

Abstract
Background
Enterococcus faecalis is one of the leading causes of nosocomial infections. Due to its innate and acquired resistance to most antibiotics, identification of new targets for antimicrobial treatment of E. faecalis is a high priority. The multiple peptide resistance factor MprF, which was first described in Staphylococcus aureus, modifies phosphatidylglycerol with lysin and reduces the negative charge of the membrane, thus increasing resistance to cationic antimicrobial peptides. We studied the effect of mprF in E. faecalis regarding influence on bacterial physiology and virulence.
Results
Two putative mprF paralogs (mprF1 and mprF2) were identified in E. faecalis by BLAST search using the well-described S. aureus gene as a lead. Two deletion mutants in E. faecalis 12030 were created by homologous recombination. Analysis of both mutants by thin-layer chromatography showed that inactivation of mprF2 abolishes the synthesis of three distinct amino-phosphatidylglycerols (PGs). In contrast, deletion of mprF1 did not interfere with the biosynthesis of amino-PG. Inactivation of mprF2 increased susceptibility against several antimicrobial peptides and resulted in a 42% increased biofilm formation compared to wild-type mprF. However, resistance to opsonic killing was increased in the mutant, while virulence in a mouse bacteremia model was unchanged.
Conclusion
Our data suggest that only mprF2 is involved in the aminoacylation of PG in enterococci, and is probably responsible for synthesis of Lys-PG, Ala-PG, and Arg-PG, while mprF1 does not seem to have a role in aminoacylation. As in other Gram-positive pathogens, aminoacylation through MprF2 increases resistance against cationic antimicrobial peptides. Unlike mprF found in other bacteria, mprF2 does not seem to be a major virulence factor in enterococci.
Introduction
Enterococcus faecalis is part of the normal flora in the gastro-intestinal tract of humans and animals. Some strains have been used as probiotics, whereas others are the cause of serious and sometimes life-threatening infections [1], [2]. Due to its innate and acquired resistance to most clinically used antibiotics, treatment of serious infections by enterococci is often limited in effect and sometimes impossible [3].
The main lipid constituents of bacterial membranes are phospholipids. The two major bacterial phospholipids are phosphatidylglycerol (PG) and diphosphatidylglycerol (DPG). Their head groups are negatively charged, thereby imparting anionic properties to the membrane surface. Many bacteria can modify negatively charged lipids with positively charged substituents, such as lysine, to form lysyl-phosphatidylglycerol (Lys-PG), reducing the negative net charge of the membrane surface. Lys-PG is synthesized by the integral membrane protein MprF, which transfers a lysyl group from lysyl-tRNA to PG and subsequently translocates Lys-PG from the inner to the outer leaflet of the cytoplasmic membrane [4]. The reduced negative net charge of the cell membrane leads to the repulsion of cationic peptides, decreasing the sensitivity against these peptides [5]. In addition, this mechanism also bestows S. aureus with resistance to cationic antibiotics such as daptomycin, vancomycin [6], and gentamicin [7]. Furthermore, MprF is also considered a virulence factor, since it allows bacteria to evade neutrophil killing and enhances the virulence of S. aureus in mice [8].
In addition to S. aureus, the mprF gene is also present in the genomes of several other clinically important pathogens, such as Mycobacterium tuberculosis, Pseudomonas aeroginosa, Listeria monocytogenes, and E. faecalis [9]. Two putative mprF genes were found in E. faecalis, Enterococcus faecium, and several other gram-positive species [10].
In the present study, we used targeted mutagenesis to inactivate the two mprF paralogs in E. faecalis to characterize the resulting changes in cell wall lipids and to investigate the contribution of phosphatidylglycerol aminoacylation to resistance, biofilm formation, and virulence.
Results
Identification and Sequence Analysis of Two mprF Paralogs in E. faecalis
Blast-search analysis of the genome sequences of E. faecalis V583 [11] and E. faecalis 12030 (unpublished results) identified genes with significant homology in these two organisms: EF_0031 and EF_1027, sharing 24% and 31% amino-acid identity (respectively) with the mprF gene of S. aureus (accession number ADJ67256.1). According to homologies with genes characterized by Roy and Ibba, we provisionally named these two genes mprF1 (EF_0031) and mprF2 (EF_1027) [9], [10]. The MprF protein of S. aureus consists of two functional domains [4]. The hydrophilic C-terminus demonstrates aminoacyl phosphatidylglycerol (PG)-synthase activity, and the hydrophobic N-terminus functions as a flippase, transferring aminoacyl-PG from the inner to the outer leaflet of the cell membrane. The homology of the two domains in S. aureus with the two mprF genes in E. faecalis 12030 was assessed: The N-terminal part (corresponding to the flippase) of mprF1 and mprF2 shows 24% and 31% identity with S. aureus, respectively. The C-terminus (synthase) of mprF1 and mprF2 demonstrates 29% and 40% identity (respectively) with the synthase of the S. aureus gene.
Growth Kinetics
The mutants 12030ΔmprF1, 12030ΔmprF2, and the respective complemented strains were no different than the wild-type (WT) regarding growth kinetics in broth culture (data not shown).
Only Deletion of mprF2 Leads to Complete Loss of Amino-phospholipids
To confirm whether these two putative enterococcal mprF genes function similarly to the mprF of S. aureus, we compared the membrane lipid composition of the wild-type and mutant strains by two-dimensional thin-layer chromatography (2D-TLC). Four prominent ninhydrin-positive spots present in the wild-type were completely absent from the mprF2 mutant, but reappeared upon complementation by knocking-in of the wild-type allele into the chromosome of the mutant, and by expression of mprF2 in trans on plasmid pMSP3535::mprF2 (see Figure 1). All spots mentioned above (A–D) were stained with molybdenum blue and ninhydrin, indicating that they represent amino-phospholipids. In E. faecalis, PG is usually acylated by two molecules of Lysin [12], [13]. Spots A and B migrate similarly to the two Lys-PG spots identified by Peschel and colleagues [8]. Spots C and D migrate similarly to Arg-PG [10] and Ala-PG [9], respectively. Deletion of mprF1 did not seem to have an effect on amino-phospholipids, because in the deletion mutant 12030ΔmprF1, all spots were identical to the wild type.
Figure 1. Lipid analysis of the wild-type and its mutants by two dimensional thin-layer chromatography.

Cell membrane total lipid extracts from E. faecalis 12030 (wild type), 12030ΔmprF1 (EF_0031), 12030ΔmprF2 (EF_1027), 12030ΔmprF2 CM (knock-in complementation), 12030ΔmprF2 p3535::mprF2. Lipids were separated using a solvent system of CHCl3/MeOH/H2O(65∶15:2, v/v/v) in the first dimension, and CHCl3/MeOH/Acetic acid/H2O (80/12/15/4, v/v/v) in the second dimension. Aminophospholipids were visualized with molybdenum stain solution, 12030 (wild-type) was also stained with ninhydrin. PG – phosphatidylgylcerol, DPG – diphosphaditylglyerol, DGlcDAG – diglycosyldiacylglycerol. MGlcDAG – monoglycosyldiacylglycerol.
mprF2 is Involved in Resistance Against Antimicrobial Peptides
The absence of Lys-PG from the membrane of S. aureus decreased the minimal inhibitory concentration (MIC) of certain cationic antimicrobial peptides (CAMP) [8]. The cationic antimicrobial peptides (CAMP) colistin, nisin, HBD-3, and polymyxin B were tested against the parental strain E. faecalis 12030 and its isogenic mutants. As shown in Table 1, 12030ΔmprF2 showed a 2-fold decreased MIC against colistin, a 4-fold decreased MIC against polymyxin B and nisin, and a >4-fold decreased MIC against HBD-3 compared to the wild-type strain. Complementation of mprF2 (12030ΔmprF2 knock-in) completely restored the wild-type phenotype. In contrast, no difference in the sensitivity of the deletion mutant 12030ΔmprF1 and the tested CAMPs was noted. E-test showed that the E. faecalis wild-type strain and mutants tested in this study were not significantly different in their sensitivity to daptomycin. The MICs of E. faecalis 12030, 12030ΔmprF1, 12030ΔmprF2, and 12030ΔmprF2 CM (knock-in), were 0.19 mg/l, 0.25 mg/l, 0.25 mg/l, and 0.25 mg/l, respectively.
