilities (dotted line), their 95% confidence interval (shaded area) and the logistic regression model’s predicted probabilities (solid line). According to the logistic models, the levels of twelve of the twenty-two mRNAs (vimentin, HGF, α-SMA, fibronectin 1, perforin, PAI1, TGFβ1, TIMP1, granzyme B, FSP1, CD103, and collagen 1A1) were significantly (P-values <0.05 with modified Bonferroni correction) associated with the diagnosis of fibrosis. Adjusted P-value for each parametric model is shown. The number of stable patients, number of fibrosis patients, and percentage of fibrosis patients within categories of the mRNA measure appear in each plot.
Receiver-Operating-Characteristic (ROC) Curve Analysis
Analysis involving ROC curve demonstrated that allograft fibrosis can be predicted accurately using urinary cell levels of mRNA for vimentin (area under the curve [AUC] and 95% confidence intervals = 0.90, 0.82–0.97), HGF (0.91, 0.84–0.98), α-SMA (0.88, 0.80–0.95), fibronectin 1 (0.83, 0.73–0.93), perforin (0.83, 0.74–0.93), TGFβ1 (0.82, 0.72- 0.92), TIMP1 (0.81, 0.71–0.90), granzyme B (0.82, 0.71–0.92), FSP1 (0.81, 0.71–0.91), PAI1 (0.79, 0.68–0.90), collagen 1A1 (0.77, 0.66–0.88) or CD103 (0.76, 0.65–0.87).
Multigene Prediction Model of Fibrosis Diagnosis in the Discovery Set
We chose to build a multigene prediction model of fibrosis around vimentin in view of biologic properties of vimentin (23), and data from pre-clinical models that vimentin is over-expressed preceding and/or during fibrosis (24,25) and the clinical observation that vimentin expression in the 3-month protocol biopsies of renal allografts is associated with fibrosis score at 12 months (26). Accordingly, we once again estimated a LOESS model and corresponding piecewise linear model for the relationship of each mRNA measure to fibrosis, this time controlling for vimentin mRNA level and the quadratic relationship of 18S rRNA level. These analyses showed that after controlling for vimentin mRNA levels, the levels of other mRNAs (HGF, TGFβ1, fibronectin 1, PAI1, FSP1, collagen 1A1, α-SMA, CD103, granzyme B or perforin) that were initially significantly associated with fibrosis were no longer significant (P>0.05), whereas the mRNAs for NKCC2 and E-cadherin became significantly associated with the diagnosis (Figure 3). Based on these findings, a 4-gene diagnostic model that included vimentin, NKCC2, E-cadherin and 18S rRNA was developed. The parameter estimates for the model, provided in Figure 3, include terms accounting for the relationships, including non-linear relationships, between the mRNAs and diagnosis.
Figure 3. Final Model Derived from the Discovery Set for the Diagnosis of Fibrosis.

The predicted probability of fibrosis (Y-axis) as a function of individual log10-transformed mRNA copy numbers (X-axis) for vimentin (A), NKCC2 (B) and E-cadherin (C) after controlling for the copy numbers for the other two mRNAs and 18S rRNA is shown. Each plot shows the LOESS model’s predicted probabilities (dotted line), their 95% confidence interval (shaded area) and the logistic regression model’s predicted probabilities (solid line). The parameter estimates for the 4-gene model including terms accounting for the relationships, including non-linear relationships, between the mRNAs and diagnosis are provided in Panel D.
The composite score based on this model was highly associated with the diagnosis of fibrosis (Figure 4A). The ROC curve (Figure 4B) shows, for various levels of this composite score, the fraction of true positive results (sensitivity) and false positive results (1-specificity) for distinguishing recipients with allograft fibrosis from recipients with normal biopsy results. The AUC was 0.95 (95%CI: 0.90 to 0.99, P<0.0001), and a significant improvement (P<0.05) over the AUC for vimentin and 18S only. Using the optimal cut-point of 4.5 (the cut-point yielding the highest combined sensitivity and specificity), the composite score predicted fibrosis with a specificity of 84.1% (95%CI: 73.3 to 94.9%) and a sensitivity of 93.8% (95%CI: 85.4.0 to 99.9%) (Figure 4B).
Figure 4. Relationship of composite score to fibrosis in the Discovery set (A), ROC curve analysis of the composite score in the Discovery set (B) and the Validation set (C) and the predicted and observed number of transplant recipients with fibrosis for each sextile of the composite score within the Discovery and Validation sets (D).

