

Abstract
Purpose of review
Highly active antiretroviral therapy (HAART) has led to a dramatic improvement in the prognosis of patients diagnosed with HIV and AIDS. This includes a significant decline in the rates of AIDS-related cancers, including Kaposi Sarcoma and Non-Hodgkin's Lymphoma. Unfortunately, rates of Non-AIDS Defining Cancers (NADCs) are on the rise, and now exceed the rates of AIDS-related cancers in patients with HIV. Treating NADCs in patients who are on HAART therapy is an open and complicated clinical question.
Recent findings
Newer targeted therapies are now available to treat cancers which were historically refractory to traditional cytotoxic chemotherapy. HAART agents are notorious for causing drug-drug interactions. The co-administration of targeted chemotherapies with HAART could well impede the efficacy or increase the toxicity of these targeted therapies. Unfortunately little is known about possible drug-drug interactions because HIV patients are typically excluded from clinical trials.
Summary
We highlight what is known about how and why HAART agents can affect drug metabolism. We then present the clinical and pharmacological data for nine recently approved targeted therapies – imatinib, dasatinib, nilotinib, erlotinib, sunitinib, lapatinib, bortezomib, sorafenib, and temsirolimus. We conclude with considerations on how to use these new agents to treat NADCs, and discuss a future research agenda to better understand and predict potential HAART-targeted therapy interactions.
Keywords: Non-AIDS Defining Cancers, Targeted Chemotherapy, HAART, Drug-Drug Interactions
INTRODUCTION
The introduction of highly active antiretroviral therapy (HAART) to treat infections caused by the Human Immunodeficiency Virus (HIV) has greatly improved the morbidity and mortality of infection patients. With the introduction of HAART, the rates of AIDS-related cancers, including Kaposi's Sarcoma and Non-Hodgkin's Lymphoma, have declined significantly. Unfortunately and for reasons that remain poorly understood, rates of Non-AIDS Defining Cancers (NADCs) are on the rise.[1-4] In fact, cases of NADCs now exceed the rates of AIDS-related cancers in patients with HIV.[5] These NADCs include cancers of the head and neck, lung, kidney, liver, gastrointestinal tract, and anus as well as Hodgkin's Lymphoma. How to treat patients on HAART once a non-AIDS cancer diagnosis is made remains poorly understood, and is complicated by the potential of drug-drug interactions between chemotherapy and HAART agents.
Great strides have been made over the past decade in developing newer agents to treat cancers which have been refractory to traditional cytotoxic chemotherapy in the past. Just over the past several years, more than a dozen new targeted therapies have garnered FDA-approval to treat diseases ranging from chronic myeloid leukemia to renal cell and hepatocellular carcinomas.
According to the National Cancer Institute, ‘targeted’ anticancer therapies are those that use drugs, small molecules, or other substances, such as monoclonal antibodies, to block the growth and spread of cancer by interfering with specific molecules involved in carcinogenesis and tumor growth.[6] While targeted agents represent a new way of treating cancers compared to the use of traditional cytotoxic agents, they nonetheless are pharmaceutical compounds that must be administered, absorbed, metabolized, and excreted in vivo. HAART agents, which are notorious for interfering with the enzymes involved in drug metabolism, may in turn interfere with the pharmacology of targeted therapies just as they can with traditional cytotoxic therapies.
In this review we will highlight what is known about how HAART agents can affect drug metabolism. Then we will present the clinical and pharmacological data for nine recently approved targeted therapies – imatinib, dasatinib, nilotinib, erlotinib, lapatinib, bortezomib, sorafenib, and temsirolimus. We will conclude with considerations on how to use these new agents in patients with HIV who are on HAART therapy, as well as discuss a future research agenda to better understand, study, and predict potential HAART-targeted therapy interactions in HIV positive patients.
CHEMOTHERAPY / HAART DRUG-DRUG INTERACTIONS
Enzymes and transporters mediate the absorption, distribution, metabolism, and excretion (ADME) of endogenous as well as exogenous metabolic and catabolic end products. Metabolic enzymes are present in the liver as well as in other peripheral tissues. Enzyme-mediated metabolic reactions can either activate prodrugs or inactivate active drugs. Enzymes can catalyze the oxidation, reduction or hydrolysis of compounds in so-called phase I metabolic reactions. The Cytochrome P450 family of enzymes is the largest source of enzymes involved in phase I metabolic reactions, especially CYP3A4. Phase II or conjugation reactions, in which a substrate is added to a drug to render it more readily eliminated in the bile or urine, can alter substrates via acetylation, glucuronidation, sulfation and methylation. Examples of phase II enzymes include UDP-glucuronosyltransferases (UGTs) and glutathione-S-transferases (GSTs).[7]
It is estimated that half of all pharmaceutical agents undergo metabolism either completely or in part by CYP3A4. A key consideration for physicians is whether patients are on drugs that may interfere with this or other enzymes, and in turn affect the pharmacology of other co-administered drugs. Drugs known to induce CYP3A4 function include dexamethasone, phenytoin, carbamazepine, rifampin, rifabutin, rifapentin, phenobarbital, and St. John's Wort. Drugs known to inhibit CYP3A4 include ketoconazole, itraconazole, voriconazole, clarithromycin, telithromycin, and nefazodone.[8]
HAART therapy typically includes a three to four drug combinations of protease inhibitors (PIs), nucleotide reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), and additional agents.[9] A significant amount of research over the past decade has investigated how these HAART drugs are metabolized, and crucial discoveries have been made in how these agents both induce and inhibit various enzymes involved in drug metabolism.[10] For example, to varying degrees all PIs inhibit CYP3A4, an enzyme that mediates the metabolism of over half of all drugs that undergo hepatic metabolism, including chemotherapy agents. This fact has been used to increase the drug delivery of other HAART agents. For example, saquinavir has been combined with low dose ritonavir (100 mg) because ritonavir's inhibition of CYP3A4 increases the systemic exposure to saquinavir. Delaviridine and efavirenz inhibit CYP3A4, with the latter also inhibiting CYP2B6 and CYP2C9/19. Nelfinavir and ritonavir inhibit CYP2B6 as well as CYP3A4, while amprenavir inhibits CYP2C19.
Other HAART drugs are known to induce increased expression of CYP450 enzymes. Enzyme induction can increase a patient's ability to eliminate other drugs which undergo similar metabolism. For example, nevirapine, efavirenz, ritonavir, and tipranavir induce CYP3A4 expression. Nevirapine also induces expression of CYP2B6. Ritonavir and nelfinavir may also induce expression of glucuronyltransferases.
Table 1 summarizes the effects that each of the commonly used HAART drugs can have on phase I and phase II metabolizing enzymes.[11-57]
Table 1.
Enzymes and HAART Agents.