Table 1. Activities of cationic antimicrobial peptides against the E. faecalis 12030 wild type, the deletion mutants and the complemented strain.
| Strains | Minimal inhibotory concentration (µg/ml) | |||
| Colistin | Polymyxin B | Nisin | HBD-3 | |
| E. faecalis 12030 | 4096 | 1024 | 4 | >512 |
| E. faecalis 12030 ΔmprF1 | 4096 | 1024 | 4 | >512 |
| E. faecalis 12030 ΔmprF2 | 2048 | 256 | 1 | 128 |
| E. faecalis 12030 ΔmprF2 CM | 4096 | 1024 | 4 | >512 |
MprF2 is Involved in Biofilm Formation and eDNA Release
We showed previously that lack of D-alanine esters on teichoic acids leads to a decrease in biofilm formation, probably due to the increase in net charge of the bacterial cell surface [14]. Inactivation of MprF2 is predicted to increase the net negative charge on bacterial cells. We compared biofilm formation on polystyrene surfaces by the wild-type strain 12030 and its isogenic mutants, 12030ΔmprF2 CM (knock-in) and 12030ΔmprF2 p3535::mprF2. Surprisingly, the mutant 12030ΔmprF2 produced significantly more biofilm than the wild type. Complementation by knock-in of mprF2 (12030ΔmprF2 CM knock-in) decreased the production of biofilm below wild-type levels, while biofilm production after complementation with a plasmid in trans (without induction by nisin) did not differ from the wild-type strain. The deletion mutant 12030ΔmprF1 and the wild type produced similar amounts of biofilm (Figure 2). As shown in Figure 3 mutant 12030ΔmprF2 released significantly more eDNA than the wild type and the mutant 12030ΔmprF1. The strain complemented by knock-in (12030ΔmprF2 CM) produced significantly less eDNA biofilm than the wild type, while the mutant complemented by plasmid partially restored eDNA levels compared to the wild type.
Figure 2. Biofilm production of the wild type and its derivate mutants.
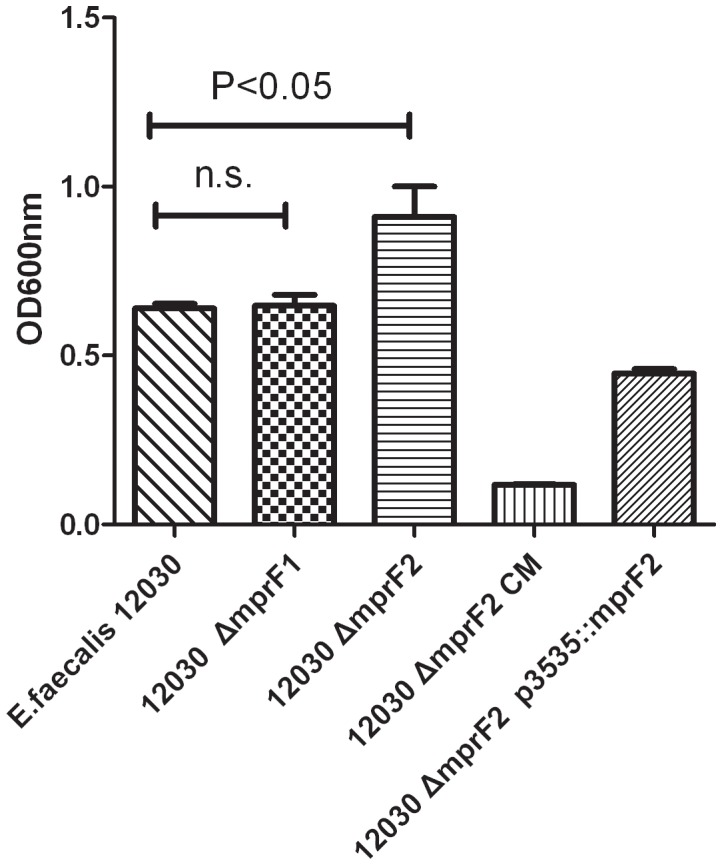
E. faecalis 12030, mutants 12030ΔmprF21 and 12030ΔmprF2 and complemented strains 12030ΔmprF2 CM and 12030ΔmprF2 P3535::mprF2 were cultivated in TSB media supplemented with 1% glucose. Bacteria were incubated for 18 h, unbound cells were removed by washing of the plates with buffer, and biofilm was stained with crystal violet. Error bars represent standard error of the mean.
Figure 3. Measurement of eDNA in biofilms.
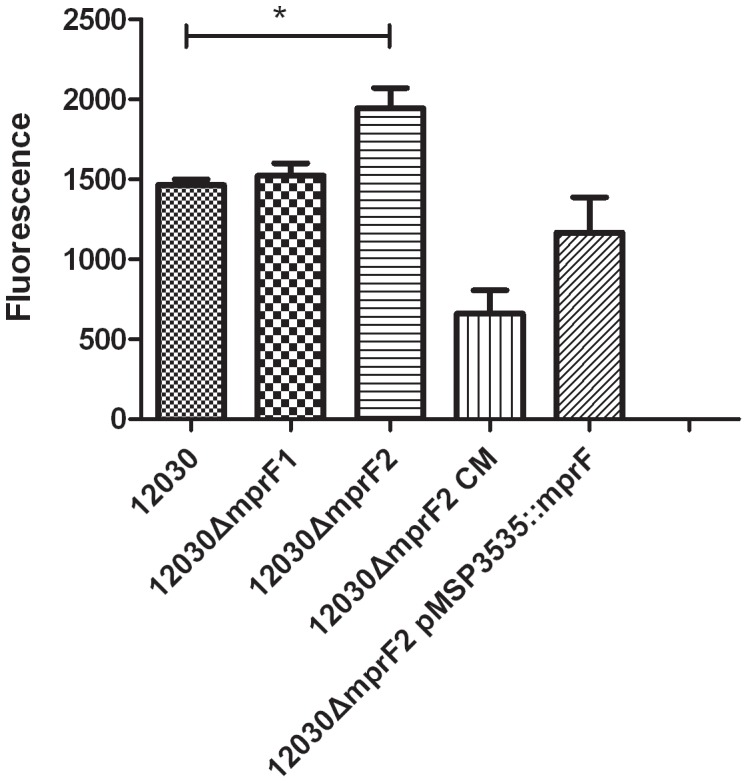
All strains (12030, 12030ΔmprF1, 12030ΔmprF2, 12030ΔmprF2 CM, 12030ΔmprF2 pMSP3535::mprF2) were cultivated overnight in TSB at 37°C and measured with excitation wavelength at 485 nm and emission wavelength at 535 nm. *indicates statistical significance at p<0.05.
Triton X-100-induced Autolysis
Mechanisms affecting the modification of the membrane net charge of the peptidoglycan structure may play a role in the modulation of autolysin activity and thus may have an impact on bacterial autolysis. Therefore we evaluated the effect of autolysis by Triton-X100 on E. faecalis 12030ΔmprF2 and the wild-type strain. No significant difference was found between them (data not shown), suggesting that mprF2 has no obvious effect on autolysis in E. faecalis.
The mprF2 Mutant is More Resistant to Opsonophagocytic Killing than the Wild-type Strain
Opsonophagocytic killing of E. faecalis 12030 wild-type and mutant 12030ΔmprF2 was compared using log-phase-grown bacteria that were opsonized with rabbit complement in conjunction with antibodies against E. faecalis LTA [15]. Subsequently, numbers of surviving bacteria were determined. The mprF2 mutant was killed significantly less than the wild type, and complementation by knock-in partially restored the killing to wild-type levels (Fig. 4). Opsonophagocytic killing in the presence of PMNs and complement alone did not differ between 12030ΔmprF2 and the wild-type strain (data not shown).