To predict fibrosis in the Discovery set, a composite score was calculated based on a logistic model, from vimentin mRNA, NKCC2 mRNA and E cadherin mRNA as well as the 18S rRNA in urine samples obtained from the 32 subjects with biopsy-confirmed fibrosis and 44 subjects with stable graft function and normal allograft biopsy. The composite score predicted fibrosis with high accuracy. (A) Figure shows the predicted probability of fibrosis (Y-axis) as a logistic function of the composite score (X-axis). The blue band represents the 95% confidence interval of the model. (B) Figure shows the receiver-operating-characteristic curve for the diagnosis of fibrosis using the composite score. The model had an area under the curve of 0.95 (95%CI: 0.90 to 0.99, P<0.0001). At a cut-point or 4.5, fibrosis was diagnosed with a specificity of 84.1% (95%CI: 73.3 to 94.9%) and a sensitivity of 93.8% (95%CI: 85.4 to 99.9%).
The final prediction equation derived from the Discovery set was used to calculate the predicted probability of fibrosis in the Validation set of 38 kidney transplant recipients; 16 with biopsy-confirmed fibrosis and 22 with stable graft function and normal allograft biopsy. (C) Figure shows the receiver-operating characteristic curve of the composite score (applying the equation from Figure 3, Panel D to the urinary cell mRNA levels of vimentin, NKCC2 and E-cadherin and 18S rRNA level of those in the Validation set) for the diagnosis of fibrosis. The area under the curve for the diagnosis of fibrosis in the Validation set was 0.89 (95%CI: 0.78 to 0.99, P<0.0001). At the composite score cut-point of 4.5 derived from the Discovery set, fibrosis was diagnosed in the Validation set with a specificity of 77.3% (95%CI: 59.8 to 94.8%) and a sensitivity of 87.5% (95% CI: 71.3 to 99.9%). (D) Figure shows the predicted and observed number of transplant recipients with fibrosis for each sextile of the composite score within the Discovery and Validation sets.
Independent Validation of the Diagnostic Signature
The final diagnostic equation predicting fibrosis in the Discovery set was then validated in an independent Validation set of 38 renal transplant recipients consisting of 16 patients with biopsy-proven fibrosis and 22 recipients with normal allograft biopsy results (Table 1). Figure 4C shows the ROC curve of this equation based on urinary cell levels of vimentin, NKCC2 and E-cadherin mRNAs and 18S rRNA level for the diagnosis of fibrosis. This 4-gene classifier could diagnose fibrosis in the Validation set with high accuracy and the AUC for the diagnosis of fibrosis in the independent Validation set was 0.89 (95%CI: 0.78 to 0.99, P<0.0001) (Figure 4C). At the composite score cut-point of 4.5 (the same cut-point used in the Discovery set), fibrosis was diagnosed in the Validation set with a specificity of 77.3% (95%CI: 59.8 to 94.8%) and a sensitivity of 87.5% (95%CI: 71.3 to 99.9%)
We also examined the fit of the predictor model by dividing the Discovery and Validation sets into sextiles of the composite score and examined the predicted and observed number of transplant recipients with fibrosis, separately for each sets, for each sextile (Figure 4D). Based on the Hosmer-Lemeshow test, the fit between the observed and the predicted number of subjects with fibrosis in each of the sextiles was excellent (P=0.69) in the Discovery set (left half of Figure 4D). For the Validation set (right half of Figure 4D), the P-value was 0.04, suggesting a good fit, given that this set was not involved in the estimation of the model.
Serum creatinine levels were higher in the fibrosis group compared to the normal biopsy group (P<0.0001, Table 1). We assessed whether our composite score independently differentiates the fibrosis and stable patient groups after controlling for serum creatinine. Our analysis showed that the composite score is statistically significant and a slightly stronger predictor of group status (Fibrosis vs. Normal) than serum creatinine (each P<0.0001, controlling for the other).
We examined whether graft dysfunction, independent of fibrosis, was associated with the composite score. The log mean composite score of the 4-gene signature was 4.58 (95%CI: 3.52 to 5.64) in the acute tubular necrosis (ATN) group with graft dysfunction (N=9 patients) and 6.49 (95%CI: 5.96 to 7.02) in the fibrosis group with graft dysfunction (N=48 patients) (P=0.01). In addition, the composite score for the ATN group was not significantly different from that of normal biopsy group (N=66) with normal graft function (P=0.12).