| Drugs | Class | Substrate | Inhibit | Induce | References |
|---|---|---|---|---|---|
| Abacavir (Ziagen) | NRTI | ALDH | 11,12 | ||
| Amprenavir (Agenerase) | PI | CYP3A4 | CYP3A4 | 13-15 | |
| Atazanavir (Reyataz) | PI | CYP3A4 | CYP3A4, UGT1A1, CYP2C8 | 16-19 | |
| Delavirdine (Rescriptor) | NNRTI | CYP3A4 CYP2D6 |
CYP3A4 CYP2C9 CYP2C19 CYP2D6 |
20 | |
| Didanosine (Videx) | NRTI | 21,22 | |||
| Efavirenz (Sustiva) | NNRTI | CYP2B6 | CYP2B6 | CYP3A4 | 23-26 |
| Indinavir (Crixivan) | PI | CYP3A4 | CYP3A4 UGT1A1 |
15,17, 27-29 | |
| Lamivudine (Epivir) | NRTI | 30,31 | |||
| Lopinavir | PI | CYP3A4 | 32-34 | ||
| Maraviroc (Selzentry) | C5I | CYP3A4 | CYP2D6 | 35,36 | |
| Nelfinavir (Viracept) | PI | CYP3A4, CYP2C19 | CYP3A4 CYP2B6 |
15,37,38 | |
| Nevirapine (Viramune) | NNRTI | CYP3A4 CYP2B6 |
CYP3A4 CYP2B6 |
39,40 | |
| Raltegravir (Isentress) | IGI | UGT1A1 | 41-43 | ||
| Ritonavir (Norvir) | PI | CYP3A4 CYP2D6 |
CYP3A4 CYP2D6 CYP2B6 |
CYP2C9 | 28,44,45 |
| Saquinavir (Invirase) | PI | CYP3A4 | CYP3A4 | 15, 46-48 | |
| Stavudine (Zerit) | NRTI | 49 | |||
| Tenofovir (Viread) | NtRTI | CYP3A4 | 50,51 | ||
| Tipranavir (Aptivus) | PI | CYP3A4 | CYP1A2 CYP2C9, CYP2C19, CYP2D6 |
52,53 | |
| Zalcitabine (ddC) | NRTI | 54 | |||
| Zidovudine (AZT) | NRTI | UGTB7 | 55-57 |
Enzymes that are known to metabolize commonly used HAART agents are listed, as well as enzymes that are inhibited or induced by each agent. If a HAART drug inhibits a metabolizing enzyme, then blood levels of other drugs that are substrates for that same enzyme may be increased. Likewise, if a HAART agent inducing the expression or function of an enzyme, than drugs that are substrates for that same enzyme will be more rapidly metabolized and eliminated.
Abbreviations : NNRTI - non-nucleoside reverse transcriptase inhibitor; NRTI - nucloeside reverse transcriptase inhibitor; PI - protease inhibitor; IGI - integrase inhibitor; C5I - CCR5 nhibitor; ALDH - Alcohol dehydrogenase.
TARGETED ANTI-CANCER AGENTS
While only recently approved, the following targeted therapies have undergone extensive pre-clinical and clinical investigations, including pharmacokinetic analyses. This research has led to a reasonable understanding of their anti-cancer mechanisms of action as well as their pharmacology, both in terms of pharmacokinetics and pharmacodynamics. A brief presentation of each targeted therapy follows.
Imatinib (Gleevec, Novartis)
Imatinib is an orally available tyrosine kinase inhibitor approved for the use in treating chronic myeloid leukemia, acute lymphoblastic leukemia, mild dysplastic diseases and gastrointestinal stromal tumors. [58-61] Imatinib inhibits the bcr-abl tyrosine kinase which is constitutively active in chronic myeloid leukemia in patients who contain the Philadelphia chromosome abnormality. The drug is also an inhibitor of the receptor tyrosine kinases for platelet-derived growth factor (PDGF), stem cell factor (SCF) and c-kit.[61]
The major enzyme involved in the metabolism of imatinib is CYP3A4.[62] Other enzymes involved in the drug metabolism and which play a minor role include CYP1A2, CYP2D6, CYP2C9 and CYP2C19.[61,62] Its main active metabolite is formed by CYP3A4. Imatinib has also been found to be a potent inhibitor of CYP2C9, CYP2D6 and CYP3A4/5.[62]
The FDA-approved Package Insert recommends dose reduction by 50% if patients are on an inhibitor of CYP3A4. It also recommends the consideration of dose adjustment if patients are on a drug known to induce the activity of CYP3A4. Finally, if patients are on drugs known to be metabolized by CYP2C9, CYP2D6 and CYP3A4, caution is recommended given imatinib's inhibition of these crucial enzymes. In healthy subjects receiving imatinib and the CYP3A4 inhibiting drug ketoconazole, blood levels of imatinib (AUC) increased by 40%.[63] Further, serum AUC of the CYP3A4 substrate simvastatin was increased by 3.5 fold when given with imatinib.[64] Of note, in an 11-patient study by van Erp et al, researchers did not find a significant increase in imatinib pharmacokinetics in cancer patients treated with the drug after a three day administration of ritonavir.[65] The authors proposed this is due to alternative metabolic pathways which imatinib may undergo in the face of CYP3A4 inhibition. Whether this finding has relevance in patients on chronic HAART therapy including ritonavir is unknown.
Dasatinib (Sprycel, Bristol Myers Squibb)
Dasatinib is an orally available tyrosine kinase inhibitor which has been found to inhibit the bcr-abl, c-kit, PDGFR data and the SRC family of tyrosine kinases.[66,67] It has been approved for use in the treatment of chronic accelerated myeloid or lymphoid blast phase chronic myeloid leukemia in patients who are resistant to prior therapies.[66,68,69] It is undergoing extensive clinical investigation for its possible role in treating many additional solid organ and hematological malignancies.[70] The standard dose is either 70 or 100 mg q.d.
Dasatinib is metabolized primarily by CYP3A4 into its active metabolite.[66,71] This metabolite is equally active compared to the parent compound. Other enzymes found to be involved in the drug metabolism include flavin-containing monooxygenase (FMO-3) and uridine diphosphate-glucuronosyltransferase (UGT).[66,71] In addition, dasatinib has been found to inhibit the activity of CYP3A4 but does not inhibit other CYP family enzymes, nor does it induce this class of enzymes.[66,72] In patients who are on drugs known to inhibit CYP3A4, dasatinib drug levels may be increased by as much as 500%, while co-administrations with an inducer of CYP3A4 led to a 82% decrease in dasatinib levels.[66] The Package Insert recommends that use of CYP3A4 inhibitors should be avoided. If this cannot be done a dose decrease to 20 mg per day should be considered -- or reduction of 80% of the standard dose. For patients on a CYP3A4 inducing agent, a dose increase of dasatinib should be considered, but no specific dose is recommended in the Package Insert.
Nilotinib (Tasigna, Novartis)
Nilotinib is an orally available inhibitor of the bcr-abl kinase and is approved for the treatment of imatinib-resistant chronic myelogenous leukemia.[73,74] It is undergoing extensive clinical investigation in a variety of other malignancies.[75] The standard dose is 400mg bid.
Nilotinib is apparently metabolized by CYP3A4.[74] Co-administrated with the inhibitor ketoconazole led to a 3-fold increase in drug levels. If patients must be on a drug known to strongly inhibit CYP3A4, a 50% dose reduction should be done to 400mg once a day (instead of twice a day) based on expected effects to the drug's pharmacokinetics and AUC. This recommendation is not based on clinical data. When co-administered with the inducer rifampicin, an 80% decrease in nilotinib levels were observed.[74] For patients on a strong CYP3A4 inhibitor, the dose of nilotinib may need to be increased but again no recommendation on what that higher dose should be is included in the Package Insert.[74]
Nilotinib inhibits in vitro CYP3A4, CYP2C8, CYP2C9, CYP2D6, and UGT1A1.[74,76] Administration of nilotinib with the CYP3A4 substrate midazolam in healthy volunteers led to a 30% increase in midazolam levels.[74] Warfarin, as a substrate of both CYP2C9 and CYP3A4, should be avoided. Nilotinib also induces CYP2B6, CYP2C8, and CYP2C9, and may decrease drug concentration of substrates for these enzymes.[74]
Erlotinib (Tarceva, OSI Pharmaceuticals)
Erlotinib is an orally available tyrosine kinase inhibitor of the epidermal growth factor receptor (EGFR) used in the treatment of non-small-cell lung cancer as well as pancreatic cancer.[77-79] The drug inhibits the EGFR tyrosine kinase on the intracellular component down regulating this critical pathway.