Figure 4. Effect of the deletion of MprF2 on resistance to opsonophagocytosis.
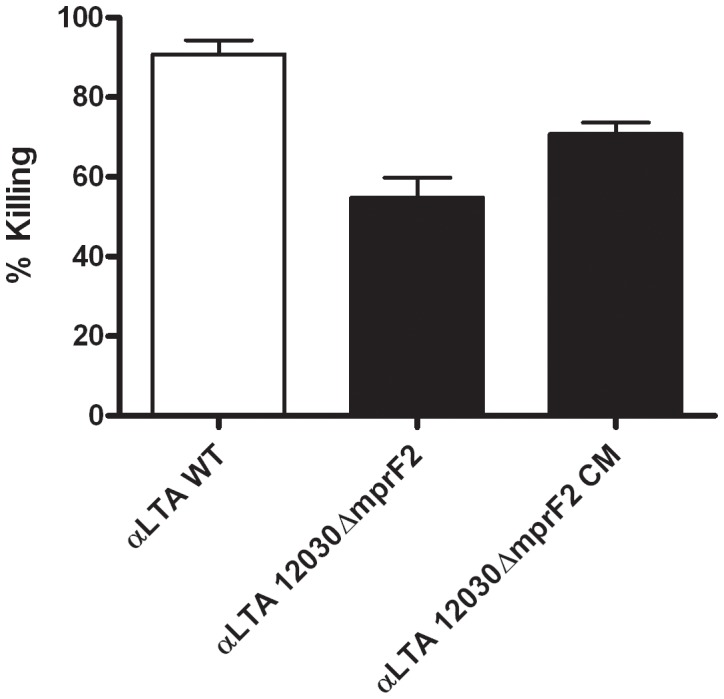
Opsonophagocytic killing of the wild type (12030), 12030ΔmprF2 and 12030ΔmprF2 CM with serum against anti-LTA (serum-dilution of 1∶1,200).
Mouse Bacteremia Model
Virulence of the mprF2 mutant was assessed in a mouse bacteremia model as described previously [16]. The number of bacteria recovered from the liver, kidney, and spleen of mice infected with the mprF2 mutant was not significantly different from those recovered from animals infected with wild-type (Figure 5).
Figure 5. Effect of the deletion of the MprF2 on virulence in mice.

1.5×108 CFU bacteria were injected in the tail vein of mice. Animals were sacrificed after 48 h, and colony counts were enumerated in liver, kidney and spleen.
Discussion
Bacteria modulate the electrostatic properties of their cell envelope to protect themselves against the innate defense systems of the host, especially antimicrobial peptides [5]. Two different mechanisms that reduce the negative net charge of the bacterial cell wall have been identified. The first is modification of teichoic acid with D-alanine through the dlt operon (dltABCD) [17]. Inactivation of genes within this operon causes complete absence or reduction of teichoic acid D-alanine esters. This results in a higher negative net charge on the bacterial surface, because D-alanine esters neutralize the negative charge of teichoic acids [14]. The second charge-reducing mechanism is modification of membrane lipid phosphatidylglycerol (PG) with positively or neutrally charged amino-groups by mprF(multiple peptides resistance factor) [8], sometimes also described as aminoacylphosphatidylglycerol synthases (aaPGSs) [10].
The earliest report described the modification of PG with lysine in S. aureus [8]. However, It has long been known that certain bacteria produce Ala-PG (e.g., Pseudomonas aeroginosa [18]) instead of Lys-PG, while others produce both Lys-PG and Ala-PG (e.g., Clostridium perfringens, Bacillus subtilis, and Enterococcus faecium; Roy & Ibba 2009 [19]). Although Ala-PG has a neutral net charge, this modification has been demonstrated to increase bacterial resistance to certain CAMPs [18]. These observations suggest that mprF-mediated CAMPs resistance not only decreases the charge of the membrane but probably also modulates some biophysical properties of the membrane, such as fluidity and permeability [20].
The presence of two putative mprF paralogs in E. faecium DO was previously reported by Roy and Ibba [9], [10], and we confirmed this finding using translated BLAST (tblastn) of the well-characterized mprF gene of S. aureus against the genome of E. faecalis 12030 [21] and V583 [11]. Two genes were identified, with one (EF_1027) showing a higher homology than the other (EF_0031). Comparing these two putative mprF genes with the two aaPGs described by Roy and Ibba [10] showed that mprF1 (EF_0031) and mprF2 (EF_1027) share 57% and 62% identities of amino acids with the sequences of aaPGs1 and aaPGs2, respectively.
Lipid analysis of the deletion mutants in genes mprF1 and mprF2 in E. faecalis 12030 indicated that mutant ΔmprF2 lacks aminoacyl phosphatidylglycerol, whereas no difference in lipid composition was seen between the mutant ΔmprF1 and the wild type. To confirm these findings, we analyzed the composition of phospholipids of insertion mutants of mprF1 and mprF2 in a second strain, E. faecalis V583. Insertional inactivation of these genes in E. faecalis V583 produced the same phenotype and aminophospholipid patterns as noted for E. faecalis 12030 (data not shown). Therefore, we assume that mprF1 (EF_0031) is not involved in the aminoacylatylation of PG, while mprF2 (EF_1027) seems to be the only functional mprF gene in E. faecalis.
Roy and Ibba studied the aminoacylation of recombinantly expressed MprF1 and MprF2 from Clostridium perfringens in E. coli. They found that MprF1 was specific for Ala-PG, while only membrane extracts of E. coli expressing MprF2 were able to aminoacylate PG with Lys [9]. They also found that aaPGS1 from E. faecium DO (corresponding to MprF1) cannot use Lys-tRNALys and Arg-tRNA Arg as aminoacyl group donors; however, results for Ala-PG were not reported. Using aaPGS2 of E. faecium and Lys-tRNALys, Ala-tRNAAla, and Arg-tRNA Arg as donors, the final products Lys-PG, Ala-PG, and Arg-PG could be isolated from the of E. coli membranes [10].
Similar to previous reports [4], in E. faecalis 12030 we found that deletion of both mprF genes has no effect on growth (data not shown). Also similar to previously published results in S. aureus and other species, we found that the mutant 12030ΔmprF2 (EF_1027) displayed a decrease in the MIC against nisin, colistin, polymyxin B, and HBD-3. Inactivation of MprF2 in E. faecalis had a lesser effect on the MIC of nisin (4-fold difference) than in S. aureus, where mutation of MprF resulted in a 28-fold drop of the MIC for this CAMP [8]. In contrast to S. aureus, however, there was no effect on daptomycin resistance. Interestingly, two recent reports identified other mutations in the synthetic pathway of phospholipids (i.e., diphosphatidylglycerol synthesis) being involved in daptomycin resistance of enterococci [22], [23], and this mechanism may be more important for resistance against the above-mentioned antimicrobial peptides. Our results suggest that Lys-PG, Ala-PG, and Arg-PG are probably not major factors involved in resistance to colistin, polymyxin B, nisin, or HBD-3 in enterococci. A Bacillus anthracis mprF mutant showed hyper-susceptibility to certain CAMPs (e.g., protamine, HNP-1, and LL-37) but exhibited only weak or no change in resistance to nisin [24], suggesting that MprF may be an important resistance mechanism for some CAMPs, but not for others. In S. aureus it has been reported that, depending on the individual strain, MprF may increase [25] or decrease [6] resistance to vancomycin, or may have no effect [26], with the respective phenotype probably being dependent on the genetic background of the isolate [6]. A great variety of additional resistance mechanisms and regulators are used by bacteria to circumvent the action of CAMPs [27]–[29].
It has been reported that in certain species (such as Bacillus subtilis, Lactococcus lactis, and Streptococcus pyogenes) the dlt operon affects autolysis by incorporation of D-Ala into lipoteichoic acids. Decreasing the net charge of the cell membrane through reducing the amount of alanyl-ester leads to increased binding of autolysins and ultimately increases autolysis [27]–[29]. Point mutations in the mprF gene were found in S. aureus strains that were resistant to daptomycin and defective in autolysis [30]. However, the mprF2 mutant in E. faecalis 12030 did not show increased autolysis compared with the wild-type (data not shown), indicating that the aminoacylation of PG has no effect on autolysis in enterococcus.
