

Abstract
Runx1 is expressed in skeletal elements, but its role in fracture repair has not been analyzed. We created mice with a hypomorphic Runx1 allele (Runx1L148A) and generated Runx1L148A/− mice in which >50% of Runx1 activity was abrogated. Runx1L148A/− mice were viable but runted. Their growth plates had extended proliferating and hypertrophic zones, and the percentages of Sox9, Runx2 and Runx3 positive cells were decreased. Femoral fracture experiments revealed delayed cartilaginous callus formation and the expression of chondrogenic markers was decreased. Conditional ablation of Runx1 in the mesenchymal progenitor cells of the limb with Prx1-Cre conferred no obvious limb phenotype; however cartilaginous callus formation was delayed following fracture. Embryonic limb bud-derived mesenchymal cells showed delayed chondrogenesis when the Runx1 allele was deleted ex vivo with adenoviral-expressed Cre. Collectively, our data suggest that Runx1 is required for commitment and differentiation of chondroprogenitor cells into the chondrogenic lineage.
Keywords: AML1, Chondrocytes, Cartilage Development, Fracture Repair
Introduction
The regenerative capacity of the adult skeleton depends on the re-induction of cellular and molecular cues that occur during embryonic development (1). Fracture repair of long bones involves an inflammatory step followed by the formation of a cartilaginous callus. This callus is then replaced with bone, and excess bone callus is finally remodeled to achieve bony union (2). In the developing limb, mesenchymal condensations undergo a series of physiological changes to form cartilage and bone, while post-natally the formation of cartilaginous callus during fracture repair requires the recruitment of periosteal progenitor cells and their differentiation into chondrocytes and osteoblasts (3-5). Failure to form a proper cartilaginous callus results in impaired fracture healing (6-8). Understanding the molecular mechanisms of progenitor cell differentiation into chondrocytes during fracture healing is important for developing novel therapeutic approaches to prevent delayed healing and reverse non-unions.
The Runx proteins (Runx1, Runx2 & Runx3) are the mammalian homologues of the Drosophila runt and lozenge transcription factors (9,10). The Runx proteins are expressed in a variety of tissues, and play key functions ranging from organogenesis to pathogenesis. Runx2 is essential for bone development (11,12) and Runx3 is important during nerve cell differentiation (13,14). Genetic evidence indicates Runx1 is required for definitive hematopoiesis, neuronal differentiation and survival, T cell development, and formation of the sternum (15-20). Similarly, conditional deletion of Runx1 in mouse bone marrow causes impairments in megakaryote and lymphocyte development leading to thrombocytopenia and lymphopenia in mice (21). In humans, haploinsufficiency of Runx1 results in an autosomal dominant syndrome called familial platelet disorder with propensity to acute myeloid leukemia (22).
The overlapping expression of Runx proteins in skeletal elements suggested multiple, perhaps overlapping roles for all three Runx proteins during bone and cartilage development. For example, Runx1 is expressed in immature chondrocytes, perichondrium, periosteum, rib, vertebrae, limb, mandible, and maxilla (23-26). Furthermore, our in vitro studies suggest that Runx1 mediates mesenchymal stem cell commitment to the chondrogenic lineage (26). However, the role of Runx1 during skeletal development in vivo is not fully understood, and its potential role in skeletal repair remains unknown. Thus, the early embryonic lethality of the Runx1 global knockout mice (15,16) warranted the development of other strategies to explore Runx1’s potential role during cartilage development and fracture repair. Here we describe the generation of a hypomorphic Runx1 allele, which we used to generate in vivo models with incremental loss of Runx1 function. We also executed a conditional deletion of Runx1 in mesenchymal chondro/osteoprogenitor cells using a floxed allele (21) in conjunction with Prx1-Cre (27). Using these animal models, we determined the in vivo function of Runx1 during skeletal development and repair.
Materials and Methods
Animals
The Runx1L148A allele was generated by homologous recombination as described for other mutations in exon 4 sequences (28). Runx1+/+, Runx1L148A/+, Runx1L148A/L148A, Runx1+/− (Runx1− = Runx1tm1Spe)(16), Runx1L148A/−, and Runx1L148A/R174Q (Runx1R174Q = Runx1tm5Spe (28) were generated by intercrossing the appropriate mouse strains. Runx1F/+, Prx1-Cre;Runx1F/+ and Prx1-Cre;Runx1F/F were generated from the breeding of Runx1F/F (Runx1F = Runx1tm3Spe) (21) and Prx1-Cre mice (27). Mice were housed at the University of Connecticut Health Center (UCHC) and Dartmouth Medical School animal facilities according to state and federal law requirements.
