

Abstract
A variety of microbial communities and their genes (microbiome) exist throughout the human body, playing fundamental roles in human health and disease. The NIH funded Human Microbiome Project (HMP) Consortium has established a population-scale framework which catalyzed significant development of metagenomic protocols resulting in a broad range of quality-controlled resources and data including standardized methods for creating, processing and interpreting distinct types of high-throughput metagenomic data available to the scientific community. Here we present resources from a population of 242 healthy adults sampled at 15 to 18 body sites up to three times, which to date, have generated 5,177 microbial taxonomic profiles from 16S rRNA genes and over 3.5 Tb of metagenomic sequence. In parallel, approximately 800 human-associated reference genomes have been sequenced. Collectively, these data represent the largest resource to date describing the abundance and variety of the human microbiome, while providing a platform for current and future studies.
Introduction
Advances in sequencing technologies coupled with novel bioinformatic developments have allowed the scientific community to commence assessment of the uncultivated majority; the microbes that inhabit our oceans, soils, human body and other locations 1. Microbes associated with the human body include eukaryotes, archaea, bacteria, and viruses, with bacteria alone estimated to outnumber human cells within an individual by an order of magnitude. Our knowledge of these communities and their gene content, referred to collectively as the human microbiome, has to date been limited by a lack of population-scale data detailing their composition and function.
The US National Institutes of Health (NIH) funded Human Microbiome Project (HMP) Consortium allied a broad collection of scientific experts to explore these microbial communities and their relationships with their human hosts. As such, the HMP 2 has focused on reference genomes (viral, bacterial and eukaryotic) which provide a critical framework for subsequent metagenomic annotation and analysis, and on generating a baseline of microbial community structure and function from an adult cohort defined by a carefully delineated set of clinical inclusion and exclusion criteria which we term “healthy” in this study (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/GetPdf.cgi?id=phd002854.2). Investigations of the microbiome from this cohort incorporated several complementary analyses including: 16S rRNA gene sequence (16S) and taxonomic profiles, whole genome shogun (WGS) or metagenomic sequencing of whole community DNA, and alignment of the assembled sequences to the reference microbial genomes from the human body 3, 4. Thus, the HMP complements other large-scale sequence-based human microbiome projects such as the MetaHIT project 5 which focused on examination of the gut microbiome using WGS data including samples from cohorts exhibiting a wide range of health status and physiological characteristics.
Additional projects supported by the HMP are investigating the association of specific components and dynamics of the microbiome with a variety of disease conditions, developing tools and technology including isolating and sequencing uncultured organisms, and studying the ethical, legal, and social implications of human microbiome research (http://commonfund.nih.gov/hmp/fundedresearch.aspx). A comprehensive list of current publications from HMP projects is available at http://commonfund.nih.gov/hmp/publications.aspx.
Here we detail the resources created to date by the HMP initiative including: clinical specimens (samples), reference genomes, sequencing and annotation protocols, methods and analyses. We describe the thousands of samples obtained from 15 to 18 distinct body sites from 242 donors over multiple time points that were processed at two clinical centers (Baylor College of Medicine (BCM) and Washington University School of Medicine). We also describe the laboratory and computational protocols developed for reliably generating and interpreting the human microbiome data. HMP resources include both protocols for, and the subsequent data generated from, 16S and metagenomic sequencing of human microbiome samples. During this study, these protocols were rigorously standardized and quality controlled for simultaneous use across four sequencing centers (BCM Human Genome Sequencing Center, The Broad Institute of Harvard and MIT, J. Craig Venter Institute, The Genome Institute at Washington University). In particular, we focus on the production of the first phase of metagenomic data sets (Phase I) used for subsequent in depth analyses and we summarize standards and recommendations based on our experiences generating and analyzing these data. An additional set of publications (many included in this communication’s references and in 4) will describe in further detail the microbial ecology and microbiological implications of these data. Collectively these resources and analyses represent an important framework for human microbiome research.
Main
HMP resource organization
Supplementary Figure 1 summarizes organization of the HMP, including the data processing and analytical steps, and the scientific entities gathered to conduct the project. An overview of available HMP data sets and additional resources are provided in Supplementary Tables 1, 2 and 3. Donors were recruited and enrolled into the HMP through the two clinical centers. Over 240 adults were carefully screened and phenotyped prior to sampling one to three times at 15 (male) or 18 (female) body sites using a common sampling protocol (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/GetPdf.cgi?id=phd003190.2). All included subjects were between the ages of 18 and 40 years and had passed a screening for systemic health based on oral, cutaneous, and body mass exclusion criteria (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/GetPdf.cgi?id=phd002854.2) 6.