In vitro assays have shown that erlotinib is primarily metabolized by CYP3A4.[79-81] CYP1A2 plays a lesser role including the extrahepatic isoform of CYP1 as well as the extrahepatic form of CYP1A1.[80,82] The administration of the CYP3A4-inhibitor ketoconazole led to a 67% increase in blood levels of erlotinib.[79] The Package Insert recommends that in patients who are on a drug known to inhibit CYP3A4 a dose reduction of erlotinib should be considered, though there is no recommendation of what this lower dose should be. In addition, in patients who are on agents known to induce CYP3A4, significantly lower drug levels of erlotinib may be expected. In fact, the area under the concentration curve (AUC) of erlotinib may be reduced by 67% to 80% when patients are on drugs such as rifampicin and other known CYP3A4 inducers.[79] The Package Insert recommends that patients seek alternative treatment to the drug known to induce CYP3A4 activity if they are going to be placed on erlotinib. If that is not clinically possible, the dose of erlotinib may be increased from 150 mg q.d. to up to 450 mg q.d. Finally, the co-administration of drugs known to be metabolized by CYP3A4 when given in conjunction with erlotinib may be effected by erlotinib-mediated inhibition of CYP3A4.[81] The administration of erlotinib with midazolam led to a 24% increase in midazolam blood levels.[79]
Sunitinib (Sutent, Pfizer)
Sunitinib is an orally available multi-targeted tyrosine kinase inhibitor approved for the treatment of renal cell carcinoma and imatinib-refractory gastrointestinal stromal tumor.[83-85] The drug inhibits PDGFR, VEGFR1 and 3, KIT, FLT3, the receptor stem cell factor (SCF), and RET.[83,86-88] In pre-clinical studies, sunitinib and its equally active metabolite SU012662 inhibit Flk1/KDR activity and PDGFR activity.
Sunitinib is metabolized by CYP3A4 to produce the active metabolite SU012662.[85] No other major metabolites have been identified. Sunitinib did inhibit the metabolic activity, or to induce expression, of CYP3A4 or any other member of the CYP family of enzmes in vitro. Concomitant administration of sunitinib with ketoconazole resulted in a 74% increase in blood levels of sunitinib and a 12% decrease in SU12662, with a net increase of 51% of total drug. When given with the CYP3A4 inducing drug rifampin, a 46% decrease in total drug blood levels was observed. The Package Insert recommends the consideration of dose reduction if patients are on a drug known to strongly inhibit CYP3A4, and to increase the dose if they are on a drug that strongly induces CYP3A4, but no specific dose recommendations are included.[85]
Lapatinib (Tykerb, GlaxoSmithKline)
Lapatinib is an orally available multi-tyrosine kinase inhibitor approved for the treatment of patients with advanced or metastatic breast cancers that overexpress HER2 when given with capecitabine (Xeloda).[89,90] When given with capaecitabine (Xeloda) for the treatement of breast cancer, the recommended dose of lapatinib is 1,250mg q.d. with no days off of treatment.
Lapatinib is metabolized extensively by CYP3A4 and CYP3A5, with CYP2C19 and CYP2C8 playing a minor role in parent drug metabolism.[90] Lapatinib also inhibits CYP3A4 and CYP2C8 in vitro at clinically relevant dose levels, and thus, lapatinib may increase concentration of drugs metabolized by these enzymes. Drug inhibitors and inducers of CYP3A4 should be avoided while taking lapatinib. Ketoconazole caused a 3.6-fold increase in lapatinib blood levels in healthy subjects, whereas the CYP3A4-inducing drug carbamazepine caused a 72% increase in blood levels.[91] According to the Package Insert, a dose reduction in lapatinib should be considered in patients on a known CYP3A4-inhibitor to 500mg per day (or a dose reduction of 60%). If on a CYP3A4 inducing agent, the Package Insert recommends a gradual dose increase up to 4,500mg/day – or an increase of 360% -- as tolerated by the patient. These recommendations are based on pharmacological modeling and not on clinical data.
Bortezomib (Velcade, Millennium Pharmaceuticals)
Bortezomib is a proteosome inhibitor that is approved for the treatment of multiple myeloma and mantle cell lymphoma.[92-94] The drug is administered intravenously with the recommended dose being 1.3mg/m2 given twice weekly for two weeks, on a three week cycle.
Bortezomib is metabolized by CYP3A4, CYP2C19, and CYP1A2, with minor contributions made by CYP2D6 and CYP2C9, into inactive metabolites.[95] Administration of ketoconazole with bortezomib led to a 35% increase in bortezomib levels, whereas co-administration with the CYP2C19 inhibitor omeprazole had no effect on the pharmacokinetics of bortezomib.[94,96] There is no reported data on the effects of CYP3A4 inducing agents on bortezomib's pharmacokinetics. The Package Insert does not make any recommendations to alter dosing if patients are on concomitant drugs that affect CYP3A4 function. Finally, bortezomib is a poor inhibitor of CYP enzymes 1A2, 2C9, 2D6 and 3A4; however, it may inhibit CYP2C19 at clinically relevant dosages, and in turn effect levels of drugs that are substrates for this liver enzyme.[97]
Sorafenib (Nexavar, Bayer)
Sorafenib is an orally available multi-kinase inhibitor approved for the use in treating unresectable hepatocellular carcinoma and renal cell carcinoma and is being extensively studied in a wide range of malignancies.[98-101] The drug inhibits intracellular CRAF, BRAF, and mutant BRAF, as well as membrane kinases including KIT, FLT-3, RET, VEGFR-1, VEGFR-2, VEGFR-3, and PDGFR-beta.[100,102] The standard dosing is 400mg twice daily.
Sorafenib is metabolized by CYP3A4 and UGT1A9.[100] Interestingly, when given to healthy volunteers along with the CYP3A4 inhibitor ketoconazole, no change was seen in plasma sorafenib levels, likely due to alternative metabolic pathways available to disposition including via UGT1A9.[103] Therefore, dose adjustment is unlikely to be needed in patients on a CYP3A4 inhibiting drug. In hepatocyte preclinical studies, sorafenib was not found to induce CYP3A4 or CYP1A2.[100] When co-administered with the CYP3A4 inducing agent rifampicin, a 37% decrease in sorafenib drug levels was observed.[100] Thus, increasing the standard dose should be considered if patients are on a known CYP3A4 inducing agent, though no clinical data exists on what dose should be used.
Sorafenib inhibits glucuronidation by the UGT1A1 and UGT1A9 enzymes, and caution is recommended if patients need to be on agents metabolized by these critical phase II enzymes. [100] In a clinical study using sorafenib and irinotecan -- which is metabolized by UGT1A1 -- an increase of 67 to 120% was seen in the active metabolite of irinotecan, SN-38.[100,104,105] Sorafenib also inhibits CYP2B6 and CYP2C8, which in turn could lead to higher serum levels of drugs metabolized by these two enzymes.[100] While in vitro human hepatocyte experiments found that sorafenib inhibited CYP2C19, CYP2D6, and CYP3A4, clinical studies found no effect on the pharmacokinetics of substrates of these enzymes (omeprazole, dextromethorphan, and midazolam, respectively).[100] Additional clinical studies found increases in drug levels of docetaxel, doxorubicin, and fluoruracil when given with sorafenib.[100,106] The pharmacokinetics of gemcitabine, oxaliplatin, gefitinib, and erlotinib where not affected by concomitant use of sorafenib.[107-110]
Temsirolimus (Torisel, Wyeth)
Temsirolimus is an mTOR inhibitor and approved for the treatment of refractory renal cell carcinoma.[111,112] mTOR (Mammalian Target of Rapamycin) is a critical protein in the PI3 kinase/AKT pathway, and blockage of mTOR activity results in reduced levels of HIF-1 and HIF-2 alpha as well as vascular endothelial growth factor. The mTOR target may have wide ranging clinical benefit in solid and hematologic malignancies.[113] Temsirolimus is being testing in a variety of malignancies.[114] The drug is administered as an intravenous infusion, with a set dose of 25mg per week given to patients - a dose which is not based on body surface area.[111]
Temsirolimus is metabolized by CYP3A4 into five metabolites, the predominant one being the active metabolite sirolimus.[115] Co-administration of ketoconazole did not effect blood levels of temsirolimus but increased sirolimus levels by 3.1 fold.[116] The Package Insert recommends a dose reduction of 50% to 12.5 mg/week in patients on a CYP3A4 inhibitor, though no clinical data supports this recommendation. Co-administration with the CYP3A4 inducer rifampin did not effect temsirolimus blood levels but reduced sirolimus levels by 43% to 56%.[117] If patients are on a CYP3A4-inducer, the Package Insert recommends a dose increase by 100% to 50mg/week should be considered – though again this is based on pharmacokinetic modeling and not on clinical research. A phase I clinical study combining sunitinib and temsirolimus resulting in dose limiting toxicities in two of the 3 patients treated at the lowest dose levels (sunitinib 25mg and temsirolimus 15mg), and the trial was closed.[118] In vitro studies indicated that temsirolimus inhibited CYP3A4 and CYP2D6, but clinical studies showed no effect on the pharmacokinetics of the CYP2D6 substrate desipramine.[111]
TREATING HIV-POSITIVE PATIENTS WITH TARGETED AGENTS
The development of these and other new targeted therapies has significantly improved our ability to treat many hematologic and solid tumor malignancies. These cancers are now occurring at high rates in men and women with HIV, and targeted therapies can and should be employed to treat Non-AIDS Defining Cancers in HIV-positive patients. However, significant interactions between HAART agents and these targeted therapies is possible and even likely. Since past clinical studies testing these newer agents typically excluded patients with HIV, there are no guidelines and little clinical experience in how to use these agents in the setting of HIV and HAART. How to treat patients with HIV on HAART with newly-diagnosed NADCs using targeted therapies is an open clinical question.