Yeast one- and modified one-hybrid assays
The yeast one- and modified one-hybrid assays were conducted as described previously (28-30).
Equilibrium binding constants
The Runt domain was purified from bacteria, and quantitative electrophoretic mobility shift assays were performed as described previously using the Core site from the SL3–3 murine leukemia virus enhancer (GGATATCTGTGGTTAAGCA) as the DNA probe 1 (28,30,31).
Clinical blood cell counts
Retro-orbital blood was collected from 6- to 10-week-old mice, transferred to tubes coated with EDTA (Startsedt), and counted on a Drew Scientific Hemavet 850.
Osteocalcin promoter luciferase activities
Bone marrow cells were isolated from the long bones of eight-week-old mice. Cells were maintained in α Minimum Essential Media (αMEM) containing 10% fetal calf serum and antibiotics in the presence of 30 ng/ml of M-CSF. Cells were co-transfected with 400 ng of the osteocalcin promoter plasmid and 100 ng of renilla luciferease vector using the FuGENE® HD transfection reagent (Roche, Indianapolis, IN) and incubated for 30 hrs. Luciferase activity was measured using a dual-luciferase assay kit (Promega, Madison, WI), and luminescence was detected with a luminometer (Lumat LB 9507, Berthold Technologies, Oak Ridge, TN).
Femoral fracture procedure
Animals were anesthetized and given analgesic prior to surgical procedure following the principles and procedures approved by the Animal Care Committee at UCHC. Femoral fractures were performed using an Einhorn device (32) on the left limb of seven to nine week old female mice as previously described (7,33,34). Fractures were radiographed by a Faxitron x-ray system (Faxitron, X-ray, Wheeling, IL) under anesthesia to monitor initial pin fixation as well as ongoing callus formation at days 7, 14, and 21 post fracture. Mice were sacrificed on 7, 14, and 21 days and fractured legs were collected for micro-computed tomographic (μCT), histological and histomorphometric analyses. Fracture callus were also harvested at day 7 for total RNA extraction and immunohistochemistry.
Micro-computed tomography (μCT) analyses
Upon sacrifice, fractured femurs were disarticulated at the hip and knee, dissected of soft tissue, and fixed in 10% neutral buffered formation overnight at room temperature. Callus morphometry was measured using conebeam micro-focus X-ray computed tomography (μCT40, Scanco Medical AG, Brüttisellen, Switzerland) as previously described (7). Standard algorithms describing trabecular morphometry were applied to obtain direct measures of both total and mineralized callus.
Histological and histomorphometric analyses
Tibiae and fractured femurs were harvested for histological assessments as previously published (7,34). For histomorphometic analyses, sections were stained with H&E and the lengths of the proliferating and hypertrophic zones determined as previously described (35). The perimeters of stromal, cartilaginous and bony tissues were outlined in the fracture callus and these regions were quantified using NIS-Elements BR 3.0 (Nikon, Melville, NY).
Immunohistochemical analyses
Sections from paraffin-embedded tibias and fractured femurs were deparaffinized and sequentially incubated with either 4N HCl (anti-Sox9 and anti-Runx3) or citrate buffer, pH 6.0 (anti-Runx2) for antigen retrieval (anti-Sox9 [ab59265], anti-Runx2 [ab54868] and anti-Runx3 [ab68938] from AbCam, Cambridge, MA). Incubation and detection was performed following our previously published protocol (33,36). Image acquisition was performed using a Q-Imaging Retig 200R camera connected to a Nikon Eclipse 50i microscope and image analyses performed using an NIS-Elements BR 3.0 software (Nikon, Melville, NY).
Gene expression analysis
Total RNA from fracture calluses were extracted for gene expression analyses via Q-RT-PCR as previously described (7,34). Specific primers were used for murine Runx1, Runx2, Runx3, Sox9, Sox5, Sox6, Type II collagen, and Type X collagen (see Table 1). Gene expression was normalized to that of β-actin. PCR products were analyzed and quantified using the StepOne™Software (Applied Biosystems, Foster City, CA).
Table 1.