A Data Analysis and Coordination Center (DACC) was created to serve as the central repository for all HMP WGS, 16S, and reference genome sequence information generated by the four sequencing centers. The DACC supports access to analysis software, biological samples, clinical protocols, news, publication announcements and project statistics, and performed centralized analysis of HMP reference genome and WGS annotation in cooperation with the sequencing centers. Unless otherwise noted, all data sets and protocols described here are available to the scientific community at the DACC (http://hmpdacc.org).
Phase I 16S and WGS sequencing overview
A set of 5,298 samples were collected from 242 adults 6 (Table 1, Supplementary Table 4) from which 16S and WGS data were generated for a total of 5,177 taxonomically characterized communities (16S) and 681 WGS samples describing the microbial communities from habitats within the human airways, skin, oral cavity, gut, and vagina. For a subset of 560 samples, both data types were generated (Table 1). These efforts constitute our initial primary metagenomic data sets (Phase I) described in more detail below. Additional efforts are ongoing to sequence and analyze the remaining samples from the complete HMP collection (11,174 primary specimens in total from 300 individuals sampled up to three times over 22 months) 6.
Table 1.
HMP donor samples examined by 16S and WGS
| Body Region | Body Site | Total Samples | Total 16S Samples | V13 Samples | V13 Read Depth (M)* | V35 Samples | V35 Read depth (M)* | Samples V13&V35 | Total WGS Samples | Total Read Depth (G)† | % Filtered Reads± | % Human Reads¥ | Remaining Read Depth (G)† | Samples 16S&WGS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gut | Stool | 352 | 337 | 193 | 1.4 | 328 | 2.38 | 184 | 139 | 1720.7 | 15 | 1 | 1450.6 | 124 |
| Buccal mucosa | 346 | 330 | 184 | 1.3 | 314 | 1.74 | 168 | 107 | 1438 | 9 | 82 | 136.68 | 91 | |
| Hard palate | 325 | 325 | 179 | 1.2 | 310 | 1.67 | 164 | 1 | 10.87 | 20 | 25 | 5.92 | 1 | |
| Keratinized gingiva | 335 | 329 | 183 | 1.3 | 319 | 1.74 | 173 | 6 | 72.3 | 5 | 47 | 34.42 | 0 | |
| Palatine Tonsils | 337 | 332 | 189 | 1.2 | 315 | 1.87 | 172 | 6 | 74.75 | 2 | 80 | 13.45 | 1 | |
| Oral Cavity | Saliva | 315 | 310 | 166 | 0.9 | 292 | 1.45 | 148 | 5 | 55.69 | 1 | 91 | 4.24 | 0 |
| Subgingival plaque | 334 | 328 | 186 | 1.2 | 314 | 1.84 | 172 | 7 | 92.06 | 5 | 79 | 15.29 | 1 | |
| Supragingival plaque | 345 | 331 | 192 | 1.3 | 316 | 1.88 | 177 | 115 | 1500.7 | 15 | 40 | 674.81 | 101 | |
| Throat | 331 | 325 | 176 | 1 | 312 | 1.67 | 163 | 7 | 78.78 | 4 | 79 | 13.57 | 1 | |
| Tongue dorsum | 348 | 332 | 193 | 1.3 | 320 | 2.04 | 181 | 122 | 1620.1 | 15 | 19 | 1084.3 | 106 | |
| Airway | Anterior nares | 316 | 302 | 169 | 1 | 283 | 1.17 | 150 | 84 | 1129.9 | 3 | 96 | 14.31 | 70 |
| Left Antecubital fossa | 269 | 269 | 158 | 0.7 | 221 | 0.47 | 110 | 0 | na | na | na | 0 | na | |
| Left Retroauricular crease | 313 | 312 | 188 | 1.6 | 295 | 1.46 | 171 | 9 | 126.34 | 9 | 73 | 22.07 | 8 | |
| Skin | Right Antecubital fossa | 274 | 274 | 158 | 0.7 | 229 | 0.52 | 113 | 0 | na | na | na | 0 | na |
| Right Retroauricular crease | 319 | 316 | 190 | 1.4 | 304 | 1.56 | 178 | 15 | 181.94 | 18 | 59 | 42.38 | 12 | |
| Mid vagina | 145 | 143 | 91 | 0.6 | 140 | 0.96 | 88 | 2 | 22.58 | 0 | 99 | 0.18 | 0 | |
| Vagina | Posterior fornix | 152 | 142 | 89 | 0.6 | 136 | 0.98 | 83 | 53 | 702.13 | 6 | 90 | 25.24 | 43 |
| Vaginal introitus | 142 | 140 | 87 | 0.6 | 131 | 0.85 | 78 | 3 | 36.48 | 1 | 98 | 0.58 | 1 | |
|
| ||||||||||||||
| total | 5298 | 5177 | 2971 | 19 | 4879 | 26.3 | 2673 | 681 | 8863.3 | 11 | 49 | 3538.1 | 560 | |
1×106 reads post-processing with the mothur pipeline (Supplementary Information)
1×109 reads post-processing with the mothur pipeline (Supplementary Information)
Fraction of reads with low quality bases that were removed (Supplementary Information)
Fraction of human reads that were removed (Supplementary Information)
16S standards development and sequencing