Nevertheless, based on what is known about the pharmacology of HAART agents and targeted anti-cancer drugs, it is possible to posit treatment considerations for medical oncologists facing this clinical dillema. Table 2 is a heat map-like grid that cross references HAART agents and targeted compounds in terms of potential interactions that may alter the targeted drugs’ pharmacokinetics, and in turn either increase drug toxicity or reduce drug efficicay. Blocks in red indicate a HAART-targeted therapy combination that poses a high risk for interaction, and dose reduction of the targeted therapy from the start of therapy should be considered. Lighter red, lined blocks are those were an interaction may occur, and careful clinical observation and possible dose reductions after therapy is started should be considered. Blocks in green are for a HAART-targeted therapy combination in which HAART-caused enzyme induction may lead to reduced levels of the targeted therapy and a dose adjustment upwards should be considered.
Table 2.
Potential Effects on Targeted Anticancer Drugs by HAART Agents in Heat Map Format.
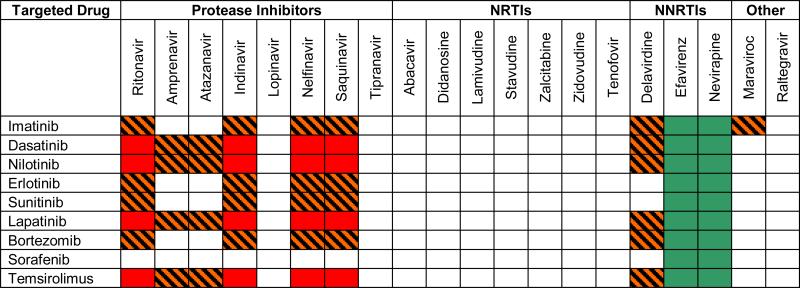 |
Potential drug-drug interactions are highlighted by color code. A red block indicates a potential HAART-targeted therapy combination that poses a high risk for interaction leading to high pharmacokinetic levels of the targeted agent. Lighter red, lined blocks indicate an interaction that may occur. Blocks in green indicate a HAART-targeted therapy combination in which HAART-caused enzyme induction may lead to reduced levels of the targeted therapy.
Table 3 summarizes the effects on the pharmacokinetics of targeted therapies casued by either the inhibition of, or the induction of, the crucial metabolizing enzyme CYP3A4. As can be seen, though many of these targeted therapies are substrates for this enzyme, there is a great variability in terms of how much blood levels in patients may be affected by inhibition or induction of this enzyme. For those targeted therapies in which a significant effect is seen in its pharmacokinietics (i.e. an increase of more than 100% or a decrease by more than 50%), then great caution should be observed in dosing these drugs if patients are on HAART agents known to inhibit or induce CYP3A4. For these agents, following the recommendations contained in the FDA Package Insert is critical, including dose alterations from the very beginning of treatment, and titrating therapy based on patient tolerance and response.
Table 3.
Effect on targeted drug pharmacokinetics by CYP3A4 Inhibition and Induction
| Drug | Pharmacokinetic Change in Drug Levels (AUC) (%) | |
|---|---|---|
| Inhibitor | Inducer | |
| Imatinib | 40 | -380 (RMP) |
| Dasatinib | 500 | - 82 (RMP) |
| Nilotinib | 300 | - 80 (RPC) |
| Erlotinib | 67 | - 67 to - 80 (RPC) |
| Sunitinib | 51 | - 46 (RMP) |
| Lapatinib | 360 | - 72 (CBZ) |
| Bortezomib | 35 | n/a |
| Sorafenib | 0 | -37 (RPC) |
| Temsirolimus | 310 * | -56 * (RMP) |
Percent change in pharmacokinetics of targeted agents as measured by Area Under Concentration (AUC) curve by the co-administration of drugs known to inhibit or induce the CYP3A4 metabolizing enzyme. A CYP3A4 inhibitor prevents metabolism of the drug and leads to higher serum drug levels as reflected in drug AUC, whereas an enzyme inducer would lead to decreased blood levels. Ketoconazole was used as the inhibiting agent, whereas rifampin (RMP), rifampicin (RPC), or carbamazepine (CBZ) were used as inducing agents. References and discussion are included in the text.
For temsirolimus, the parent drug was unaffected but the active metabolite sirolimus was affected by the inhibiting and inducing agents.
Finally, just as HAART agents may interfere with the metabolism of targeted therapies, so too can targeted drugs interfere with the metabolism of HAART drugs. Table 4 highlights potential combinations that can lead to altered blood levels of HAART agents. Red blocks indicate combinations of that may lead to higher blood levels of the HAART drug in question due to inhibition of its metabolism by a targeted drug. In these cases, higher toxicities associated with the HAART drug may be observed. Green blocks indicate a combination that can lead to reduced levels of the HAART drug due to enzyme induction by the targeted therapy. In this later case, immune parameters would need to be carefully monitored, and a slow titration up of the HAART drug may be considered.
Table 4.
Potential Effects on HAART drugs by Targeted Anticancer Drugs in Heat Map Format.
| Targeted Agents | Protease Inhibitors | NRTIs | NNRTIs | Other | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ritonavir | Amprenavir | Atazanavir | Indinavir | Lopinavir | Nelfinavir | Saquinavir | Tipranavir | Abacavir | Didanosine | Lamivudine | Stavudine | Zalcitabine | Zidovudine | Tenofovir | Delavirdine | Efavirenz | Nevirapine | Maraviroc | Raltegravir | |
| Imatinib | ||||||||||||||||||||
| Dasatinib | ||||||||||||||||||||
| Nilotinib | ||||||||||||||||||||
| Erlotinib | ||||||||||||||||||||
| Sunitinib | ||||||||||||||||||||
| Lapatinib | ||||||||||||||||||||
| Bortezomib | ||||||||||||||||||||
| Sorafenib | ||||||||||||||||||||
| Temsirolimus | ||||||||||||||||||||
Targeted therapies can also affect drug metabolizing enzymes by inhibiting and inducing their function. Blocks marked with red indicate a targeted therapy/HAART drug combination which may lead to higher HAART agent levels due to enzyme inhibition by the targeted agent. Blocks in green indicate a potential interaction in which patients may experience lower levels of the HAART drug due to enzyme induction by the targeted anticancer drug.