Formation of biofilm is frequently associated with virulence [31] and poses a clinical challenge, especially in foreign body infections [32]. Although E. faecalis 12030 is already a strong biofilm producer, the mprF2 deletion increased biofilm formation about 42% compared to the wild type. This may be caused by pleiotropic or compensatory effects, or by down-regulation of specific biofilm regulators [33], but the exact mechanism has not yet been elucidated. In the complemented mutant, gene mprF2 was sequenced and was found to contain 3 amino acids changes compared to the wild-type, which may be the reason that the complementation of 12030ΔmprF2 cannot completely restore the phenotype to the level of the wild type. That the complementation of ΔmprF2 with vector pMSP3535::mprF2 did only partly restore the lipid content of the mutant compared to the wild type level may be explained by the fact that expression of the gene could not be induced. Vector pMSP3535 contains a strong nisin promoter, which is capable to over-express the gene cloned into the vector when nisin is added. However, nisin is also a cationic antimicrobial peptide that influences by itself the lipid composition of cell membrane (data not shown). Therefore, nisin was not added for the biofilm formation assay leading to only base-line expression of the gene cloned into pMSP3535 (i.e mprF2). This could explain the lower lipid contents extracted from 12030ΔmprF2 pMSP3535::mprF2 compared to the wild-type.
Extracellular DNA was measured in biofilms to assess whether the increased biofilm formation in the 12030ΔmprF2 mutant is caused by eDNA, because it has been previously observed that eDNA may serve as an important matrix component of microbial biofilms. For the major autolysins (AtlE of Staphylococcus epidermidis and muramidase 2 of E. faecalis) several authors confirmed a role in biofilm formation [34]–[37] and Qin et al [34] described eDNA as an integral component during biofilm formation. The measurement of eDNA release confirmed our biofilm results (see Figure 3), i.e. the 12030ΔmprF2 mutant showed increased biofilm production and increased eDNA release. While eDNA has been shown previously to be primarily a by-product of cell lysis, the 12030ΔmprF2 mutant was not significantly different regarding autolysis compared to the wild type in our experiments. This suggests that in Enterococcus, similar to the results by Grande [38], accumulation of eDNA may be not caused only by autolysis but also by other, so far unknown mechanisms.
Although biofilm formation is usually considered a virulence factor, there was no effect seen on the pathogenicity of the isogenic mutant compared to the wild type in a mouse bacteremia model. The increased adherence of the mutant to polystyren may rely on different mechanism and therefore does probably not correspond to an increased adherence to eukaryotic cells in the host. Alternatively, compensatory mechanisms (such as the increased susceptibility of the mutant against antimicrobial peptides) may counteract the increased adherence in vivo. Comparing an mprF mutant and a dltA mutant in a rabbit endocarditis model, Weidenmeier and colleagues observed that the reduction in virulence of the dltA mutation was more pronounced than the mprF mutation [39]. Using a mouse bacteremia model, we could detect a significant reduction in virulence in the dltA mutant [14] but were not able to observe this effect in the mprF mutant. We therefore conclude that, contrary to the observations in S. aureus [8], [40], Listeria monocytogenes [41], and Mycobacterium tuberculosis [42], MprF is probably not a major virulence factor in E. faecalis.
Previous studies have shown that a mprF mutant of S. aureus is killed more readily by neutrophils due to increased susceptibility of phagocytosed bacteria to defensins like HNP1-3 or cathelicidin LL-37 that are stored in the azurophilic granules [8], [40]. Our results are not comparable to these studies, because enterococcus is not killed by complement and phagocytes alone [43]. Only the addition of opsonic antibodies, e.g., those against LTA [15] or capsular polysaccharides [44], results in effective killing. The thick layer of polysaccharide material in enterococci [45] may confer additional resistance against neutrophil killing and probably diminishes the specific effects of MprF. Our findings suggest that bactericidal mechanisms other than CAMPs are more important in the killing of enterococci by neutrophils. Streptococcus pneumoniae, for example, is killed even by neutrophils that lack HNP1-3, suggesting a minor role of defensins in the killing of this gram-positive pathogen. Instead, the neutrophil serine proteases elastase, cathepsin G, and proteinase 3 are critical for the intracellular killing of S. pneumoniae by neutrophils [46]. Hence, nonoxidative mechanisms such as serine proteases or reactive oxidative species may be of greater importance than CAMPs in the killing of E. faecalis by neutrophils.
We investigated the role of mprF2 as a virulence factor in vivo in a mouse bacteremia model. Our results suggest that the expression of aminoacyl-PG does not affect bacterial survival during bloodstream infection. In contrast, impaired aminoacylation of phosphatidylglyercerol in S. aureus increases intracellular killing by neutrophils, associated with a reduced bacterial burden during bacteremia and endocarditis [8], [39]. In bloodstream infections, neutrophils are the first line of defense against invading pathogens, and it is therefore not surprising that MprF is an important virulence factor in this model. Since opsonophagocytic killing was not increased in the E. faecalis mprF2 mutant, it seems plausible that virulence was also not altered during bloodstream infection. However, we cannot exclude the possibility that inactivation of mprF2 impairs virulence in other modes of infection, e.g., during colonization or biofilm infection. For example, skin expression of the antimicrobial protein RNase 7 has an important role in the protection of human skin against E. faecium colonization [47].The function of mprF1 is not clear from our results or from the data presented by Roy and Ibba [9], [10]; there seems to be no obvious effect on the cell wall lipids, and the 12030ΔmprF1 mutant has been tested also by OPA, in the mouse sepsis and in the autolysis assay. However, in none of these tests there was a statistically significant difference between this mutant and the wild-type (data not shown). Whether this protein functions as a sensor or regulator for the expression of mprF2 must be the subject of future studies.
Analysis of insertional mutants in genes mprF1 (ef0031) and mprF2 (ef1027) in a plasmid-cured Enterococcus faecalis V583-derivative strain (VE14089) has been reported by Rigottier-Gois et al. [43]. This study showed that there was no difference in growth kinetics and resistance to antibiotics for the single cross-over mutants SCO ef0031 and SCO ef1027 The SCO ef1027 mutant was killed by PMNs and complement without the addition of serum. In contrast, we observed that E. faecalis 12030 ΔmprF2 was efficiently killed only by a combination of PMNs with complement and specific antibody. Furthermore, the SCO ef1027 mutant in E. faecalis VE14089 showed decreased virulence in a Galleria mellonella model while our ΔmprF2 mutant in E. faecalis 12030 was not significantly affected in virulence in a mouse bacteremia model. While the reasons for these differences have not been studied yet, the different virulence model systems and the different strain background may explain these contrasting results.
In conclusion, our data demonstrate that mprF2 (EF_1027) seems to be the only functional aminoacyl-phosphatidylglycerol synthase in E. faecalis in the conditions tested by us. It is responsible for synthesis of three distinct amino-PGs, most likely Lys-PG, Ala-PG, and Arg-PG. MprF2 is involved in resistance against CAMPs but is unnecessary for autolysis, killing by neutrophils, or bacteremia in mice.
Materials and Methods
Bacterial Strains, Plasmids, and Growth Conditions
The bacterial strains and plasmids used are listed in Table 2. E. faecalis strain 12030 was grown in 37°C tryptic soy broth (TSB; CASO broth; Merck) or on tryptic soy agar plates (TSA; CASO agar; Merck). When required, erythromycin (100 or 150 µg/ml) or kanamycin (1000 µg/ml) were added. Escherichia coli strains were cultured under vigorous shaking at 37°C in Luria-Bertani broth (LB; Merck) with ampicillin (100 µg/ml), kanamycin (50 µg/ml), or erythromycin (100 µg/ml) when required. All antibiotics were purchased from Sigma Chemicals.