The goals of the HMP required that 16S sequences and profiles from data produced at the four participating sequencing centers be comparable in a variety of downstream analyses however, no suitable methodology was available at the commencement of the project. While establishing 16S protocols, we determined that many components of data production and processing can contribute errors and artifacts. We investigated methods that avert these errors and their subsequent effects on taxonomic classification and OTU-based community structure. The results are discussed in detail in Supplementary Information and 7. Thus, multiple evaluations of 16S protocols were undertaken before adopting a single standardized protocol that ensured consistency in the high throughput production.
To maximize accuracy and consistency, protocols were evaluated primarily using a synthetic mock community (MC) of 21 known organisms (Supplementary Table 5, 7). Additional testing of the protocol was carried out on a subset of HMP samples (Supplementary Table 1). Collectively, these efforts resulted in adoption of a protocol to amplify and sequence samples using the Roche-454 FLX Titanium platform (7, http://www.hmpdacc.org/doc/HMP_MDG_454_16S_Protocol_V4_2_102109.pdf). The HMP created both cell mixtures and genomic DNA extracts of the MC (Supplementary Table 3). A large body of metagenomic data (both 16S and WGS) (RES:HMMC) from these and other calibration experiments are available to the community to facilitate further benchmarking of new molecular and analytical approaches (Supplementary Table 2).
The majority of the sample collection was targeted for 16S sequencing using the 454 FLX Titanium based strategy 7. The nucleotide sequence of the 16S rRNA gene consists of regions of highly conserved sequence which alternate with nine regions or windows of variable nucleotide sequence that constitute the most informative portions of the gene sequence for use in taxonomic classification. A window covering the number three (V3) through five (V5) variable regions (V35) of the 16S rRNA gene was chosen as the target for 4,879 samples. Sequence of a V1 to V3 (V13) window was also included for a subset of 2,971 samples to provide a complementary view of taxonomic profiles (RES:HMR16S, Supplementary Figs. 2 and 3, Supplementary Information, 7).
Following adoption of the 16S protocol including removal of multiple sources of potential artifacts or bias generated by 16S sequencing using pyrosequencing 8, 9, a variety of approaches for accurate diversity estimation were developed and compared 10. A 16S data processing pipeline was established using the mothur software package 11 (Supplementary Information) that includes two optional low and high stringency approaches. The former provides an output favoring longer read lengths tailored towards taxonomic classification, the latter an output with more aggressive sequence error reduction tailored towards Operational Taxonomic Unit (OTU) construction (RES:HMMCP). A third complementary pipeline was also developed using the QIIME software package 12 (Supplementary Information), which processes these data using an OTU-binning strategy to which taxonomic classification is added (RES:HMQCP). All pipelines result in highly comparable views of the human microbiome.
Metagenomic assembly and gene cataloging
Approximately 749 samples representing targeted body sites were chosen for WGS sequencing using the Illumina GAIIx platform with 101 bp paired-end reads. From a high quality set of 681 samples an average depth of 13 Gb (±4.3) was achieved per sample, collectively producing a total of 8.8 Tb (RES:HMIWGS) (Table 1). Theoretically, these per sample data are sufficient to cover a 3Mb bacterial genome present at only 0.8% abundance with a probability of 90% 13. In addition, 12 stool samples were simultaneously sequenced using the 454 FLX Titanium platform (RES:HM4WGS). Comparisons between the centers demonstrated high consistency of target sequencing depth and success rates 4. Following development of a protocol for removing reads resulting from human DNA contamination (Supplementary Information), 49% of the reads were targeted for removal as human (for information on authorized access to these reads see Supplementary Information). Samples collected from soft tissue tended to have higher human contamination (e.g., mid vagina (96%), anterior nares (82%), throat (75%)). Preparations from saliva were also high in human DNA sequence (80%), while stool contained a relatively low abundance of human reads (up to 1%) (Supplementary Fig. 4).