CONCLUSION
New advances in the treatment of cancers through the use of targeted therapies has improved the clinical outcomes of HIV-negative patients, an improvement that can and should be extended to HIV-positive patients diagnosed with NADCs. However, a better knowledge of specific drug-drug interactions between HAART and targeted agents, both at the molecular level and clinically, would aid in establishing clear guidelines for how specific NADCs should be treated in patients with HIV. A new research focus on the part of the National Cancer Institute as well as the AIDS Malignancy Consortium may help elucidate these potential interactions and hopefully lead to the development of treatment guidelines. The first of a number of planned clinical trials will investigate drug-drug interactions between sunitinib and differing HAART combinations. Given the growing incidence of NADCs in HIV-positive patients, and the need for such patients to remain on HAART, this pharmacologic and clinical research is timely and needed. If successful, this research should aid medical oncologists in choosing the right targeted therapy, at the right dose, to treat their patient. Only though this and other clinical and translational research will we have the same success in addressing the NADC epidemic in HIV-positive patients has we have had over the past decade in treating AIDS-defining cancers, as well as HIV infection itself.
REFERENCES
- 1.Herida M, Mary-Krause M, Kaphan R, et al. Incidence of non-AIDS-defining cancers before and during the highly active antiretroviral therapy era in a cohort of human immunodeficiency virus-infected patients. J Clin Oncol. 2003;21(18):3447–53. doi: 10.1200/JCO.2003.01.096. [DOI] [PubMed] [Google Scholar]
- 2.Clifford GM, Polesel J, Rickenbach M, et al. for the Swiss HIV Cohort Cancer risk in the Swiss HIV Cohort Study: associations with immunodeficiency, smoking, and highly active antiretroviral therapy. J Natl Cancer Inst. 2005;97(6):425–32. doi: 10.1093/jnci/dji072. [DOI] [PubMed] [Google Scholar]
- 3.Engels EA, Pfeiffer RM, Goedert JJ, et al. Trends in cancer among people with AIDS in the United States 1980-2002. AIDS. 2006;20(12):1645–54. doi: 10.1097/01.aids.0000238411.75324.59. [DOI] [PubMed] [Google Scholar]
- 4*.Powles T, Robinson D, Stebbing J, et al. Highly active antiretroviral therapy and the incidence of non-AIDS defining cancers in people with HIV infection. J Clin Oncol. 2009;27(6):884–90. doi: 10.1200/JCO.2008.19.6626. [The most recent epidemiology study confirming the cross-national rise in Non AIDS-Defining Cancers in HIV-positive patients.] [DOI] [PubMed] [Google Scholar]
- 5.Mitsuyasu R. Non-AIDS defining malignancies in HIV. Top HIV Med. 2008;16:117–121. [PubMed] [Google Scholar]
- 6.National Cancer Institute [April 7, 2009];Targeted Cancer Therapies: Questions and Answers. http://www.cancer.gov/cancertopics/factsheet/Therapy/targeted.
- 7.Gonzales FJ, Tukey RH. Drug metabolism. In: Brunton LL, editor. Goodman & Gilman's The Pharmacological Basis of Therapeutics. 11th edition McGraw Hill; New York: 2006. [Google Scholar]
- 8*.Flockhart D. P450 Drug Interaction Table. http://medicine.iupui.edu/clinpharm/ddis/table.asp. [An extensive and easy to use reference on the metabolism of dozens of commonly used FDA-approved pharmaceutical agents.]
- 9.Hammer SM, Saag MS, Schechter M, Montaner JS, Schooley RT, Jacobsen DM, Thompson MA, Carpenter CC, Fischl MA, Gazzard BG, Gatell JM, Hirsch MS, Katzenstein DA, Richman DD, Vella S, Yeni PG, Volberding PA, International AIDS Society-USA panel International AIDS Society-USA panel. Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society-USA panel. JAMA. 2006;296(7):827–43. doi: 10.1001/jama.296.7.827. [DOI] [PubMed] [Google Scholar]
- 10.Antoniou T, Tseng AL. Interactions between antiretrovirals and antineoplastic drug therapy. Clin Pharmacokinet. 2005;44(2):111–45. doi: 10.2165/00003088-200544020-00001. [DOI] [PubMed] [Google Scholar]
- 11.O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, Cornelissen JJ, Fischer T, Hochhaus A, Hughes T, Lechner K, Nielsen JL, Rousselot P, Reiffers J, Saglio G, Shepherd J, Simonsson B, Gratwohl A, Goldman JM, Kantarjian H, Taylor K, Verhoef G, Bolton AE, Capdeville R, Druker BJ, IRIS Investigators Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 12.Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, Tanaka M, Hecht JR, Heinrich MC, Fletcher CD, Crowley JJ, Borden EC. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626–32. doi: 10.1200/JCO.2007.13.4452. [DOI] [PubMed] [Google Scholar]
- 13.Ottmann OG, Druker BJ, Sawyers CL, Goldman JM, Reiffers J, Silver RT, Tura S, Fischer T, Deininger MW, Schiffer CA, Baccarani M, Gratwohl A, Hochhaus A, Hoelzer D, Fernandes-Reese S, Gathmann I, Capdeville R, O'Brien SG. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood. 2002;100(6):1965–71. doi: 10.1182/blood-2001-12-0181. [DOI] [PubMed] [Google Scholar]
- 14.Gleevec Package Insert. http://www.fda.gov/cder/foi/label/2006/021588s009lbl.pdf.
- 15.Peng B, Lloyd P, Schran H. Clinical pharmacokinetics of imatinib. Clin Pharmacokinet. 2005;44(9):879–94. doi: 10.2165/00003088-200544090-00001. [DOI] [PubMed] [Google Scholar]
- 16.Dutreix C, Peng B, Mehring G, Hayes M, Capdeville R, Pokorny R, Seiberling M. Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects. Cancer Chemother Pharmacol. 2004;54(4):290–4. doi: 10.1007/s00280-004-0832-z. [DOI] [PubMed] [Google Scholar]
- 17.O'Brien SG, Meinhardt P, Bond E, Beck J, Peng B, Dutreix C, Mehring G, Milosavljev S, Huber C, Capdeville R, Fischer T. Effects of imatinib mesylate (STI571, Glivec) on the pharmacokinetics of simvastatin, a cytochrome p450 3A4 substrate, in patients with chronic myeloid leukaemia. Br J Cancer. 2003;89(10):1855–9. doi: 10.1038/sj.bjc.6601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Erp NP, Gelderblom H, Karlsson MO, Li J, Zhao M, Ouwerkerk J, Nortier JW, Guchelaar HJ, Baker SD, Sparreboom A. Influence of CYP3A4 inhibition on the steady-state pharmacokinetics of imatinib. Clin Cancer Res. 2007;13(24):7394–400. doi: 10.1158/1078-0432.CCR-07-0346. [DOI] [PubMed] [Google Scholar]
- 19.Sprycel Package Insert. http://packageinserts.bms.com/pi/pi_sprycel.pdf.
- 20.Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM, Fairchild C, Hunt JT, Inigo I, Johnston K, Kamath A, Kan D, Klei H, Marathe P, Pang S, Peterson R, Pitt S, Schieven GL, Schmidt RJ, Tokarski J, Wen ML, Wityak J, Borzilleri RM. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658–61. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 21.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O'Brien S, Nicaise C, Bleickardt E, Blackwood-Chirchir MA, Iyer V, Chen TT, Huang F, Decillis AP, Sawyers CL. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–41. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 22.Cortes J, Rousselot P, Kim DW, Ritchie E, Hamerschlak N, Coutre S, Hochhaus A, Guilhot F, Saglio G, Apperley J, Ottmann O, Shah N, Erben P, Branford S, Agarwal P, Gollerkeri A, Baccarani M. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood. 2007;109(8):3207–13. doi: 10.1182/blood-2006-09-046888. [DOI] [PubMed] [Google Scholar]
- 23.Tomillero A, Moral MA. Gateways to clinical trials. December 2008. Methods Find Exp Clin Pharmacol. 2008;30(10):761–82. [PubMed] [Google Scholar]
- 24.Wang L, Christopher LJ, Cui D, Li W, Iyer R, Humphreys WG, Zhang D. Identification of the human enzymes involved in the oxidative metabolism of dasatinib: an effective approach for determining metabolite formation kinetics. Drug Metab Dispos. 2008;36(9):1828–39. doi: 10.1124/dmd.107.020255. [DOI] [PubMed] [Google Scholar]
- 25.Li X, He Y, Ruiz CH, Koenig M, Cameron MD. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37(6):1242–50. doi: 10.1124/dmd.108.025932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kantarjian HM, Giles F, Gattermann N, Bhalla K, Alimena G, Palandri F, Ossenkoppele GJ, Nicolini FE, O'Brien SG, Litzow M, Bhatia R, Cervantes F, Haque A, Shou Y, Resta DJ, Weitzman A, Hochhaus A, le Coutre P. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood. 2007;110(10):3540–6. doi: 10.1182/blood-2007-03-080689. [DOI] [PubMed] [Google Scholar]
- 27.Tasigna Package Insert. http://www.pharma.us.novartis.com/product/pi/pdf/tasigna.pdf.