We investigated whether the time to biopsy was associated with the diagnostic signature (composite score). This analyses showed that there was no significant association between the diagnostic signature and time to biopsy; Pearson correlation coefficient r=0.17, P=0.24 in the fibrosis biopsy group (N=48) and r=0.23, P=0.07 in the normal biopsy group (N=66).
Fibrosis Grades and the 4-Gene Composite Score
We investigated whether our 4-gene composite score could strongly discriminate patients with differing degrees of fibrosis from patients with no evidence of fibrosis. Our analysis revealed that the log mean composite score derived from urinary cell vimentin, NKCC2 and E-cadherin mRNA levels and 18S rRNA level was significantly different among the four groups (fibrosis grades I [<25% of cortical area], II [26–50%], and III [>50%] and those with no evidence of fibrosis, P<0.0001, one-way ANOVA) (SDC Figure 2). Pair-wise comparisons revealed that the mean composite score of normal biopsies were significantly different from that of grade I fibrosis (P=0.0002), grade II fibrosis (P<0.0001) and grade III fibrosis (P<0.0001). The mean composite score however did not differ significantly among the three grades of fibrosis (P=0.58).
Allograft Fibrosis with Concurrent Inflammation and the 4-Gene Composite Score
Among the 48 patients with allograft fibrosis, 32 biopsies from 32 patients showed no inflammation and 16 biopsies from 16 patients displayed both fibrosis and inflammation. The log mean composite score was 7.5 ± 2.3 in the 16 urine samples from patients with both fibrosis and inflammation and 5.9 ± 1.3 score in the 32 urine samples from patients with fibrosis only and without concurrent inflammation (P=0.003).
DISCUSSION
We have discovered and validated an mRNA signature for the noninvasive diagnosis of human renal allograft fibrosis. The area under the curve for the defined signature was 0.93 (95% CI: 0.88 to 0.97, P<0.0001) when all 114 samples (fibrosis biopsy group=48 and normal biopsy group, N=66) were included, and at the composite score cut-point of 4.5 (the same cut-point used in the Discovery and Validation sets), fibrosis was diagnosed with a specificity of 81.8% (95%CI: 72.5 to 91.1%) and a sensitivity of 91.7% (95%CI: 83.8% to 99.5%). These estimates however are somewhat upwardly biased (due to the cutpoint being selected to optimize sensitivity and specificity within the discovery set). An important attribute of the defined signature is that early fibrosis can be distinguished from biopsies without any fibrosis, and a weakness is that the diagnostic signature does not distinguish the different grades of fibrosis.
A number of features of our study may have contributed to our development of a noninvasive test for allograft fibrosis. First, we measured absolute levels of mRNA copy number using the standard curve method rather than relative levels of gene expression calculated using the delta-delta Ct method; the absolute quantification approach avoids some of the ambiguities inherent to the delta-delta Ct method of quantification of mRNA copy numbers since it is not always clear what should be used as the “control” for the “disease” studied. Second, we used a Discovery set to develop a prediction equation and identify the composite score cut-point, and then used the same equation and cut-point to validate the diagnostic accuracy of the urinary cell mRNA signature in an independent cohort of renal allograft recipients. Third, we gave consideration to the potential for non-linear relationships of gene expression measures to renal allograft diagnosis, and our approach for the discovery phase of the analysis used LOESS methods to examine the relationship of the mRNA measures to diagnosis (Fibrosis vs. Normal). The predicted probability plots, illustrated in Figures 2 and 3, capture well the threshold effects of mRNA copy number to the renal allograft diagnosis, and the lack of a simple linear relationship between mRNA abundance and allograft status; these issues would have been missed if the transcript levels were summarized as group means. Another contributor to our successful testing of the hypothesis that urinary cell mRNA profiles distinguish allografts with fibrosis from allografts without fibrosis is our use of urine samples from patients with protocol biopsies with normal biopsy findings and without fibrosis as the control group. Had we used urine samples from renal allograft recipients with acute rejection, calcineurin toxicity or BK virus nephropathy as the control group, the development of a robust biomarker that distinguishes biopsies with fibrosis from biopsies without fibrosis may have been compromised since each of these conditions may be associated with some degree of fibrosis.