After application of a quality control protocol that includes human sequence removal, quality filtering and trimming of reads, (Supplementary Information), the remaining 3.5 Tb from 681 samples were subjected to a three-tiered complementary analysis strategy (Supplementary Information), of reference genome mapping (that was able to use ~57% of the data), assembly and gene prediction (~50% of the data), and metabolic reconstruction (~36% of the data). This combined strategy facilitated the extraction of maximal organismal and functional information.
Metagenomic assemblies were generated for all available samples using an optimized SOAPdenovo protocol with parameters designed to produce substrates for downstream analyses such as gene and function prediction, resulting in a total of 41 million contigs (RES:HMASM) (Supplementary Information). Reads that remained unassembled were pooled across individual body sites and re-assembled using the same approach, resulting in an additional 4,200,672 contigs (RES:HMBSA). These body site-specific assemblies are aimed at reconstructing organisms that represent too small a fraction in any individual sample to assemble but are found among many individuals. For 12 stool samples both Illumina and 454 FLX Titanium data (RES:HM4WGS) were generated allowing a hybrid assembly approach, using Newbler (Supplementary Information) (RES:HMHASM). Overall, the assembly statistics recovered varied substantially by body site and community complexity (Supplementary Fig. 5). However, our results indicate that, for the assembly strategy we employed, metagenomic assembly quality plateaus at approximately 6 Gb of microbial sequence coverage for a sample possessing a microbial community structure similar to that of stool samples (Supplementary Fig. 6).
A WGS based perspective of community membership was obtained by aligning the reads to a set of 1,742 finished bacterial, 131 archaeal, 3,683 viral, and 326 lower eukaryotic reference genomes (RES:HMREFG) (Supplementary Information, 14) representing a broad taxonomic range from each of these four domains. A total of 57.6% of the high quality microbial reads could be associated with a known genome (ranging from 33 to 77%, for anterior nares and posterior fornix, respectively) (RES:HMSCP). The overwhelming majority of mapped sequences originated from bacteria (99.7%), while the remaining reads mapped to microeukaryotes (0.3%) or archaea (< 0.01%) (Supplementary Information).
Two complementary approaches were used to summarize overall function and metabolism of the human microbiome producing two primary data sets of annotations (Supplementary Information, RES:HMMRC, RES:HMGI) and additional secondary analyses (Supplementary Information, RES:HMGS, HMHGI, HMGC, HMGOI) available to the community for further interrogation. The first primary data set of annotations was produced by mapping individual shotgun reads to characterized protein families (RES:HMMRC) 15. The second, was produced from functionally annotated gene predictions generated from the metagenomic assemblies (RES:HMGI) which were subsequently grouped according to high-level biological processes and to selected additional processes specific to metabolism and regulation (RES:HMGS) 16 (Supplementary Tables 6 and 7, Supplementary Fig. 7).
HMP data generation and analysis lessons
A key manner in which the HMP resources will serve to guide future studies of the microbiome is by enabling informed decisions regarding sampling protocols and genomic DNA preparation 6, sequencing depth, 13 statistical power, 17 and metagenomic data type. As indicated in Table 1, the consortium successfully amplified 16S sequences to our target depth at all 18 body sites, with the fewest sequences recovered consistently from the antecubital fossae. The amount of host human DNA recovered and the finest level of OTU resolution varied for 16S sequences among body sites (Supplementary Figs. 3-4, 7).
From our WGS investigations, a series of protocols (http://hmpdacc.org/tools_protocols/tools_protocols.php) have been established to process large volumes of short read WGS data and to annotate and examine these data through both a multi-tiered assembly approach and as single reads 18. An investigator’s choice of metagenomic technologies can thus be guided not only by a 16S versus WGS dichotomy, but also by the expected fraction of host sequence and appropriate 16S region targeting the dominant taxa at each body site (Supplementary Figs. 2-6, 8).