- 28.Tomillero A, Moral MA. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2008;30(7):543–88. [PubMed] [Google Scholar]
- 29.Deremer DL, Ustun C, Natarajan K. Nilotinib: a second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clin Ther. 2008;30(11):1956–75. doi: 10.1016/j.clinthera.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 30.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabárbara P, Seymour L, National Cancer Institute of Canada Clinical Trials Group Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 31.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W, National Cancer Institute of Canada Clinical Trials Group Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–6. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 32.Tarceva Package Insert. http://www.gene.com/gene/products/information/pdf/tarceva-prescribing.pdf.
- 33.Ling J, Johnson KA, Miao Z, et al. Metabolism and excretion of erlotinib, a small molecule inhibitor of epidermal growth factor receptor tyrosine kinase, in healthy male volunteers. Drug Metab Dispos. 2006;34:420–6. doi: 10.1124/dmd.105.007765. [DOI] [PubMed] [Google Scholar]
- 34.Li J, Zhao M, He P, Hidalgo M, Baker SD. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007;13(12):3731–7. doi: 10.1158/1078-0432.CCR-07-0088. [DOI] [PubMed] [Google Scholar]
- 35.Rakhit A, Pantze MP, Fettner S, Jones HM, Charoin JE, Riek M, Lum BL, Hamilton M. The effects of CYP3A4 inhibition on erlotinib pharmacokinetics: computer-based simulation (SimCYP) predicts in vivo metabolic inhibition. Eur J Clin Pharmacol. 2008;64(1):31–41. doi: 10.1007/s00228-007-0396-z. [DOI] [PubMed] [Google Scholar]
- 36.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–24. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 37.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–38. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 38.Sutent Package Insert. http://www.pfizer.com/files/products/uspi_sutent.pdf.
- 39.Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2(5):471–8. [PubMed] [Google Scholar]
- 40.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–37. [PubMed] [Google Scholar]
- 41.Schueneman AJ, Himmelfarb E, Geng L, Tan J, Donnelly E, Mendel D, McMahon G, Hallahan DE. SU11248 maintenance therapy prevents tumor regrowth after fractionated irradiation of murine tumor models. Cancer Res. 2003;63(14):4009–16. [PubMed] [Google Scholar]
- 42.Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, Berger M, Oliva C, Rubin SD, Stein S, Cameron D. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–43. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 43.Tykerb Package Insert. http://us.gsk.com/products/assets/us_tykerb.pdf.
- 44.Smith DA, Koch KM, Arya N, Bowen CJ, Herendeen JM, Beelen A. Effects of ketoconazole and carbamazepine on lapatinib pharmacokinetics in healthy subjects. Br J Clin Pharmacol. 2009;67(4):421–6. doi: 10.1111/j.1365-2125.2009.03370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Bladé J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC, Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–98. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 46.Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Stadtmauer EA, O'Connor OA, Shi H, Boral AL, Goy A. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24(30):4867–74. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 47.Velcade Package Insert. http://www.velcade.com/pdf/VELCADE_PRESCRIBING_INFORMATION.pdf.
- 48.Uttamsingh V, Lu C, Miwa G, Gan LS. Relative contributions of the five major human cytochromes P450, 1A2, 2C9, 2C19, 2D6, and 3A4, to the hepatic metabolism of the proteasome inhibitor bortezomib. Drug Metab Dispos. 2005;33(11):1723–8. doi: 10.1124/dmd.105.005710. [DOI] [PubMed] [Google Scholar]
- 49.Quinn DI, Nemunaitis J, Fuloria J, Britten CD, Gabrail N, Yee L, Acharya M, Chan K, Cohen N, Dudov A. Effect of the Cytochrome P450 2C19 Inhibitor Omeprazole on the Pharmacokinetics and Safety Profile of Bortezomib in Patients with Advanced Solid Tumours, Non-Hodgkin's Lymphoma or Multiple Myeloma. Clin Pharmacokinet. 2009;48(3):199–209. doi: 10.2165/00003088-200948030-00006. [DOI] [PubMed] [Google Scholar]
- 50.Lu C, Gallegos R, Li P, Xia CQ, Pusalkar S, Uttamsingh V, Nix D, Miwa GT, Gan LS. Investigation of drug-drug interaction potential of bortezomib in vivo in female Sprague-Dawley rats and in vitro in human liver microsomes. Drug Metab Dispos. 2006;34(4):702–8. doi: 10.1124/dmd.105.008060. [DOI] [PubMed] [Google Scholar]
- 51.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM, TARGET Study Group Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–34. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 52.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Häussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J, SHARP Investigators Study Group Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 53.Nexavar Package Insert. http://berlex.bayerhealthcare.com/html/products/pi/Nexavar_PI.pdf.
- 54.Tomillero A, Moral MA. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2009;31(2):107–46. [PubMed] [Google Scholar]
- 55.Wilhelm S, Chien DS. BAY 43-9006: preclinical data. Curr Pharm Des. 2002;8(25):2255–7. doi: 10.2174/1381612023393026. [DOI] [PubMed] [Google Scholar]
- 56.Lathia C, Lettieri J, Cihon F, Gallentine M, Radtke M, Sundaresan P. Lack of effect of ketoconazole-mediated CYP3A inhibition on sorafenib clinical pharmacokinetics. Cancer Chemother Pharmacol. 2006;57(5):685–92. doi: 10.1007/s00280-005-0068-6. [DOI] [PubMed] [Google Scholar]
- 57.Mross K, Steinbild S, Baas F, Gmehling D, Radtke M, Voliotis D, Brendel E, Christensen O, Unger C. Results from an in vitro and a clinical/pharmacological phase I study with the combination irinotecan and sorafenib. Eur J Cancer. 2007;43(1):55–63. doi: 10.1016/j.ejca.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 58.Mross K, Steinbild S, Baas F, Reil M, Buss P, Mersmann S, Voliotis D, Schwartz B, Brendel E. Drug-drug interaction pharmacokinetic study with the Raf kinase inhibitor (RKI) BAY 43-9006 administered in combination with irinotecan (CPT-11) in patients with solid tumors. Int J Clin Pharmacol Ther. 2003;41(12):618–9. doi: 10.5414/cpp41618. [DOI] [PubMed] [Google Scholar]
- 59.Richly H, Schultheis B, Adamietz IA, Kupsch P, Grubert M, Hilger RA, Ludwig M, Brendel E, Christensen O, Strumberg D. Combination of sorafenib and doxorubicin in patients with advanced hepatocellular carcinoma: results from a phase I extension trial. Eur J Cancer. 2009;45(4):579–87. doi: 10.1016/j.ejca.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 60.Siu LL, Awada A, Takimoto CH, Piccart M, Schwartz B, Giannaris T, Lathia C, Petrenciuc O, Moore MJ. Phase I trial of sorafenib and gemcitabine in advanced solid tumors with an expanded cohort in advanced pancreatic cancer. Clin Cancer Res. 2006;12(1):144–51. doi: 10.1158/1078-0432.CCR-05-1571. [DOI] [PubMed] [Google Scholar]
- 61.Kupsch P, Henning BF, Passarge K, Richly H, Wiesemann K, Hilger RA, Scheulen ME, Christensen O, Brendel E, Schwartz B, Hofstra E, Voigtmann R, Seeber S, Strumberg D. Results of a phase I trial of sorafenib (BAY 43-9006) in combination with oxaliplatin in patients with refractory solid tumors, including colorectal cancer. Clin Colorectal Cancer. 2005;5(3):188–96. doi: 10.3816/ccc.2005.n.030. [DOI] [PubMed] [Google Scholar]
- 62.Adjei AA, Molina JR, Mandrekar SJ, Marks R, Reid JR, Croghan G, Hanson LJ, Jett JR, Xia C, Lathia C, Simantov R. Phase I trial of sorafenib in combination with gefitinib in patients with refractory or recurrent non-small cell lung cancer. Clin Cancer Res. 2007;13(9):2684–91. doi: 10.1158/1078-0432.CCR-06-2889. [DOI] [PubMed] [Google Scholar]
- 63.Duran I, Hotté SJ, Hirte H, Chen EX, MacLean M, Turner S, Duan L, Pond GR, Lathia C, Walsh S, Wright JJ, Dancey J, Siu LL. Phase I targeted combination trial of sorafenib and erlotinib in patients with advanced solid tumors. Clin Cancer Res. 2007;13(16):4849–57. doi: 10.1158/1078-0432.CCR-07-0382. [DOI] [PubMed] [Google Scholar]
- 64.Torisel Package Insert. http://www.wyeth.com/content/showlabeling.asp?id=490.