Table 2. Enterococcal strains and plasmids used in this study.
| Strain or plasmid | Characterization | Reference or source |
| Strains | ||
| E.faecalis V583 | Reference strain, fully sequenced | [11] |
| E.faecalis 12030 | Clinical isolate | [21] |
| E.faecalis 12030ΔmprF1 | mprF1 (EF_0031) mutant | This study |
| E.faecalis 12303ΔmprF2 | mprF2 (EF_1027) mutant | This study |
| E.faecalis 12030ΔmprF2 CM | Strain complemented with knock in of mprF2 gene | This study |
| E.faecalis 12030ΔmprF2/pMSP3535::mprF2 | Strain complemented with mprF2 gene by plasmid pMSP | This study |
| E.coli TOP10 F | Gram-negative cloning host | Invitrogen |
| Plsmids | ||
| pCASPER | Gram-positive, temp-sensitive mutagenesis vector | [57] |
| pMAD | Gram-positive, temp-sensitive mutagenesis vector | [51] |
| pMAD - ΔmprF1 | pMAD carrying mprF1 deleted | This study |
| pCASPER - ΔmprF2 | pCAPER carrying mprF2 deleted | This study |
| pMSP3535 | Emr, pAMb1 and ColE1 replicons, nisRK, PnisA | [54] |
| pMSP3535::mprF2 | Expression vector carrying the mprF2 gene | This study |
| pCRII-TOPO | Gram-negative cloning vector | Invitrogen |
General Molecular Techniques
Chromosomal DNA from enterococci was prepared using the DNeasy Tissue kit (Qiagen) according to the manufacturer’s instructions. Plasmids were purified using the Wizard Plus SV Miniprep System (Promega). PCR was carried out in a reaction volume of 25 µl with about 100 ng of chromosomal DNA of E. faecalis 12030 and Platinum Taq DNA polymerase (Invitrogen); the annealing temperature depended on the calculated melting temperature of primers. In general, 30 cycles were performed, and PCR products were subsequently purified using the QIAquick PCR purification Kit (Qiagen Hilden, Germany). Primers used for this study are listed in Table 3. Custom primers were manufactured by Invitrogen and Sigma. Restriction and modifying enzymes were obtained from New England Biolabs and Fermentas. Electrocompetent enterococci were prepared as described previously [48]. All the other methods (DNA ligation, eletrophoresis, and transformation of competent E. coli) used standard techniques [49].
Table 3. Primers used in this study.
| No. | Name | Sequence (5′–3′) a |
| 1 | EF0031DMF EcoRI | CTGTCGAATTCCATCAGCGCTTAGGAATAATTG |
| 2 | EF0031DMR SmaI | CTGTCCCCGGGCAACATAACGTAGCCAAAGAG |
| 3 | EF0031 inside 1 | CAATAATTTAACGACTACATAGTC |
| 4 | EF0031 inside 2 | GTCACTAGTTGGCAACCAC |
| 5 | EF1027 delF | CAGCAATTGGGTTTCTTTGAA |
| 6 | EF1027 delR | TTTGATGAGATTCCGCTATGG |
| 7 | EF1027 OEL | ACTAGCGCGGCCGCTTGCTCC CCAAGTTGGTGAGTTTCCAGA |
| 8 | EF1027 OER | GGAGCAAGCGGCCGCGCTAGT AGCAATCCCAATAATCGAAGC |
| 9 | pMSP1027 BamHI | CCTGTCGGATCCGGAAATGAAGGTGTCTAAATGAA |
| 10 | pMSP1027 PstI | CCTGTCCTGCAGAATTGAGCTTCTTTTTGTTAGTC |
Linkers are underlined.
Construction of Deletion Mutants Delta mprF1 and mprF2
A non-polar deletion mutant 12030ΔmprF1 was constructed. A part of gene mprF1 (EF_0031 in E. faecalis V583; GenBank accession no. NP_813841) was deleted from aminoacid 84 to 817, i.e. a total of 733 aa was deleted using the method described by Le Jeune et al. [50] with the following modification. Briefly, two fragments of approximately 900 bp, corresponding to the flanking regions of the target gene, were amplified by PCR using primers shown in Table 3. The DNA fragments were purified, digested with restriction enzymes, and ligated into vector pMAD [51] (Table 2).
A deletion mutant of mprF2 (EF_1027 in the E. faecalis V583 genome) was created using the method described by Cieslewicz et al. [52]. Briefly, two fragments of approximately 600 bp, corresponding to the flanking regions of the target gene, were amplified by PCR using primers shown in Table 3. This resulted in the deletion of a fragment from nucleotide position 738 to 1538, i.e. 800 bp were deleted. The resulting fragments were cloned into the gram-negative cloning vector pCRII-TOPO (Invitrogen) and excised with restriction enzyme EcoRI.
The mprF2 fragment was inserted into the gram-positive vector pCASPER (Table 2), which contains a temperature-sensitive origin of replication [53].
The ligation mixtures of the two plasmids pMAD::ΔmprF1 and pCASPER::ΔmprF2 were transformed by electroporation into E. coli Top10F cells. After selection and verification, the generated recombinant plasmids were used to transform electro-competent E. faecalis 12030 cells [14], and gene replacement was performed via double cross-over as described previously [14], [50].
Complementation of the Deletion Mutants
The entire mprF2 with flanking regions was amplified using primer-pairs 5/6 (Table 3). The PCR product was cloned into pMAD, and the resulting plasmid pMAD::mprF2 was transformed into E. faecalis 12030ΔmprF2 by electroporation. Double cross-over and selection of mutants was performed subsequently as described above (knock-in mutant).
The mprF2 gene was also cloned into expression vector pMSP3535 (Table 2) [54] using primers 9 and 10 to amplify the entire gene mprF2 including the RBS, start codon (ATG), and stop codon (TGA). The PCR product and plasmid pMSP3535 were digested with restriction enzyme BamHI and PstI and ligated using T4 DNA ligase. The chimeric plasmid pMSP3535::mprF2 was transformed in E. coli Top10, and correct inserts were confirmed by PCR. Plasmid pMSP3535::mprF2 was extracted from E. coli and transformed into E. faecalis 12030ΔmprF2 by electroporation; transformants were selected on TSA plates containing erythromycin.
Growth Kinetics
Growth curves of the wild-type E. faecalis 12030, its isogenic derivative mutants, and the complemented mutants were compared in TSB. An overnight culture was diluted 1∶50 and incubated at 37°C, while the OD600 was measured every hour.
Membrane Lipid Extraction
Lipids were extracted from E. faecalis 12030, its isogenic derivative mutants, as well as the complemented mutants using a modified Bligh-Dyer method [55]. For isolation of these strains’ membrane lipids, 500 ml overnight TSB culture grown at 37°C was used. Cultures were cooled on ice for 30 to 60 min, and bacteria were collected by centrifugation and washed with 0.1 M citrate buffer (pH 4.7). The bacterial suspension was mixed with an equal volume of glass beads, and bacterial cells were lysed with a bead beater (Biospec Products, Inc.) by vigorous shaking for 2 min three times at 4°C. Bacteria were cooled on ice between each run for 5 min. Glass beads were sedimented by centrifugation for 1 min at 200×g, and bacterial cell membranes were removed with the supernatant. The remaining bacterial debris was again sedimented by centrifugation at 12,000×g for 20 min. The pellets were washed with 40 ml of 0.1 M citrate buffer (pH 4.7), the wet weight was determined, and samples were stored frozen at –20°C. For lipid extraction, frozen pellets were resuspended in 0.1 M citrate buffer (pH 4.7). Chloroform and methanol were added to obtain a final chloroform/methanol/buffer ratio of 2∶1∶0.8. Lipids were extracted for 2 h at room temperature with vigorous vortexing. Insoluble material was removed by centrifugation at 2,600×g for 20 min, and the extracted lipids were transferred with the supernatant into new tubes. The extraction was repeated as described above; chloroform and buffer were added to the combined extracts to obtain a methanol/chloroform/buffer ratio of 1∶1∶0.8. Following vigorous vortexing, samples were centrifuged at 2,600×g for 20 min, and the chloroform phase containing lipids was transferred to a new tube. Lipids were dried under a stream of nitrogen, and the dry weight was determined. Lipids were then resuspended in methanol-chloroform (1∶1) at a concentration of 10 mg/ml and stored at –20°C, and 100-µg samples were analyzed by thin-layer chromatography (TLC).