Together, these datasets represent comprehensive and complementary views of the human microbiome as shown by comparing organismal (Fig. 1a) and gene (Fig. 1b) catalogues, and the ratio of genes contributed per OTU (Fig. 1c). The discovery rate of new gene clusters (as determined by annotation of assembled WGS data) is in general detected more slowly relative to organismal discovery (as determined by OTU data) due to the fragmentary nature of these community reads and assemblies despite high sequence depth (Fig. 1a and b, Supplementary Fig. 9) and the number of genes contributed per OTU varies by body site (Fig. 1c, Supplementary Information). However in general, these results highlight an important point for consideration of further microbiome investigations using these data sets as they suggest that the majority of the common taxa and genes present in this reference population have been detected.
Figure 1. Rates of gene and OTU discovery from HMP taxonomic and metagenomic data.
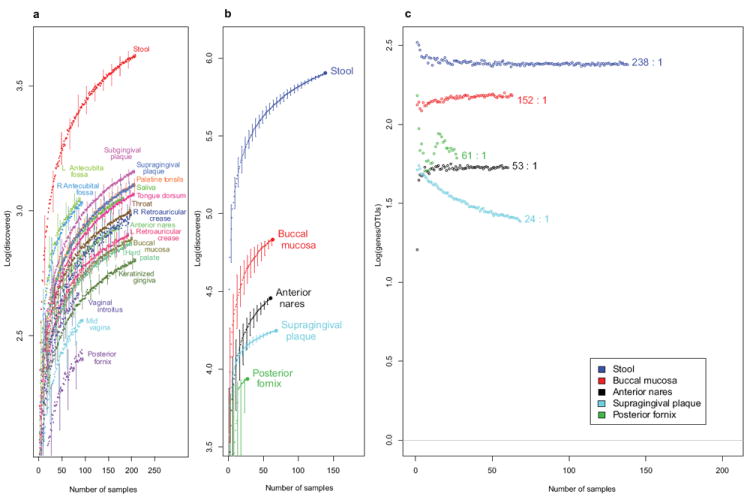
Accumulation curves for a, OTU counts from 16S data (all body sites) b, clustered gene index counts from metagenomic data (all applicable body sites) and c, the ratio of average unique genes contributed versus unique OTUs encountered with increasing sample counts (Supplementary Information). Ratios given for each curve in c represent the average number of unique genes contributed per unique OTU at the final sample count. Curves for stool, buccal mucosa and anterior nares suggest that the proportion of gene-to-taxa discovery has stabilized. In contrast, the curve for supragingival plaque suggests relatively fewer new genes are being contributed per additional OTU. Error bars represent 95% confidence intervals.
We additionally compared the gut community gene catalogue sampled by the HMP with that of MetaHIT in terms of total detected gene counts. The HMP recovered more total non-redundant gene counts (5,140,472) than reported by MetaHIT (3,299,822) 5 likely reflecting a combination of the increased sequence depth obtained by the HMP (11.7 Gb HMP, 4.5Gb MetaHIT on average) and differences in data generation and processing 5.
The two non-redundant sets of gene sequences were subsequently combined and compared by matches to a database of orthologous groups 19 of functionally annotated genes. Approximately 57% of the orthologous groups recovered by this method overlapped between the data sets, while an additional 34% versus 10% were unique to the HMP and MetaHIT, respectively (Supplementary Fig. 10, Supplementary Table 8, Supplementary Information). After removal of genes that received any orthologous group assignment, the remaining novel genes were subsequently clustered 20. Approximately 79% of the HMP-derived novel gene clusters were orthologous to one or more clusters in MetaHIT, while an additional 16% were unique to this study versus 5% for MetaHIT-derived data (Supplementary Fig. 11, Supplementary Table 8, Supplementary Information, 5). These results suggest that for this body habitat, relatively similar gene catalogues were recovered despite differences in experimental design and protocols. However, a greater proportion of both annotated and unique novel genes were detected in the HMP data set, emphasizing the utility of sequencing depth in recovering gene function and in particular, deriving rare function. These results further underscore the importance of large-scale sequenced based studies of the microbiome to better characterize its gene content and diversity.
Human microbiome reference genomes
The current goal for the reference genome component of the HMP is to sequence at least 3,000 reference microbial genomes associated with the human body. Thus far, more than 800 genomes have been sequenced and are available from NCBI and the DACC (http://hmpdacc.org/HMRGD). From an alignment of WGS reads to reference genomes (RES:HMREFG), approximately 26% from the total read set (46% of all reads that could be aligned) were matched to a subset of 223 HMP reference genomes (Supplementary Information, Supplementary Data).
We continue to solicit community feedback for strains that will best benefit our attempts at understanding the breadth of human microbiome diversity. For example, a prioritized list of the “most wanted” HMP taxa is currently being maintained (http://hmpdacc.org/most_wanted/) with the goal of targeting these difficult to obtain organisms using both culture-based and single-cell approaches.