- 65.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A, RECORD-1 Study Group Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372(9637):449–56. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 66.Figlin RA, Brown E, Armstrong AJ, Akerley W, Benson AB, 3rd, Burstein HJ, Ettinger DS, Febbo PG, Fury MG, Hudes GR, Kies MS, Kwak EL, Morgan RJ, Jr, Mortimer J, Reckamp K, Venook AP, Worden F, Yen Y. NCCN Task Force Report: mTOR inhibition in solid tumors. J Natl Compr Canc Netw. 2008;(Suppl 5):6, S1–S20. [PubMed] [Google Scholar]
- 67.Tomillero A, Moral MA. Gateways to Clinical Trials. Methods Find Exp Clin Pharmacol. 2008;30(5):383–408. [PubMed] [Google Scholar]
- 68.Cai P, Tsao R, Ruppen ME. In vitro metabolic study of temsirolimus: preparation, isolation, and identification of the metabolites. Drug Metab Dispos. 2007;35(9):1554–63. doi: 10.1124/dmd.107.014746. [DOI] [PubMed] [Google Scholar]
- 69.Boni JP, Leister C, Burns J, Hug B. Differential effects of ketoconazole on exposure to temsirolimus following intravenous infusion of temsirolimus. Br J Cancer. 2008;98(11):1797–802. doi: 10.1038/sj.bjc.6604376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boni J, Leister C, Burns J, Cincotta M, Hug B, Moore L. Pharmacokinetic profile of temsirolimus with concomitant administration of cytochrome p450-inducing medications. J Clin Pharmacol. 2007;47(11):1430–9. doi: 10.1177/0091270007306957. [DOI] [PubMed] [Google Scholar]
- 71.Patel PH, Senico PL, Curiel RE, Motzer RJ. Phase I study combining treatment with temsirolimus and sunitinib malate in patients with advanced renal cell carcinoma. Clin Genitourin Cancer. 2009;7(1):24–7. doi: 10.3816/CGC.2009.n.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walsh JS, Reese MJ, Thurmond LM. The metabolic activation of abacavir by human liver cytosol and expressed human alcohol dehydrogenase isozymes. Chem Biol Interact. 2002;142(1-2):135–54. doi: 10.1016/s0009-2797(02)00059-5. [DOI] [PubMed] [Google Scholar]
- 73.Ziagen Package Insert. http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020977s019,020978s022lbl.pdf.
- 74.Decker CJ, Laitinen LM, Bridson GW, Raybuck SA, Tung RD, Chaturvedi PR. Metabolism of amprenavir in liver microsomes: role of CYP3A4 inhibition for drug interactions. J Pharm Sci. 1998;87(7):803–7. doi: 10.1021/js980029p. [DOI] [PubMed] [Google Scholar]
- 75.Agenerase Package Insert. http://www.accessdata.fda.gov/drugsatfda_docs/label/2005/021007s017lbl.pdf.
- 76.Granfors MT, Wang JS, Kajosaari LI, Laitila J, Neuvonen PJ, Backman JT. Differential inhibition of cytochrome P450 3A4, 3A5 and 3A7 by five human immunodeficiency virus (HIV) protease inhibitors in vitro. Basic Clin Pharmacol Toxicol. 2006;98(1):79–85. doi: 10.1111/j.1742-7843.2006.pto_249.x. [DOI] [PubMed] [Google Scholar]
- 77.Colombo S, Buclin T, Cavassini M, Décosterd LA, Telenti A, Biollaz J, Csajka C. Population pharmacokinetics of atazanavir in patients with human immunodeficiency virus infection. Antimicrob Agents Chemother. 2006;50(11):3801–8. doi: 10.1128/AAC.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang D, Chando TJ, Everett DW, Patten CJ, Dehal SS, Humphreys WG. In vitro inhibition of UDP glucuronosyltransferases by atazanavir and other HIV protease inhibitors and the relationship of this property to in vivo bilirubin glucuronidation. Drug Metab Dispos. 2005;33(11):1729–39. doi: 10.1124/dmd.105.005447. [DOI] [PubMed] [Google Scholar]
- 79.Atazanivir Package Insert. http://www.fda.gov/cder/foi/label/2006/021567s009lbl.pdf.
- 80.Reyataz Package Insert. http://www.fda.gov/cder/foi/label/2006/021567s009lbl.pdf.
- 81.Rescriptor Package Insert. http://www.pfizer.com/files/products/uspi_rescriptor.pdf.
- 82.Videx Package Insert. http://packageinserts.bms.com/pi/pi_videx.pdf.
- 83.Hartman NR, Yarchoan R, Pluda JM, Thomas RV, Marczyk KS, Broder S, Johns DG. Pharmacokinetics of 2',3'-dideoxyadenosine and 2',3'-dideoxyinosine in patients with severe human immunodeficiency virus infection. Clin Pharmacol Ther. 1990;47(5):647–54. doi: 10.1038/clpt.1990.86. [DOI] [PubMed] [Google Scholar]
- 84.Mouly S, Lown KS, Kornhauser D, Joseph JL, Fiske WD, Benedek IH, Watkins PB. Hepatic but not intestinal CYP3A4 displays dose-dependent induction by efavirenz in humans. Clin Pharmacol Ther. 2002;72(1):1–9. doi: 10.1067/mcp.2002.124519. [DOI] [PubMed] [Google Scholar]
- 85.Hesse LM, von Moltke LL, Shader RI, Greenblatt DJ. Ritonavir, efavirenz, and nelfinavir inhibit CYP2B6 activity in vitro: potential drug interactions with bupropion. Drug Metab Dispos. 2001;29(2):100–2. [PubMed] [Google Scholar]
- 86.Kwara A, Lartey M, Sagoe KW, Xexemeku F, Kenu E, Oliver-Commey J, Boima V, Sagoe A, Boamah I, Greenblatt DJ, Court MH. Pharmacokinetics of efavirenz when co-administered with rifampin in TB/HIV co-infected patients: pharmacogenetic effect of CYP2B6 variation. J Clin Pharmacol. 2008;48(9):1032–40. doi: 10.1177/0091270008321790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sustiva Package Insert. http://www.fda.gov/cder/foi/label/1998/20972lbl.pdf.