Our use of protocol biopsies, performed primarily in the early post-transplantation period, as controls for the fibrosis biopsy group resulted in a significant difference (P<0.0001) in the time to biopsy between the fibrosis biopsy group and the normal biopsy group. This raised the interesting question whether the difference in the time to biopsy rather than allograft biopsy findings (fibrosis present vs. fibrosis absent) was responsible for the diagnostic signature. While this possibility cannot be conclusively excluded, the composite score being time dependent appears unlikely since there was no significant association (P>0.05) between the score and time to biopsy within the fibrosis or normal biopsy group.
In the 4-gene diagnostic signature defined in this study, vimentin had the strongest association with the allograft fibrosis diagnosis. Vimentin is a major intermediate filament protein expressed by mesenchymal cells. Ivaska et al. (23) have reviewed the dynamic nature of vimentin expression and the role of this evolutionarily conserved protein in cell adhesion, migration and signaling. Whereas healthy renal tubular cells are reported not to express vimentin protein, injured ones are decorated by vimentin. Vimentin expressing regenerating renal tubular cells have been reported by Nakatsuji et al. (24) and vimentin over-expression has also been reported in a folic acid-induced tubulointerstitial model (25). Hertig et al. reported that renal allograft recipients with greater than 10% of renal tubular cells expressing vimentin in their 3-month protocol biopsy have a higher tubulointerstitial fibrosis score in their 12-month biopsies.
Urinary cell levels of several other mRNAs such as TGFβ1 and HGF were also significantly higher in the urine from the fibrosis biopsy group compared to urine from the normal biopsy group. Their levels however were no longer significantly (P>0.05) associated with allograft fibrosis after controlling for vimentin mRNA levels, and did not contribute to the diagnostic accuracy of the composite score. Nevertheless, we discuss below their potential role(s) because of the biologic plausibility of their contributing to fibrosis/EMT (27,28) and/or their association with allograft fibrosis (29–31).
TGFβ1, a fibrogenic cytokine, may be responsible not only for the fibrosis but also for tubular cell atrophy that is a consistent “companion” of interstitial fibrosis. While there is an ongoing debate regarding whether the renal tubular epithelial cells indeed give rise to the interstitial fibroblasts/myofibroblasts (32, 33), the experimental findings of Koesters et al. (34) that in-vivo over-expression of TGFβ1 in renal tubules results in peritubular fibrosis, tubular dedifferentiation and decomposition by autophagy proffers an explanation for the invariable co-existence of tubular atrophy and interstitial fibrosis in native kidneys and renal allografts.
HGF can block TGFβ1-induced EMT, enhance matrix degradation in vitro and reverse fibrosis in animal models of chronic renal injury (35–38). In accordance with our results, HGF is over-expressed in vivo in studies of acute kidney injury (38, 39) and in most forms of chronic kidney diseases in animal models (40–42), and in the serum of patients with end-stage renal failure (43). HGF induction may serve as a protective, counter-regulatory mechanism since HGF blockade promotes tissue fibrosis and renal dysfunction (40, 41, 44). The heightened expression of HGF in patients with allograft fibrosis is reminiscent of our earlier findings that mRNA for immunosuppressive cytokine IL-10 (45) and mRNA for regulatory T cell specification factor FoxP3 (11) are present at high levels during an episode of acute rejection.
Emerging data suggest that the renal allografts with fibrosis and concurrent inflammation fare less well compared to grafts with fibrosis and without inflammation (20). Our findings that the urinary cell 4-gene composite score is significantly higher in those with biopsies showing both fibrosis and inflammation compared to those with biopsies showing fibrosis without concurrent inflammation suggest that the score may also be useful to distinguish those with fibrosis and concurrent inflammation from those with fibrosis alone.
A weakness inherent to our cross-sectional study design is that the temporal relationship between the urinary cell mRNA signature (the composite score of the 4-gene signature) and histological detection of allograft fibrosis cannot be resolved. Whether the urinary cell mRNA signature is of diagnostic value only (that is the composite score threshold is reached only when the allograft shows fibrosis) or whether it is also anticipatory of allograft fibrosis (that is the composite score threshold is reached days or weeks prior to biopsies showing allograft fibrosis) cannot be addressed with our study design. It is also possible that an elevated composite score is an intrinsic feature of patients who will eventually develop fibrosis; that is, the patients who over-express mRNAs such as vimentin due to genomic and/or non-genomic reasons are at increased risk for developing fibrosis. We speculate that the diagnostic signature defined in this study may serve also as an anticipatory biomarker and this speculation is based on the recent findings from the CTOT-04 Trial that urinary cell mRNA profiles of longitudinally collected urine specimens predict acute rejection days to weeks prior to biopsy diagnosis (46). This hypothesis however needs to be tested using a longitudinal study design.