Mouse primers used for real-time RT-PCR experiments
| Genes | Forward primer | Reverse primer | Accession # |
|---|---|---|---|
| Runx1 | GCATGGTGGAGGTACTAGCTG | GCCGTCCACTGTGATTTTG | NM009821 |
| Runx2 | GACATCCCCATCCATCCACT | TGAGAGAGGAAGGCCAGAGG | D14636 |
| Runx3 | CTCCAGCCCGAGACTACAAG | GGGATGCACAGCTAGAGAGG | AF155880 |
| Sox9 | AGGAAGCTGGCAGACCAGTA | CGTTCT TCACCGACT TCCTC | AF421878 |
| Sox5 | ACCTCAGAAGGCGGAAGAAG | CTTCAGGGTGTCCACCACAT | NM011444 |
| Sox6 | ACACCTGAGAGCCTTGCAGA | TGCTGCCAGCTTTTTCTGTT | NM011445 |
| Type II collagen | ACTGGTAAGTGGGGCAAGAC | CCACACCAAATTCCTGTTCA | BC052326 |
| Type X collagen | CTTTGTGTGCCTTTCAATCG | GTGAGGTACAGCCTACCAGTTTT | X67348 |
| β-actin | AGATGTGGATCAGCAAGCAG | GCGCAAGTTAGGTTTTGTCA | NM007393 |
Limb bud micromass cultures
Forelimbs and hindlimbs were harvested from Runx1F/F embryos at E12.5 and limb bud cells isolated as previously described (26,37). Cell suspensions (2 × 107/ml) were transduced with either Cre adenovirus or the GFP control virus at 100 MOI in growth media. Infected cells seeded at a density of 2 × 105 cells per 10μl for two hours to allow adherence. Micromass cultures were then submerged in media supplemented with 25 μg/ml ascorbic acid and 54 μg/ml beta-glycerol phosphate. Media were changed every 2-3 days and cells harvested at days 4, 7 and 14 for alcian blue staining, Western blotting and ChIP assays.
Western blotting
Ten μg of nuclear proteins harvested from micromass cultures were subjected to SDS-PAGE as previously published (36,37). Western blotting was accomplished using antibodies for Runx1 (Calbiochem, La Jolla, CA), Runx2, Runx3, Sox9 (anti-Sox9 [ab59265], anti-Runx2 [ab54868] and anti-Runx3 [ab49117] from AbCam, Cambridge, MA), or actin ([A2668] Sigma, St. Louis, MO) and corresponding secondary antibodies ([anti-mouse IgG 7076], [anti-rabbit IgG 7074] Cell Signaling Technology, Danvers, MA). Chemiluminescent detection was performed using Pierce femto reagent (Pierce, Rockford, IL). Density of each band was analyzed using the NIH Image J analysis software.
Chromatin immunoprecipitation assays
Limb bud micromass were prepared as described above. Cells were harvested at days 4, 7, 10, and 14 and chromatin was isolated using the ChIP-IT kit (Active Motif, Carlsbad, CA) for immunoprecipitation (ChIP) assay following the manufacturer’s instructions. Ten μg of sheared fragments were immunoprecipitated with 5μg anti-RUNX1 antibody (ab23980, Abcam,Cambridge, MA) or the same amount of IgG antibody as negative control. Immunoprecipitated and input DNA were analyzed by real time-PCR using primers, which embrace Runx1 responsive elements on Sox9, Sox5, and Sox6 promoter regions (Table 2).
Table 2.
Mouse primers which embrace Runx1 responsive elements of each Sox9, Sox5, and Sox6 promoters
| Promoter | Forward primer | Reverse primer |
|---|---|---|
| Sox9-site1 | TCGGAAGAAAACGAGAGGAA | CCTCGGAACCACAAACAAAC |
| Sox9-site2 | CAGGAGGCAAGAAGCAGAAC | ACGAAGCTGGTGTGGTGACT |
| Sox5 | CCCTCCCAGTCCCTTTTATT | AGAGGGAGCAAAGCAATCAG |
| Sox6-site1 | CATAGGGAACCACATGAGCA | TGTGAAACATGGGCTGCTAC |
| Sox6-site2 | CGTGCTTGACATCCTCTCTG | CACCACAGCATGAGGAACTG |
Results
Creation of a hypomorphic Runx1 allele
Runx1 interacts with DNA through a DNA-binding sequence called Runt domain, which also interacts with Runx1 partner protein, CBFß (30). To generate a hypomorphic Runx1 allele, previously generated Runt domain mutants targeting exon 4-encoded sequences (amino acids 141-177) were analyzed to identify mutations that result in disruption of DNA binding (28,30). Semi-quantitative yeast one-hybrid analyses using a liquid β-galactosidase assay was employed to measure the ability of Runt domain proteins, containing one of five different mutations: K144A, L148A, V152A, P156A, A160G (29,30), to bind DNA. All of these mutations decreased DNA binding in the range of 4 to 13 fold (Fig. 1A). A modified yeast one-hybrid assay was also used to measure the binding of the Runt domain:CBFβ heterodimer to DNA (28,30). The mutations decreased DNA binding of the heterodimer by 2 to 6 fold (Fig. 1B). Interestingly, several of the mutations (L148A, P156A, A160G) negatively impact DNA binding of the isolated Runt domain more extensively than they impaired DNA binding by the Runt domain:CBFβ heterodimer complex. All of the mutations were predicted to perturb the fold of the Runt domain (or in the case of K144A a salt bond) (30),. CBFβ stabilizes some of the mutant Runt domains (L148A, P156A, A160G) to a greater extent than others (K144A, V152A), ameliorating the effects of the mutations on DNA binding activity.