A catalogue of all HMP reference genomes along with custom filtering, viewing, graphing, and download options can be found at the DACC Project Catalogue (http://www.hmpdacc-resources.org/hmp_catalog/main.cgi). In addition, comparative analyses of reference genomes are provided by the data warehouse and analytical systems, IMG/HMP (http://www.hmpdacc-resources.org/cgi-bin/imgm_hmp/main.cgi). Cultures of all HMP reference strains are required to be made publicly available through, the Biodefense and Emerging Infections Research Resources Repository (BEI). Information on strain acquisition can be found at the DACC (http://hmpdacc.org/reference_genomes/reference_genomes.php) and BEI (http://www.beiresources.org/tabid/1901/stabid/1901/CollectionLinkID/4/Default.aspx).
Conclusion
An overarching goal of this multi-year, multi-center project is the generation of a community resource to advance research efforts related to the microbiome. The result is a collection of 11,174 primary biological specimens representing the human microbiome as well as corresponding blood samples from the human donors which are being reserved for sequencing at a future date and from which cell lines will be developed. A variety of new protocols were developed to enable a project of this scope; these include methods for donor recruitment, laboratory and sequence processing, and analysis of 16S and WGS sequence and profiles. These resources serve as models to guide the design of similar projects. Studies with a primary focus on disease can use this reference for comparative purposes including detecting shifts in microbial taxonomic and functional profiles, or identification of new species not present in healthy cohorts that appear under disease conditions. The catalogue described in this study is the largest reference set to date of human microbiome data associated with healthy individuals. Collectively the data represents a treasure trove that can be continually mined to identify new organisms, gene functions, and metabolic and regulatory networks as well as correlations between microbial community structure and health and disease 4. Among other future benefits, this resource may promote the development of novel prophylactic strategies such as the application of prebiotics and probiotics to foster human health.
Supplementary Material
Institutional Review Board Statement.
As a part of a multi-institutional collaboration, the Human Microbiome Project human subjects study was reviewed by the Institutional Review Boards at each sampling site: Baylor College of Medicine (IRB protocols H-22895 (IRB #00001021) and H-22035 (IRB #00002649)); Washington University School of Medicine (IRB protocol HMP-07-001 (IRB #201105198)); and St. Louis University (IRB #15778). The study was also reviewed by the J. Craig Venter Institute under IRB protocol 2008-084 (IRB #00003721), and at the Broad Institute the study was determined to be exempt from IRB review. All study participants gave their written informed consent before sampling and the study was conducted using the Human Microbiome Project Core Sampling Protocol A. Each IRB has a federal wide assurance and follows the regulations established at 45 CFR Part 46. The study was conducted in accordance with the ethical principles expressed in the Declaration of Helsinki and the requirements of applicable federal regulations
Acknowledgments
The Consortium would like to thank our external scientific advisory board: Richard Blumberg, Julian Davies, Robert Holt, Pilar Ossorio, Francis Ouellette, Gary Schoolnik, and Alan Williamson. We would also like to thank our collaborators throughout the International Human Microbiome Consortium, particularly the investigators of the MetaHIT project, for advancing human microbiome research. Data repository management was provided by the National Center for Biotechnology Information and the Intramural Research Program of the NIH National Library of Medicine. We especially appreciate the generous participation of the