METHODS
Study cohorts
We examined 114 urine samples from 114 kidney transplant recipients who had undergone either a diagnostic (for-cause) renal allograft biopsy or a scheduled (protocol) biopsy. The biopsies were examined for the presence or absence of tubulointerstitial fibrosis, inflammation as well as classified according to the Banff schema (21) by a pathologist (SVS) blinded to the mRNA results. The institutional review board at the Weill Cornell Medical College in New York approved the study, and each patient gave written informed consent (see SDC for additional information).
Quantitation of mRNAs
Urine was centrifuged at 2,000g for 30 minutes within 4 hours of collection. RNA was extracted from the pellet using the RNeasy mini kit (Qiagen) and reverse-transcribed to cDNA using TaqMan® Reverse Transcription Reagents (Applied Biosystems). We designed oligonucleotide primers and fluorogenic probes for the measurement of levels of mRNAs (SDC Table 1). PCR analysis involved a preamplification step followed by quantification of mRNA with an ABI Prism 7500 Fast detection system. Transcript levels were calculated by a standard curve method (47). (See SDC for additional information).
Statistical analysis
The 114 patients (48 recipients with allograft fibrosis and 66 recipients with normal biopsies) were rank ordered within group by the copy number of 18S rRNA and partitioned into consecutive triplets. Within each triplet, the first and third patients were assigned to the Discovery set and the second patient was assigned to the Validation set, resulting in the two sets being exactly matched on fibrosis status and very closely matched on 18S. Twice as many patients were assigned to the Discovery set in order to enhance statistical power for the exploratory analyses which included a procedure to protect against the risk of a Type I error.
The distribution of each mRNA, as well as 18S rRNA, exhibited considerable positive skewness, which was substantially reduced by use of a log transformation. LOESS methods were used to examine the relationship of the mRNA measures to diagnosis (Fibrosis vs. Normal). An initial LOESS model revealed a U-shaped relationship of 18S to diagnosis that was well represented by a quadratic function. We then used a GAM (generalized additive model) (48, 49) procedure to fit an additive LOESS model of the relationship of each individual mRNA measure with diagnosis while statistically controlling for the quadratic effect of 18S. The smoothing parameter for the LOESS model was determined using the generalized cross validation criterion, but restricted to DF<5). After reviewing the smoothed relationship, we next fit a piece-wise linear logistic regression spline model that closely approximated the LOESS-smoothed relationship. We present plots where the parametric model of the relationship of mRNA level to the probability of being in the Fibrosis group is superimposed on the LOESS model. We also present the AUC and its 95% confidence interval for each logistic model. Significance levels of the 22 parametric models were adjusted for the experiment-wise risk of a Type I error using Holm’s modified (27) Bonferroni method. Based on the results, we chose one mRNA to be definitely included in the final model, and then repeated the above process for the remaining 21 mRNA measures to determine which if any could further improve the prediction of fibrosis diagnosis. This stepwise process was repeated until, after 3 steps, no further mRNA measures significantly improved the prediction model. The ROC curve for the final model and its AUC are presented.
In the Validation phase, the final prediction equation from the Discovery phase was used to calculate composite scores for those in the Validation set. A logistic regression analysis predicting fibrosis diagnosis from this single composite score was estimated to test the significance of the prediction equation. The ROC curve for the prediction equation and its AUC for the Validation set are presented. Finally, the Discovery and Validation sets were each divided into sextiles and an exact test version of the Hosmer-Lemeshow test (50) was used to assess the fit of the equation in both the Discovery and Validation sets.
All analyses were performed using SAS, version 9.2 (Cary, NC).
Supplementary Material
Acknowledgments
Supported in part by an NIAID MERIT Award (2R37-AI051652) from the National Institutes of Health and an award (NPRP 08-503-3-11) from the Qatar National Research Foundation to M. Suthanthiran; by a Clinical and Translational Science Center award (ULI RR 024996) to Weill Cornell Medical College and a Mentored Career Development Award (K08- DK087824) to T. Muthukumar from the National Institutes of Health.