Lipid Analysis by TLC
Lipids were separated by TLC using silica 60 F254 HPTLC plates (Merck) and developed with chloroform/methanol/water (65∶15∶2, by volume) in the first direction and chloroform/methanol/acetic acid/water (80∶12∶15∶4, by volume) in the second direction. For detection of phospholipids, TLC plates were stained with molybdenum blue, and aminophospholipids were stained with ninhydrin, as previously described [8]. Phosphatidylgylcerol (PG), diphosphaditylglycerol (DPG), diglycosyldiacylglycerol (DGlcDAG), and monoglycosyldiacylglycerol (MGlcDAG) were identified by commercial (purchased from Sigma-Aldrich) or internal laboratory standards [48] and used to determine the position of the PG and DPG spots in 2D-TLC.
Biofilm Assay
Biofilm formation was measured as described by Baldassarri et al. [52]. In brief 180 µl TSB supplemented with 1% glucose in a 96 well tissue culture plate (Brand) were inoculated with 20 µl of a stationary phase culture of the respective strain. Afterwards, the plate was incubated for 18 h at 37°C without shaking. To prevent the cultures from drying, the growth environment was kept humid. Bacterial growth was determined the next day by maesuring the OD of each well in a plate reader at a wavelength of 600 nm. After discarding the growth medium and washing the wells three times with PBS, the biofilm was dried for 1 h at 60°C. Subsequently, the biofilm was stained with 100 µl Huckers Crystall Violet for 2–3 min followed by washing the plate under tap water and drying of the stained biofilm. The OD600 was measured in a plate reader and the biofilm index was calculated as follows: (OD(Biofilm)×0.5)/OD (Growth).
eDNA Assay
Analysis of eDNA was carried out as described previously [36], [37]. All strains were cultivated overnight in TSB at 37°C. The culture was diluted 1∶10 in TSB with 1% glucose, and 200 µl of this cell suspension was used to inoculate a sterile black 96-well FIA- plates (Greiner Bio-one). Each strain was cultivated in triplicate. After 18 h at 37°C, wells were gently washed three times with 200 µl of phosphate-buffered saline (PBS), and dried at 60°C for 1 hour. 100 µl of DNA-specific dye SYTOX green (Invitrogen) was added to these wells at a final concentration of 1 µM in DMSO, and incubated for 10 minutes before being spectrofluorometrically measured with excitation wavelength at 485 nm and emission wavelength at 535 nm.
Triton X-100-induced Autolysis Assays Under Non-growing Conditions
Strains were grown to an OD600 of 0.8 in TSB medium and treated as previously described [56]. Briefly, cells were pelleted by centrifugation (4000 g, 10 min at 4°C), washed in the same volume of ice-cold sterile water, and resuspended in the same volume of 50 mM Tris-HCl, pH 7.5, containing 0.1% Triton X-100. The cell suspensions were then transferred into 96-well sterile microplates and incubated at 37°C without shaking. Autolysis was monitored by measuring OD600 every 10 min with an automated incubator/optical density reader (Model 680, Bio-Rad Laboratories). The results were normalized to the OD600 at time zero, i.e., percent lysis at time t = [(OD at time zero – OD at time t)/OD at time zero]×100.
Antimicrobial Susceptibility to Polymyxin B, Nisin, Colistin, HBD-3, and Daptomycin
The minimal inhibitory concentration (MIC) of polymyxin B, nisin, HBD-3, and colistin was determined in serial dilution with a modified NCCLS method [29]. Experiments were performed in TSB broth in 96-well microtiter tissue culture plates (Greiner). Wells were inoculated with 50-µl volumes of a suspension containing approximately 1×106 CFU/ml of the test organism, and concentrations were confirmed by enumeration of CFUs after serial dilution. Microtiter plates were incubated overnight at 37°C, and the MICs were defined as the lowest concentration at which visible growth was inhibited. The MIC against daptomycin was determined by E-test according to standard laboratory procedures.
Opsonophagocytic Assay
The opsonophagocytic assay was performed as described elsewhere [48] using baby rabbit serum as complement source and rabbit sera raised against purified LTA from E. faecalis 12030. Polymorphonuclear neutrophils (PMN) were freshly prepared from human blood collected from healthy adult volunteers. The institutional review board of the University of Freiburg approved the study protocol, and written informed consent was obtained from all study participants. Bacterial strains were grown to mid-log phase in TSB, adjusted to 2×107/ml, and mixed with equal volumes of serum (dilution 1∶100), absorbed baby rabbit sera as complement source (dilution 1∶15), and PMNs (adjusted to 2×107/ml). Controls included tubes from which PMNs (PMNneg), complement (c’neg), or antibody (Abneg) was omitted. The mixture was incubated on a rotor rack at 37°C for 90 min, and samples were plated in duplicate at time 0 and after 90 min. Percent killing was calculated by comparing the colony counts at 90 min (t90) of a control not containing PMNs (PMNneg) to the colony counts of a tube that contained all four components of the assay using the following formula: {[(mean CFU PMNneg at t90) - (mean CFU at t90)]/(mean CFU PMNneg at t90)}×100.
Animal Studies
The virulence of E. faecalis 12030, its isogenic derivative mutants, and the complemented mutants was evaluated in a mouse bacteremia model [48]. In summary, eight female BALB/c mice 6–8 weeks old were challenged by i.v. injection with 1.5×108 cfu of E. faecalis 12030, 12030ΔmprF2, and 12030ΔmprF2 CM (knock-in) via the tail vein. Forty-eight hours after injection the animals were sacrificed, and livers, spleens, and kidneys were removed to assess bacterial loads.