Figure 1. Generation of a hypomorphic Runx1 allele.
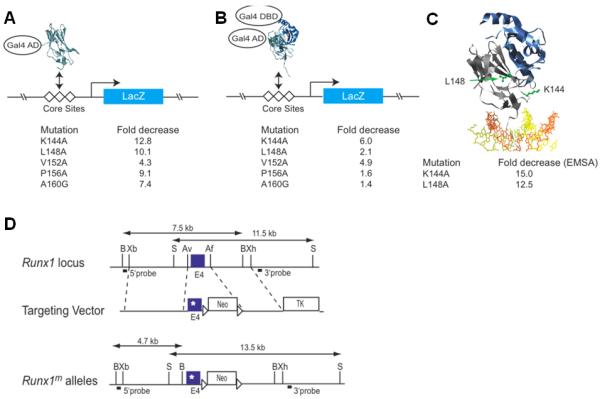
(A) Yeast one hybrid assay for Runt domain binding to three core sites driving lacZ expression. The Runt domain (RD) is fused to the GAL4 activation domain (GAL4 AD). Listed below are the relative decreases in β-galactosidase activity measured using a quantitative liquid assay. (B) Modified yeast one-hybrid assay to measure binding of the RD:CBFβ heterodimer to DNA. Although the Gal4 DNA-binding domain is fused to CBFβ in the modified yeast one-hybrid assay, there are no Gal4 binding sites on the promoter driving lacZ and therefore CBFβ’s activity is mediated only through the core sites. CBFβ increases the affinity of the RD for DNA by approximately 10-fold (44). (C) Ribbon diagram of the RD:CBFβ:DNA ternary complex (29,38) and the residues targeted for mutation. The Runt domain and CBFβ are shown in grey and blue, respectively, and DNA is orange and yellow. The side chains of K144 and L148 are shown and labeled in green. On the side are the fold decreases in DNA binding by purified Runt domain (in the absence of CBFβ) as determined by EMSA. (D) Targeting vector. Point mutations were engineered into exon 4 of Runx1. A neomycin resistance gene flanked by loxP sites is in intron 4. The location of probes and restriction length fragments from the wild type and targeted Runx1 alleles are indicated. B, BamHI; Xb, XbaI; S, SalI; Av, AvrII;, Af, AflII; Xh, XhoI.
Since K114A and L148A mutants most negatively impacted DNA and CBFβ, we specifically focused on these mutations. Neither of these residues directly contacts DNA or CBFβ. K144 is located in the βE’-F loop in close proximity to the DNA contacting residue R142 (29,38). The K144A mutation decreased binding in the yeast one-hybrid assay by 12.8 fold (Fig. 1A), and decreased DNA binding by the heterodimer (modified one-hybrid) by 6 fold (Fig. 1B). The L148 sidechain is in the βF strand, buried in the hydrophobic core of the Runt domain. The L148A mutation impaired DNA binding of the isolated Runt domain by 10.1 fold, and of the heterodimer by 2.1 fold. Differences in equilibrium dissociation constants for the Runt domain-DNA complex measured by quantitative electrophoretic mobility shift assays (28,30) were consistent with those obtained in the yeast one-hybrid analyses (Fig. 1C).