individuals from the Saint Louis, MO, and Houston, TX areas who made this study possible. This research was supported in part by National Institutes of Health grants U54HG004969 to B.W.B.; U54HG003273 to R.A.G.; U54HG004973 to R.A.G., S.K.H. and J.F.P.; U54HG003067 to E.S.L.; U54AI084844 to K.E.N.; N01AI30071 to R.L.S.; U54HG004968 to G.M.W.; U01HG004866 to O.R.W.; U54HG003079 to R.K.W.; R01HG005969 to C.H.; R01HG004872 to R.K.; R01HG004885 to M.P.; R01HG005975 to P.D.S.; R01HG004908 to Y.Y.; R01HG004900 to M.K.C. and P.L.S.; R01HG005171 to D.E.H.; R01HG004853 to A.L.M.; R01HG004856 to R.R.; R01HG004877 to R.R.S. and R.F.; R01HG005172 to P.G.S.; R01HG004857 to M.P.; R01HG004906 to T.M.S.; R21HG005811 to E.A.V; G.A.B. was supported by UH2AI083263 and UH3AI083263 (G.A.B., Cynthia N. Cornelissen, Lindon K. Eaves and Jerome F. Strauss); M.J.B. was supported by UH2AR057506, S.M.H. was supported by UH3DK083993 (Vincent B. Young, Eugene B. Chang, Folker Meyer, Thomas M. Schmidt, Mitchell L. Sogin, James M. Tiedje); K.P.R. was supported by UH2DK083990 (James Versalovic); J.A.S. and H.H.K. were supported by UH2AR057504 and UH3AR057504 (J.A.S.); DP2OD001500 to K.M.A.; N01HG62088 to the Coriell Institute for Medical Research; U01DE016937 to F.E.D.; S.K.H. was supported by RC1DE202098 and R01DE021574 (S.K.H. and Huiying Li); J.G.I. was supported by R21CA139193 (J.G.I. and Dominique S. Michaud); K.P.L. was supported by P30DE020751 (Daniel J. Smith); Army Research Office grant W911NF-11-1-0473 to C.H.; National Science Foundation grants NSF DBI-1053486 to C.H. and NSF IIS-0812111 to M.P.; The Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 for P.S.G.C.; LANL Laboratory-Directed Research and Development grant 20100034DR and the U.S. Defense Threat Reduction Agency grants B104153I and B084531I to P.S.G.C.; Research Foundation - Flanders (FWO) grant to K.F. and J.R.; R.K. is an HHMI Early Career Scientist; Gordon & Betty Moore Foundation funding and institutional funding from the J. David Gladstone Institutes to K.S.P.; A.M.S was supported by fellowships provided by the Rackham Graduate School and the NIH Molecular Mechanisms in Microbial Pathogenesis Training Grant T32AI007528; a Crohn’s and Colitis Foundation of Canada Grant in Aid of Research to E.A.V.; 2010 IBM Faculty Award to K.C.W.; Analysis of the HMP data was performed using National Energy Research Scientific Computing resources; the BluBioU Computational Resource at Rice University.
Footnotes
Author Contributions
Principal investigators: BWB, RAG, SKH, BAM, KEN, JFP, GMW, OW, RKW
Manuscript preparation: BAM, KEN, MP, HHC, MGG, DG, CH, JFP
Funding agency management: CCB, TB, VB, JLC, SC, CD, VDF, CG, MYG, RDL, JM, PM, JP, LMP, JAS, LW, CW, KAW
Project leadership: SA, JHB, BWB, ATC, HHC, AME, MGF, RSF, DG, MGG, KH, SKH, CH, EAL, RM, VM, JCM, BAM, MM, DMM, KEN, JFP, EJS, JV, GMW, OW, AMW, KCW, JRW, SKY, QZ
Analysis preparation for manuscript: MB, BLC, DG, MGG, MEH, CH, KL, BAM, XQ, JRW, MT
Data release: LA, TB, IAC, KC, HHC, NJD, DJD, AME, VMF, LF, JMG, SG, SKH, MEH, CJ, VJ, CK, AAM, VMM, TM, MM, DMM, JO, KP, JFP, CP, XQ, RKS, NS, IS, EJS, DVW, OW, KW, KCW, CY, BPY, QZ
Methods and research development: SA, HMA, MB, DMC, AME, RLE, MF, SF, MGF, DCF, DG, GG, BJH, SKH, MEH, WAK, NL, KL, VM, ERM, BAM, MM, DMM, CN, JFP, MEP, XQ, MCR, CR, EJS, SMS, DGT, DVW, GMW, YW, KMW, SY, BPY, SKY, QZ
DNA sequence production: SA, EA, TA, TB, CJB, DAB, KDD, SPD, AME, RLE, CNF, SF, CCF, LLF, RSF, BH, SKH, MEH, VJ, CLK, SLL, NL, LL, DMM, IN, CN, MO, JFP, XQ, JGR, YR, MCR, DVW, YW, BPY, YZ
Clinical sample collection: KMA, MAC, WMD, LLF, NG, HAH, ELH, JAK, WAK, TM, ALM, PM, SMP, JFP, GAS, JV, MAW, GMW
Body site experts: KMA, EAV, GA, LB, MJB, CCD, FED, LF, JI, JAK, SKH, HHK, KPL, PJM, JR, TMS, JAS, JDS, JV
Ethical, legal and social implications: RMF, DEH, WAK, NBK, CML, ALM, RR, PS, RRS, PS, LZ
Strain management: EAV, JHB, IAC, KC, SWC, HHC, TZD, ASD, AME, MGF, MGG, SKH, VJ, NCK, SLL, LL, KL, EAL, VMM, BAM, DMM, KEN, IN, IP, LS, EJS, CMT, MT, DVW, GMW, AMW, YW, KMW, BPY, LZ, YZ