Ethics Statement
All animal experiments were performed in compliance with the German animal protection law (TierSchG). Mice were housed and handled in accordance with good animal practice as defined by FELASA and the national animal welfare body GV-SOLAS. The animal welfare committees of the University of Freiburg (Regierungspräsidium Freiburg Az 35/9185.81/G-07/15) approved all animal experiments. The institutional review board of the University of Freiburg approved the study protocol, and written informed consent was obtained from all study participants.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: Supported by grants from the German Ministry of Science and Education (ERA-Net PathoGenoMics 0313933 and BMBF 01 EO 0803). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Franz CMAP, Huch M, Abriouel H, Holzapfel W, Gálvez A. Enterococci as probiotics and their implications in food safety. International Journal of Food Microbiology. doi:10.1016/j.ijfoodmicro.2011.08.014. 2011. [DOI] [PubMed]
- 2.Koch S, Hufnagel M, Huebner J. Treatment and prevention of enterococcal infections–alternative and experimental approaches. Expert opinion on biological therapy 4: 1519–1531. doi:10.1517/14712598.4.9.1519. 2004. [DOI] [PubMed]
- 3.Arias CA, Murray BE. Antibiotic-resistant bugs in the 21st century–a clinical super-challenge. N Engl J Med 360: 439–443. doi:10.1056/NEJMp0804651. 2009. [DOI] [PubMed]
- 4.Ernst CM, Peschel A. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol Microbiol 80: 290–299. doi. 2011;10(1111/j.1365–2958.2011.07576):x. doi: 10.1111/j.1365-2958.2011.07576.x. [DOI] [PubMed] [Google Scholar]
- 5.Peschel A. How do bacteria resist human antimicrobial peptides? Trends Microbiol. 2002;10:179–186. doi: 10.1016/s0966-842x(02)02333-8. [DOI] [PubMed] [Google Scholar]
- 6.Nishi H, Komatsuzawa H, Fujiwara T, McCallum N, Sugai M. Reduced content of lysyl-phosphatidylglycerol in the cytoplasmic membrane affects susceptibility to moenomycin, as well as vancomycin, gentamicin, and antimicrobial peptides, in Staphylococcus aureus. Antimicrob Agents Chemother 48: 4800–4807. doi. 2004. pp. 10.1128/AAC.48.12.4800–4807.2004. [DOI] [PMC free article] [PubMed]
- 7.Ho SW, Jung D, Calhoun JR, Lear JD, Okon M, et al. Effect of divalent cations on the structure of the antibiotic daptomycin. Eur Biophys J 37: 421–433. doi. 2008. pp. 10.1007/s00249–007-0227-2. [DOI] [PubMed]
- 8.Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med. 2001;193:1067–1076. doi: 10.1084/jem.193.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy H, Ibba M. RNA-dependent lipid remodeling by bacterial multiple peptide resistance factors. Proc Natl Acad Sci USA 105: 4667–4672. doi:10.1073/pnas.0800006105. 2008. [DOI] [PMC free article] [PubMed]
- 10.Roy H, Ibba M. Broad range amino acid specificity of RNA-dependent lipid remodeling by multiple peptide resistance factors. J Biol Chem 284: 29677–29683. doi:10.1074/jbc.M109.046367. 2009. [DOI] [PMC free article] [PubMed]
- 11.Paulsen IT, Banerjei L, Myers GSA, Nelson KE, Seshadri R, et al. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299: 2071–2074. doi:10.1126/science.1080613. 2003. [DOI] [PubMed]
- 12.Gould RM, Lennarz WJ. Biosynthesis of aminoacyl derivatives of phosphatidylglycerol. Biochemical and Biophysical Research Communications. 1967;26:512–515. doi: 10.1016/0006-291x(67)90578-5. [DOI] [PubMed] [Google Scholar]
- 13.Santos Mota dos JM, Kamp den JA, Verheij HM, van Deenen LL. Phospholipids of Streptococcus faecalis. J Bacteriol. 1970;104:611–619. doi: 10.1128/jb.104.2.611-619.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fabretti F, Theilacker C, Baldassarri L, Kaczynski Z, Kropec A, et al. Alanine esters of enterococcal lipoteichoic acid play a role in biofilm formation and resistance to antimicrobial peptides. Infect Immun 74: 4164–4171. doi. 2006. pp. 10.1128/IAI.00111–06. [DOI] [PMC free article] [PubMed]
- 15.Theilacker C, Kaczynski Z, Kropec A, Fabretti F, Sange T, et al. Opsonic antibodies to Enterococcus faecalis strain 12030 are directed against lipoteichoic acid. Infect Immun 74: 5703–5712. doi. 2006. pp. 10.1128/IAI.00570–06. [DOI] [PMC free article] [PubMed]
- 16.Huebner J, Quaas A, Krueger WA, Goldmann DA, Pier GB. Prophylactic and therapeutic efficacy of antibodies to a capsular polysaccharide shared among vancomycin-sensitive and -resistant enterococci. Infect Immun. 2000;68:4631–4636. doi: 10.1128/iai.68.8.4631-4636.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neuhaus FC, Baddiley J. A continuum of anionic charge: structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol Mol Biol Rev. 2003;67:686–723. doi: 10.1128/MMBR.67.4.686-723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein S, Lorenzo C, Hoffmann S, Walther JM, Storbeck S, et al. Adaptation of Pseudomonas aeruginosa to various conditions includes tRNA-dependent formation of alanyl-phosphatidylglycerol. Mol Microbiol 71: 551–565. doi. 2009;10(1111/j.1365–2958.2008.06562):x. doi: 10.1111/j.1365-2958.2008.06562.x. [DOI] [PubMed] [Google Scholar]
- 19.Johnston NC, Baker JK, Goldfine H. Phospholipids of Clostridium perfringens: a reexamination. FEMS Microbiol Lett 233: 65–68. doi:10.1016/j.femsle.2004.01.048. 2004. [DOI] [PubMed]
- 20.Mishra NN, Yang S-J, Sawa A, Rubio A, Nast CC, et al. Analysis of cell membrane characteristics of in vitro-selected daptomycin-resistant strains of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 53: 2312–2318. doi. 2009. pp. 10.1128/AAC.01682–08. [DOI] [PMC free article] [PubMed]
- 21.Huebner J, Wang Y, Krueger WA, Madoff LC, Martirosian G, et al. Isolation and chemical characterization of a capsular polysaccharide antigen shared by clinical isolates of Enterococcus faecalis and vancomycin-resistant Enterococcus faecium. Infect Immun. 1999;67:1213–1219. doi: 10.1128/iai.67.3.1213-1219.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer KL, Daniel A, Hardy C, Silverman J, Gilmore MS. Genetic basis for daptomycin resistance in enterococci. Antimicrob Agents Chemother 55: 3345–3356. doi. 2011. pp. 10.1128/AAC.00207–11. [DOI] [PMC free article] [PubMed]
- 23.Arias CA, Panesso D, McGrath DM, Qin X, Mojica MF, et al. Genetic basis for in vivo daptomycin resistance in enterococci. N Engl J Med 365: 892–900. doi:10.1056/NEJMoa1011138. 2011. [DOI] [PMC free article] [PubMed]
- 24.Samant S, Hsu F-F, Neyfakh AA, Lee H. The Bacillus anthracis protein MprF is required for synthesis of lysylphosphatidylglycerols and for resistance to cationic antimicrobial peptides. J Bacteriol 191: 1311–1319. doi. 2009. pp. 10.1128/JB.01345–08. [DOI] [PMC free article] [PubMed]
- 25.Ruzin A, Severin A, Moghazeh SL, Etienne J, Bradford PA, et al. Inactivation of mprF affects vancomycin susceptibility in Staphylococcus aureus. Biochim Biophys Acta. 2003;1621:117–121. doi: 10.1016/s0304-4165(03)00028-x. [DOI] [PubMed] [Google Scholar]
- 26.Komatsuzawa H, Ohta K, Fujiwara T, Choi GH, Labischinski H, et al. Cloning and sequencing of the gene, fmtC, which affects oxacillin resistance in methicillin-resistant Staphylococcus aureus. FEMS Microbiol Lett. 2001;203:49–54. doi: 10.1111/j.1574-6968.2001.tb10819.x. [DOI] [PubMed] [Google Scholar]
- 27.WECKE J, Perego M, Fischer W. d-Alanine Deprivation of Bacillus subtilisTeichoic Acids Is without Effect on Cell Growth and Morphology But Affects the Autolytic Activity. Microbial Drug Resistance 2: 123–129. doi:10.1089/mdr.1996.2.123. 1996. [DOI] [PubMed]
- 28.Steen A, Palumbo E, Deghorain M, Cocconcelli PS, Delcour J, et al. Autolysis of Lactococcus lactis is increased upon D-alanine depletion of peptidoglycan and lipoteichoic acids. J Bacteriol 187: 114–124. doi. 2005. pp. 10.1128/JB.187.1.114–124.2005. [DOI] [PMC free article] [PubMed]