Footnotes
Author Contributions
Dany Anglicheau
Participated in the performance of the research, data analysis, in the writing of the paper
No conflict of interest.
Thangamani Muthukumar
Participated in the performance of the research, data analysis, in the writing of the paper
No conflict of interest.
Aurélie Hummel
Participated in the performance of the research.
No conflict of interest.
Ruchuang Ding
Participated in the performance of the research and contributed new reagents.
No conflict of interest.
Vijay K Sharma
Participated in the performance of the research.
No conflict of interest.
Darshana Dadhania
Participated in the performance of the research.
No conflict of interest.
Surya Seshan
Participated in the performance of the research, data analysis, and in the writing of the paper.
No conflict of interest.
Joseph E. Schwartz
Participated in research design, data analysis, and in the writing of the paper.
No conflict of interest.
Manikkam Suthanthiran
Participated in research design, data analysis, and in the writing of the paper.
No conflict of interest.
References
- 1.Arias M, Serón D, Moreso F, Bestard O, Praga M. Renal allograft damage: existing challenges. Transplantation. 2011;91:4. doi: 10.1097/TP.0b013e31821792fd. [DOI] [PubMed] [Google Scholar]
- 2.Yilmaz S, Isik I, Afrouzian M, et al. Evaluating the accuracy of functional biomarkers for detecting histological changes in chronic allograft nephropathy. Transpl Int. 2007;20:608. doi: 10.1111/j.1432-2277.2007.00494.x. [DOI] [PubMed] [Google Scholar]
- 3.Huraib S, Goldberg H, Katz A, et al. Percutaneous needle biopsy of the transplanted kidney: technique and complications. Am J Kidney Dis. 1989;14:13. doi: 10.1016/s0272-6386(89)80087-3. [DOI] [PubMed] [Google Scholar]
- 4.Beckingham IJ, Nicholson ML, Bell PR. Analysis of factors associated with complications following renal transplant needle core biopsy. Br J Urol. 1994;73:13. doi: 10.1111/j.1464-410x.1994.tb07449.x. [DOI] [PubMed] [Google Scholar]
- 5.Benfield MR, Herrin J, Feld L, Rose S, Stablein D, Tejani A. Safety of kidney biopsy in pediatric transplantation: a report of the Controlled Clinical Trials in Pediatric Transplantation Trial of Induction Therapy Study Group. Transplantation. 1999;67:544. doi: 10.1097/00007890-199902270-00010. [DOI] [PubMed] [Google Scholar]
- 6.Sorof JM, Vartanian RK, Olson JL, Tomlanovich SJ, Vincenti FG, Amend WJC. Histopathological concordance of paired renal allograft biopsy cores: effect on the diagnosis and management of acute rejection. Transplantation. 1995;60:1215. [PubMed] [Google Scholar]
- 7.Colvin RB, Cohen AH, Saiontz C, et al. Evaluation of pathologic criteria for acute renal allograft rejection: reproducibility, sensitivity, and clinical correlation. J Am Soc Nephrol. 1997;8:1930. doi: 10.1681/ASN.V8121930. [DOI] [PubMed] [Google Scholar]
- 8.Nicholson ML, Wheatley TJ, Doughman TM, et al. A prospective randomized trial of three different sizes of core-cutting needle for renal transplant biopsy. Kidney Int. 2000;58:390. doi: 10.1046/j.1523-1755.2000.00177.x. [DOI] [PubMed] [Google Scholar]
- 9.Joh K, Morozumi K, Kitamura H. Symposium: evaluating the reproducibility of pathological diagnosis using the 1997 Banff classification update. Clin Transplant. 2006;20 (Suppl 15):53. doi: 10.1111/j.1399-0012.2006.00551.x. [DOI] [PubMed] [Google Scholar]
- 10.Li B, Hartono C, Ding R, et al. Noninvasive diagnosis of renal-allograft rejection by measurement of messenger RNA for perforin and granzyme B in urine. N Engl J Med. 2001;344:947. doi: 10.1056/NEJM200103293441301. [DOI] [PubMed] [Google Scholar]
- 11.Muthukumar T, Dadhania D, Ding R, et al. Messenger RNA for FOXP3 in the urine of renal-allograft recipients. N Engl J Med. 2005;353:2342. doi: 10.1056/NEJMoa051907. [DOI] [PubMed] [Google Scholar]