16S data analysis: KMA, EJA, GLA, CAA, MB, BWB, JPB, GAB, SRC, SC, JC, TZD, FED, ED, AME, RCE, MF, AAF, JF, HG, DG, BJH, TAH, SMH, CH, JI, JKJ, STK, SKH, RK, HHK, OK, PSLR, REL, KL, CAL, DM, BAM, KAM, MM, MP, JFP, MP, KSP, XQ, KPR, MCR, BR, PDS, TMS, NS, JAS, WDS, TJS, CSS, EJS, RMT, JV, TAV, ZW, DVW, GMW, JRW, KMW, YY, SY, YZ
Shotgun data processing and alignments: CJB, JCC, ED, DG, AG, MEH, HJ, DK, KCK, CLK, YL, JCM, BAM, MM, DMM, JO, JFP, XQ, JGR, RKS, NUS, IS, EJS, GGS, SMS, JW, ZW, GMW, OW, KCW, TW, SKY, LZ
Assembly: HMA, CJB, PSC, LC, YD, SPD, MGF, MEH, HJ, SK, BL, YL, CL, JCM, JMM, JRM, PJM, MM, JFP, MP, MEP, XQ, MR, RKS, MS, DDS, GGS, SMS, CMT, TJT, WW, GMW, KCW, LY, YY, SKY, LZ
Annotation: OOA, VB, CJB, IAC, ATC, KC, HHC, ASD, MGG, JMG, JG, AG, SG, BJH, KH, SKH, CH, HJ, NCK, RM, VMM, KM, TM, MM, JO, KP, MP, XQ, NS, EJS, GGS, SMS, MT, GMW, KCW, JRW, CY, SKY, QZ, LZ
WGS Metabolic Reconstruction: SA, BLC, JG, CH, JI, BAM, MM, BR, AMS, NS, MT, GMW, SY, QZ, JDZ
Accession numbers for all primary sequencing data are given in Supplementary Information.
Competing Interests Statement
The authors declare that they have no competing interests.
References
- 1.Gilbert JA, Dupont CL. Microbial metagenomics: beyond the genome. Ann Rev Mar Sci. 2011;3:347–371. doi: 10.1146/annurev-marine-120709-142811. [DOI] [PubMed] [Google Scholar]
- 2.NIH HMP Working Group. The NIH Human Microbiome Project. Genome Res. 2009;19(12):2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Human Microbiome Jumpstart Reference Strains Consortium. A catalog of reference genomes from the human microbiome. Science. 2010;328(5981):994–999. doi: 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The Human Microbiome Consortium. Structure, Function and Diversity of the Human Microbiome in an Adult Reference Population. in review. [Google Scholar]
- 5.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aagaard K, et al. A Comprehensive Strategy for Sampling the Human Microbiome. in review. [Google Scholar]
- 7.Jumpstart Consortium Human Microbiome Project Data Generation Working Group. Evaluation of 16S rDNA-based community profiling for human microbiome resaerch. PloS One. doi: 10.1371/journal.pone.0039315. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol. 2010;12(1):118–123. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- 9.Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8(7):R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;6(12):e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schloss PD, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wendl MC, Kota K, Weinstock GM, Mitreva M. A sampling theory for metagenomic DNA sequencing based on a generalization of Stevens’ theorem. doi: 10.1007/s00285-012-0586-x. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin J, Sykes, et al. Optimizing read mapping to reference genomes to determine composition and species prevalence in microbial communities. PLoS One. doi: 10.1371/journal.pone.0036427. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abubucker S, Segata N, Goll J, Schubert AM, Izard J. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comp Bio. doi: 10.1371/journal.pcbi.1002358. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.La Rosa PS, et al. Power calculations for taxonomic-based analysis of human microbiome data. doi: 10.1371/journal.pone.0052078. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goll J, Thiagarajan M, Abubucker S, Huttenhower C, Yooseph S, Methé BA. A case study for large-scale human microbiome analysis using JCVI’s Metagenomics Reports (METAREP) PLoS One. doi: 10.1371/journal.pone.0029044. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller J, Szklarczyk D, Julien P, Letunic I, Roth A, et al. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 2010;38:190–195. doi: 10.1093/nar/gkp951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinform. 2010;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.