- 29.Kristian SA, Datta V, Weidenmaier C, Kansal R, Fedtke I, et al. D-alanylation of teichoic acids promotes group a streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J Bacteriol 187: 6719–6725. doi. 2005. pp. 10.1128/JB.187.19.6719–6725.2005. [DOI] [PMC free article] [PubMed]
- 30.Patel D, Husain M, Vidaillac C, Steed ME, Rybak MJ, et al. Mechanisms of in-vitro-selected daptomycin-non-susceptibility in Staphylococcus aureus. Int J Antimicrob Agents 38: 442–446. doi:10.1016/j.ijantimicag.2011.06.010. 2011. [DOI] [PubMed]
- 31.O’Toole G, Kaplan HB, Kolter R. Biofilm formation as microbial development. Annu Rev Microbiol 54: 49–79. doi:10.1146/annurev.micro.54.1.49. 2000. [DOI] [PubMed]
- 32.Fabretti F, Huebner J. Implant infections due to enterococci: role of capsular polysaccharides and biofilm. Int J Artif Organs. 2005;28:1079–1090. doi: 10.1177/039139880502801105. [DOI] [PubMed] [Google Scholar]
- 33.Hufnagel M, Koch S, Creti R, Baldassarri L, Huebner J. A putative sugar-binding transcriptional regulator in a novel gene locus in Enterococcus faecalis contributes to production of biofilm and prolonged bacteremia in mice. J INFECT DIS 189: 420–430. doi:10.1086/381150. 2004. [DOI] [PubMed]
- 34.Qin Z, Ou Y, Yang L, Zhu Y, Tolker-Nielsen T, et al. Role of autolysin-mediated DNA release in biofilm formation of Staphylococcus epidermidis. Microbiology (Reading, Engl) 153: 2083–2092. doi. 2007. pp. 10.1099/mic.0.2007/006031–0. [DOI] [PubMed]
- 35.Mohamed JA, Huang W, Nallapareddy SR, Teng F, Murray BE. Influence of origin of isolates, especially endocarditis isolates, and various genes on biofilm formation by Enterococcus faecalis. Infect Immun 72: 3658–3663. doi. 2004. pp. 10.1128/IAI.72.6.3658–3663.2004. [DOI] [PMC free article] [PubMed]
- 36.Thomas VC, Hiromasa Y, Harms N, Thurlow L, Tomich J, et al. A fratricidal mechanism is responsible for eDNA release and contributes to biofilm development of Enterococcus faecalis. Mol Microbiol 72: 1022–1036. doi. 2009;10(1111/j.1365-2958.2009.06703):x. doi: 10.1111/j.1365-2958.2009.06703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas VC, Thurlow LR, Boyle D, Hancock LE. Regulation of autolysis-dependent extracellular DNA release by Enterococcus faecalis extracellular proteases influences biofilm development. J Bacteriol 190: 5690–5698. doi. 2008. pp. 10.1128/JB.00314–08. [DOI] [PMC free article] [PubMed]
- 38.Grande R, Di Giulio M, Bessa LJ, Di Campli E, Baffoni M, et al. Extracellular DNA in Helicobacter pylori biofilm: a backstairs rumour. J Appl Microbiol 110: 490–498. doi. 2011;10(1111/j.1365-2672.2010.04911):x. doi: 10.1111/j.1365-2672.2010.04911.x. [DOI] [PubMed] [Google Scholar]
- 39.Weidenmaier C, Peschel A, Kempf VAJ, Lucindo N, Yeaman MR, et al. DltABCD- and MprF-mediated cell envelope modifications of Staphylococcus aureus confer resistance to platelet microbicidal proteins and contribute to virulence in a rabbit endocarditis model. Infect Immun 73: 8033–8038. doi. 2005. pp. 10.1128/IAI.73.12.8033–8038.2005. [DOI] [PMC free article] [PubMed]
- 40.Kristian SA, Dürr M, Van Strijp JAG, Neumeister B, Peschel A. MprF-mediated lysinylation of phospholipids in Staphylococcus aureus leads to protection against oxygen-independent neutrophil killing. Infect Immun. 2003;71:546–549. doi: 10.1128/IAI.71.1.546-549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thedieck K, Hain T, Mohamed W, Tindall BJ, Nimtz M, et al. The MprF protein is required for lysinylation of phospholipids in listerial membranes and confers resistance to cationic antimicrobial peptides (CAMPs) on Listeria monocytogenes. Mol Microbiol 62: 1325–1339. doi. 2006;10(1111/j.1365-2958.2006.05452):x. doi: 10.1111/j.1365-2958.2006.05452.x. [DOI] [PubMed] [Google Scholar]
- 42.Maloney E, Stankowska D, Zhang J, Fol M, Cheng Q-J, et al. The two-domain LysX protein of Mycobacterium tuberculosis is required for production of lysinylated phosphatidylglycerol and resistance to cationic antimicrobial peptides. PLoS Pathog 5: e1000534. doi:10.1371/journal.ppat.1000534. 2009. [DOI] [PMC free article] [PubMed]
- 43.Rigottier-Gois L, Alberti A, Houel A, Taly J-F, Palcy P, et al. Large-Scale Screening of a Targeted Enterococcus faecalis Mutant Library Identifies Envelope Fitness Factors. PLoS ONE 6: e29023. doi:10.1371/journal.pone.0029023.t001. 2011. [DOI] [PMC free article] [PubMed]
- 44.Theilacker C, Kaczynski Z, Kropec A, Sava I, Ye L, et al. Serodiversity of Opsonic Antibodies against Enterococcus faecalis -Glycans of the Cell Wall Revisited. PLoS ONE 6: e17839. doi:10.1371/journal.pone.0017839. 2011. [DOI] [PMC free article] [PubMed]
- 45.Hufnagel M, Hancock LE, Koch S, Theilacker C, Gilmore MS, et al. Serological and genetic diversity of capsular polysaccharides in Enterococcus faecalis. J Clin Microbiol 42: 2548–2557. doi. 2004. pp. 10.1128/JCM.42.6.2548–2557.2004. [DOI] [PMC free article] [PubMed]
- 46.Standish AJ, Weiser JN. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J Immunol 183: 2602–2609. doi:10.4049/jimmunol.0900688. 2009. [DOI] [PubMed]
- 47.Köten B, Simanski M, Gläser R, Podschun R, Schröder J-M, et al. RNase 7 contributes to the cutaneous defense against Enterococcus faecium. PLoS ONE 4: e6424. doi:10.1371/journal.pone.0006424. 2009. [DOI] [PMC free article] [PubMed]
- 48.Theilacker C, Sanchez-Carballo P, Toma I, Fabretti F, Sava I, et al. Glycolipids are involved in biofilm accumulation and prolonged bacteraemia in Enterococcus faecalis. Mol Microbiol 71: 1055–1069. doi. 2009;10(1111/j.1365-2958.2009.06587):x. doi: 10.1111/j.1365-2958.2008.06587.x. [DOI] [PubMed] [Google Scholar]
- 49.Sambrook J, Russell DW. Molecular cloning. CSHL Press. 2001.
- 50.Le Jeune A, Torelli R, Sanguinetti M, Giard J-C, Hartke A, et al. The extracytoplasmic function sigma factor SigV plays a key role in the original model of lysozyme resistance and virulence of Enterococcus faecalis. PLoS ONE 5: e9658. doi:10.1371/journal.pone.0009658. 2010. [DOI] [PMC free article] [PubMed]
- 51.Arnaud M, Chastanet A, Débarbouillé M. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol 70: 6887–6891. doi. 2004. pp. 10.1128/AEM.70.11.6887–6891.2004. [DOI] [PMC free article] [PubMed]
- 52.Cieslewicz MJ, Kasper DL, Wang Y, Wessels MR. Functional analysis in type Ia group B Streptococcus of a cluster of genes involved in extracellular polysaccharide production by diverse species of streptococci. J Biol Chem 276: 139–146. doi:10.1074/jbc.M005702200. 2001. [DOI] [PubMed]
- 53.Callegan MC, Jett BD, Hancock LE, Gilmore MS. Role of hemolysin BL in the pathogenesis of extraintestinal Bacillus cereus infection assessed in an endophthalmitis model. Infect Immun. 1999;67:3357–3366. doi: 10.1128/iai.67.7.3357-3366.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bryan EM, Bae T, Kleerebezem M, Dunny GM. Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44: 183–190. doi:10.1006/plas.2000.1484. 2000. [DOI] [PubMed]
- 55.BLIGH EG, DYER WJ. A rapid method of total lipid extraction and purification. Canadian journal of biochemistry and physiology. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 56.Meyrand M, Boughammoura A, Courtin P, Mézange C, Guillot A, et al. Peptidoglycan N-acetylglucosamine deacetylation decreases autolysis in Lactococcus lactis. Microbiology (Reading, Engl) 153: 3275–3285. doi. 2007. pp. 10.1099/mic.0.2007/005835–0. [DOI] [PubMed]
- 57.Callegan MC, Booth MC, Jett BD, Gilmore MS. Pathogenesis of gram-positive bacterial endophthalmitis. Infect Immun. 1999;67:3348–3356. doi: 10.1128/iai.67.7.3348-3356.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]