- 88.Koudriakova T, Iatsimirskaia E, Utkin I, Gangl E, Vouros P, Storozhuk E, Orza D, Marinina J, Gerber N. Metabolism of the human immunodeficiency virus protease inhibitors indinavir and ritonavir by human intestinal microsomes and expressed cytochrome P4503A4/3A5: mechanism-based inactivation of cytochrome P4503A by ritonavir. Drug Metab Dispos. 1998;26(6):552–561. [PubMed] [Google Scholar]
- 89.von Moltke LL, Greenblatt DJ, Grassi JM, Granda BW, Duan SX, Fogelman SM, Daily JP, Harmatz JS, Shader RI. Protease inhibitors as inhibitors of human cytochromes P450: high risk associated with ritonavir. J Clin Pharmacol. 1998;38(2):106–11. doi: 10.1002/j.1552-4604.1998.tb04398.x. [DOI] [PubMed] [Google Scholar]
- 90.Crixivan Package Insert. http://www.merck.com/product/usa/pi_circulars/c/crixivan/crixivan_pi.pdf.
- 91.van Leeuwen R, Lange JM, Hussey EK, Donn KH, Hall ST, Harker AJ, Jonker P, Danner SA. The safety and pharmacokinetics of a reverse transcriptase inhibitor, 3TC, in patients with HIV infection: a phase I study. AIDS. 1992;6(12):1471–5. doi: 10.1097/00002030-199212000-00008. [DOI] [PubMed] [Google Scholar]
- 92.Epivir Package Insert. http://us.gsk.com/products/assets/us_epivir.pdf.
- 93.Kumar GN, Jayanti V, Lee RD, Whittern DN, Uchic J, Thomas S, Johnson P, Grabowski B, Sham H, Betebenner D, Kempf DJ, Denissen JF. In vitro metabolism of the HIV-1 protease inhibitor ABT-378: species comparison and metabolite identification. Drug Metab Dispos. 1999;27(1):86–91. [PubMed] [Google Scholar]
- 94.Kumar GN, Dykstra J, Roberts EM, Jayanti VK, Hickman D, Uchic J, Yao Y, Surber B, Thomas S, Granneman GR. Potent inhibition of the cytochrome P-450 3A-mediated human liver microsomal metabolism of a novel HIV protease inhibitor by ritonavir: A positive drug-drug interaction. Drug Metab Dispos. 1999;27(8):902–8. [PubMed] [Google Scholar]
- 95.Kaletra Package Insert. http://www.accessdata.fda.gov/drugsatfda_docs/label/2007/021226s022lbl.pdf.
- 96.Abel S, Russell D, Taylor-Worth RJ, Ridgway CE, Muirhead GJ. Effects of CYP3A4 inhibitors on the pharmacokinetics of maraviroc in healthy volunteers. Br J Clin Pharmacol. 2008;65(Suppl 1):27–37. doi: 10.1111/j.1365-2125.2008.03133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Selzentry Package Insert. http://www.pfizer.com/files/products/uspi_maraviroc.pdf.
- 98.Lillibridge JH, Liang BH, Kerr BM, Webber S, Quart B, Shetty BV, Lee CA. Characterization of the selectivity and mechanism of human cytochrome P450 inhibition by the human immunodeficiency virus-protease inhibitor nelfinavir mesylate. Drug Metab Dispos. 1998;26(7):609–16. [PubMed] [Google Scholar]
- 99.Viracept Package Insert. http://media.pfizer.com/files/products/uspi_viracept.pdf.
- 100.Erickson DA, Mather G, Trager WF, Levy RH, Keirns JJ. Characterization of the in vitro biotransformation of the HIV-1 reverse transcriptase inhibitor nevirapine by human hepatic cytochromes P-450. Drug Metab Dispos. 1999;27(12):1488–95. [PubMed] [Google Scholar]
- 101.Viramune Package Insert. http://bidocs.boehringeringelheim.com/BIWebAccess/ViewServlet.ser?docBase=renetnt&folderPath=/Prescribing+Information/PIs/Viramune/Viramune.pdf.
- 102.Kassahun K, McIntosh I, Cui D, Hreniuk D, Merschman S, Lasseter K, Azrolan N, Iwamoto M, Wagner JA, Wenning LA. Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab Dispos. 2007;35(9):1657–63. doi: 10.1124/dmd.107.016196. [DOI] [PubMed] [Google Scholar]
- 103.Isentress Package Insert. http://www.merck.com/product/usa/pi_circulars/i/isentress/isentress_pi.pdf.
- 104.Iwamoto M, Kassahun K, Troyer MD, Hanley WD, Lu P, Rhoton A, Petry AS, Ghosh K, Mangin E, DeNoia EP, Wenning LA, Stone JA, Gottesdiener KM, Wagner JA. Lack of a pharmacokinetic effect of raltegravir on midazolam: in vitro/in vivo correlation. J Clin Pharmacol. 2008;48(2):209–14. doi: 10.1177/0091270007310382. [DOI] [PubMed] [Google Scholar]
- 105.Kumar GN, Rodrigues AD, Buko AM, Denissen JF. Cytochrome P450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J Pharmacol Exp Ther. 1996;277(1):423–431. [PubMed] [Google Scholar]
- 106.Lim ML, Min SS, Eron JJ, Bertz RJ, Robinson M, Gaedigk A, Kashuba AD. Coadministration of lopinavir/ritonavir and phenytoin results in two-way drug interaction through cytochrome P-450 induction. J Acquir Immune Defic Syndr. 2004;36(5):1034–40. doi: 10.1097/00126334-200408150-00006. [DOI] [PubMed] [Google Scholar]
- 107.Fitzsimmons ME, Collins JM. Selective biotransformation of the human immunodeficiency virus protease inhibitor saquinavir by human small-intestinal cytochrome P4503A4: potential contribution to high first-pass metabolism. Drug Metab Dispos. 1997;25(2):256–66. [PubMed] [Google Scholar]
- 108.Farrar G, Mitchel AM, Hooper H, Stewart F, Malcolm SL. Prediction of potential drug interactions of saquinavir (Ro 31-8959) from in vitro data. Br. J. Clin. Pharmacol. 1994;38:162. [Google Scholar]
- 109.Invirase Package Insert. http://www.rocheusa.com/products/invirase/pi.pdf.
- 110.Zerit Package Insert. http://packageinserts.bms.com/pi/pi_zerit.pdf.
- 111.Matal J, Orság J, Nekvindová J, Anzenbacherová E, Veinlichová A, Anzenbacher P, Zídek Z, Holý A. Experimental approaches to studies on drug metabolism and drug interactions in man: interaction of acyclic nucleoside phosphonates with human liver cytochromes P450 and flavin-containing monooxygenase 3. Neuro Endocrinol Lett. 2006;(Suppl 2):27–30. [PubMed] [Google Scholar]
- 112.Viread Package Insert. http://www.gilead.com/pdf/viread_pi.pdf.
- 113.Vourvahis M, Kashuba AD. Mechanisms of pharmacokinetic and pharmacodynamic drug interactions associated with ritonavir-enhanced tipranavir. Pharmacotherapy. 2007;27(6):888–909. doi: 10.1592/phco.27.6.888. [DOI] [PubMed] [Google Scholar]
- 114.Aptivus Package Insert. http://www.fda.gov/cder/foi/label/2005/021814lbl.pdf.
- 115.Hivid Package Insert. http://www.rocheusa.com/products/hivid/pi.pdf.
- 116.Mano Y, Usui T, Kamimura H. Comparison of inhibition potentials of drugs against zidovudine glucuronidation in rat hepatocytes and liver microsomes. Drug Metab Dispos. 2007;35(4):602–6. doi: 10.1124/dmd.106.014225. [DOI] [PubMed] [Google Scholar]
- 117.Cretton EM, Waterhous DV, Bevan R, Sommadossi JP. Glucuronidation of 3'-azido-3'-deoxythymidine by rat and human liver microsomes. Drug Metab Dispos. 1990;18:369–372. [PubMed] [Google Scholar]
- 118.Retrovir Package Insert. http://us.gsk.com/products/assets/us_retrovir.pdf.