- 12.Anglicheau D, Suthanthiran M. Noninvasive prediction of organ graft rejection and outcome using gene expression patterns. Transplantation. 2008;86:192. doi: 10.1097/TP.0b013e31817eef7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maluf DG, Mas VR, Archer KJ, et al. Molecular pathways involved in loss of kidney graft function with tubular atrophy and interstitial fibrosis. Mol Med. 2008;14:276. doi: 10.2119/2007-00111.Maluf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strutz F. Pathogenesis of tubulointerstitial fibrosis in chronic allograft dysfunction. Clin Transplant. 2009;23 (Suppl 21):26. doi: 10.1111/j.1399-0012.2009.01106.x. [DOI] [PubMed] [Google Scholar]
- 15.Boor P, Ostendorf T, Floege J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol. 2010;6:643. doi: 10.1038/nrneph.2010.120. [DOI] [PubMed] [Google Scholar]
- 16.Zeisberg M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21:1819. doi: 10.1681/ASN.2010080793. [DOI] [PubMed] [Google Scholar]
- 17.Park W, Griffin M, Grande JP, Cosio F, Stegall MD. Molecular evidence of injury and inflammation in normal and fibrotic renal allografts one year posttransplant. Transplantation. 2007;83:1466. doi: 10.1097/01.tp.0000265501.33362.d3. [DOI] [PubMed] [Google Scholar]
- 18.Scherer A, Gwinner W, Mengel M, et al. Transcriptome changes in renal allograft protocol biopsies at 3 months precede the onset of interstitial fibrosis/tubular atrophy (IF/TA) at 6 months. Nephrol Dial Transplant. 2009;24:2567. doi: 10.1093/ndt/gfp183. [DOI] [PubMed] [Google Scholar]
- 19.Seron D, Moreso F. Protocol biopsies in renal transplantation: prognostic value of structural monitoring. Kidney Int. 2007;72:690. doi: 10.1038/sj.ki.5002396. [DOI] [PubMed] [Google Scholar]
- 20.Park WD, Griffin MD, Cornell LD, Cosio FG, Stegall MD. Fibrosis with inflammation at one year predicts transplant functional decline. J Am Soc Nephrol. 2010;21:1987. doi: 10.1681/ASN.2010010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solez K, Colvin RB, Racusen LC, et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant. 2008;8:753. doi: 10.1111/j.1600-6143.2008.02159.x. [DOI] [PubMed] [Google Scholar]
- 22.Holm S. A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics. 1979;6:65. [Google Scholar]
- 23.Ivaska J, Pallari HM, Nevo J, Eriksson JE. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp Cell Res. 2007;313:2050. doi: 10.1016/j.yexcr.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 24.Nakatsuji S, Yamate J, Sakuma S. Relationship between vimentin expressing renal tubules and interstitial fibrosis in chronic progressive nephropathy in aged rats. Virchows Arch. 1998;433:359. doi: 10.1007/s004280050260. [DOI] [PubMed] [Google Scholar]
- 25.Bielesz B, Sirin Y, Si H, et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest. 2010;120:4040. doi: 10.1172/JCI43025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hertig A, Anglicheau D, Verine J, et al. Early epithelial phenotypic changes predict graft fibrosis. J Am Soc Nephrol. 2008;19:1584. doi: 10.1681/ASN.2007101160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeisberg M, Soubasakos MA, Kalluri R. Animal models of renal fibrosis. Methods Mol Med. 2005;117:261. doi: 10.1385/1-59259-940-0:261. [DOI] [PubMed] [Google Scholar]
- 28.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma VK, Bologa RM, Xu GP, et al. Intragraft TGF-beta 1 mRNA: a correlate of interstitial fibrosis and chronic allograft nephropathy. Kidney Int. 1996;49:1297. doi: 10.1038/ki.1996.185. [DOI] [PubMed] [Google Scholar]
- 30.Hotchkiss H, Chu TT, Hancock WW, et al. Differential expression of profibrotic and growth factors in chronic allograft nephropathy. Transplantation. 2006;81:342. doi: 10.1097/01.tp.0000195773.24217.95. [DOI] [PubMed] [Google Scholar]
- 31.Mas V, Maluf D, Archer K, et al. Establishing the molecular pathways involved in chronic allograft nephropathy for testing new noninvasive diagnostic markers. Transplantation. 2007;83:448. doi: 10.1097/01.tp.0000251373.17997.9a. [DOI] [PubMed] [Google Scholar]
- 32.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? J Clin Invest. 2011;121:468. doi: 10.1172/JCI44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koesters R, Kaissling B, Lehir M, et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 2010;177:632. doi: 10.2353/ajpath.2010.091012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J, Liu Y. Blockage of tubular epithelial to myofibroblast transition by hepatocyte growth factor prevents renal interstitial fibrosis. J Am Soc Nephrol. 2002;13:96. doi: 10.1681/ASN.V13196. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Dai C, Liu Y. Hepatocyte growth factor gene therapy and angiotensin II blockade synergistically attenuate renal interstitial fibrosis in mice. J Am Soc Nephrol. 2002;13:2464. doi: 10.1097/01.asn.0000031827.16102.c1. [DOI] [PubMed] [Google Scholar]
- 37.Yang J, Dai C, Liu Y. A novel mechanism by which hepatocyte growth factor blocks tubular epithelial to mesenchymal transition. J Am Soc Nephrol. 2005;16:68. doi: 10.1681/ASN.2003090795. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Tolbert EM, Lin L, et al. Up-regulation of hepatocyte growth factor receptor: an amplification and targeting mechanism for hepatocyte growth factor action in acute renal failure. Kidney Int. 1999;55:442. doi: 10.1046/j.1523-1755.1999.00267.x. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y. Hepatocyte growth factor and the kidney. Curr Opin Nephrol Hypertens. 2002;11:23. doi: 10.1097/00041552-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Rajur K, Tolbert E, Dworkin LD. Endogenous hepatocyte growth factor ameliorates chronic renal injury by activating matrix degradation pathways. Kidney Int. 2000;58:2028. doi: 10.1111/j.1523-1755.2000.00375.x. [DOI] [PubMed] [Google Scholar]
- 41.Mizuno S, Matsumoto K, Kurosawa T, Mizuno-Horikawa Y, Nakamura T. Reciprocal balance of hepatocyte growth factor and transforming growth factor-beta 1 in renal fibrosis in mice. Kidney Int. 2000;57:937. doi: 10.1038/sj.ki.4491416. [DOI] [PubMed] [Google Scholar]
- 42.Mizuno S, Matsumoto K, Nakamura T. Hepatocyte growth factor suppresses interstitial fibrosis in a mouse model of obstructive nephropathy. Kidney Int. 2001;59:1304. doi: 10.1046/j.1523-1755.2001.0590041304.x. [DOI] [PubMed] [Google Scholar]
- 43.Lohr JW, Lee TP, Farooqui M, Mookerjee BK. Increased levels of serum hepatocyte growth factor in patients with end-stage renal disease. J Med. 2000;31:131. [PubMed] [Google Scholar]
- 44.Mizuno S, Matsumoto K, Nakamura T. HGF as a renotrophic and anti-fibrotic regulator in chronic renal disease. Front Biosci. 2008;13:7072. doi: 10.2741/3211. [DOI] [PubMed] [Google Scholar]
- 45.Xu GP, Sharma VK, Li B, et al. Intragraft expression of IL-10 messenger RNA: A novel correlate of renal allograft rejection. Kidney Int. 1995;48:1504. doi: 10.1038/ki.1995.440. [DOI] [PubMed] [Google Scholar]
- 46.Suthanthiran M, Ding R, Sharma V, et al. Urinary Cell Messenger RNA Expression Signatures Anticipate Acute Cellular Rejection: A Report from CTOT-04. Am J Transplant. 2011;11 (Suppl 2):29. [Google Scholar]
- 47.Anglicheau D, Sharma VK, Ding R, et al. MicroRNA expression profiles predictive of human renal allograft status. Proc Natl Acad Sci U S A. 2009;106:5330. doi: 10.1073/pnas.0813121106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hastie TJ, Tibshirani RJ. Generalized additive models (with discussion) Statistical Science. 1986;1:297. [Google Scholar]
- 49.Hastie TJ, Tibshirani RJ, editors. Generalized Additive Models. New York: Chapman & Hall; 1990. [Google Scholar]
- 50.Hosmer DW, Jr, Lemeshow S. Applied Logistic Regression. New York: John Wiley & Sons; 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.