The K144A and L148A mutations were targeted into exon 4 of the Runx1 locus using the strategy described previously (28) (Fig. 1D). Mice heterozygous for the K144A and L148A mutations appeared grossly normal. Multiple litters from Runx1K144A/+ intercrosses yielded only one live Runx1K144A/K144A pup out of a total of 51, indicating that homozygosity for this mutation was lethal in most progeny. Runx1L148A/L148A mice, on the other hand, were viable, and indistinguishable from their wild type littermates. However, when the Runx1L148A allele was carried over a nonfunctional (Runx1−) or weakly dominant negative Runx1 allele (Runx1R174Q) (28) the frequency of viable offspring was reduced (Supplementary Table 1). Clinical blood counts from mice homozygous for the Runx1L148A allele, or Runx1L148A/− mice revealed significantly lower numbers of red blood cells, hemoglobin, hematocrit levels, lymphocytes, platelets and neutrophils compared to wild type mice, whereas most of these parameters (with the exception of platelet numbers) were equivalent in wild type and Runx1+/− mice (Supplementary Table 2). Altogether the data indicate that Runx1L148A was a hypomorphic Runx1 allele.
Increased loss of Runx1 function results in growth abnormalities
A hypomorphic allelic series was generated consisting of wild type, Runx1L148A/+, Runx1+/−, and Runx1L148A/− mice to study the role of Runx1 in skeletal development and fracture repair. Examination of the gross phenotypes during the exponential growth phase (4 weeks postnatal) showed that mice with the highest loss of Runx1 function (Runx1L148A/−) were runted (Fig. 2A). This is further evidenced by X-ray analyses showing shorter femurs in Runx1L148A/− mice compared to their Runx1+/+, Runx1L148A/+, and Runx1+/− littermates (Fig. 2A). This runted phenotype correlated with lower body mass of Runx1L148A/− mutants compared to other genotypes for up to 11 weeks of age (data not shown). Marrow cells were isolated from each genotype and transfected with the osteocalcin promoter reporter, which contains three Runx binding sites to measure Runx1 activity. While promoter activity was reduced by 50% in Runx1+/− mice, it was further decreased by 70% in the Runx1L148A/− mutants (Fig. 2B).
Figure 2. Characterization of mice with the incremental loss of Runx1 function.
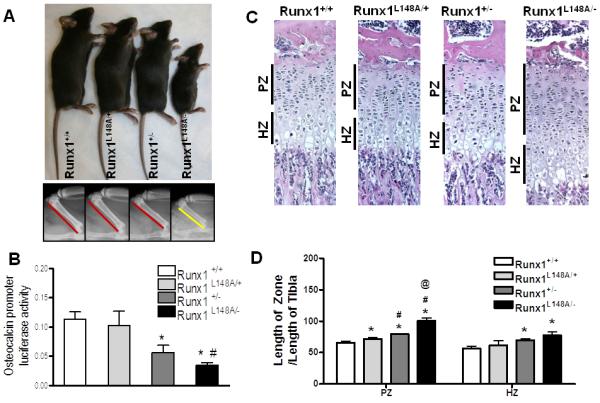
Runx1 +/+, Runx1 L148A/+, Runx1 +/−, and Runx1 L148A(*/− 4 week old age mice were characterized by (A) global appearance (Top panel) and representative radiographs of four-week-old mice femurs. (Lower panel). (B) Activity of osteocalcin promoter in mouse bone marrow cells isolated from limbs of Runx1 +/+, Runx1 L148A/+, Runx1 +/−, and Runx1 L148A/− mice was measured. (C-D) Proliferating (PZ), and hypertrophic (HZ) zones of tibial growth plates at 4 week old age from all genotypes were categorized based on H&E and relative lengths were quantified by histomorphemtric analysis. n=5 mice per group. *p<0.05 vs Runx1 +/+; #p<0.05 vs Runx1 L148A/+; and @p<0.05 vs Runx1 +/−.
Because of the impaired skeletal growth in Runx1L148A/− mice, histological examination of growth plates was performed at 4 weeks of age. Figure 2C shows that while the incremental loss of Runx1 function does not impair the organization of growth plate, the lengths of the proliferating and hypertrophic zones were enhanced with increasing loss of Runx1 function. Histomorphometric analyses showed that the ratio of the length of the proliferating zone divided by the length of the Runx1L148A/− tibia was increased by 9%, 21% and 53% in Runx1+/−, Runx1L148A/+, and Runx1+/+ mice, respectively. Runx1L148A/− and Runx1+/− mice also displayed significantly longer hypertrophic zones compared to Runx1+/+ (37% and 24%, respectively) (Fig. 2D). These changes were not observed during embryonic development when we examined the gross morphology of long bones at E16.5 indicating that these effects are most likely post-natal (data not shown).
Decreased expression of chondrogenic markers in growth plate chondrocytes of mice with decreased Runx1 activity
We examined protein expression of Sox9, Runx2, and Runx3 in the proliferating zones of the growth plates of all genotypes by immunohistochemistry (Supplementary Figure 1). As expected, while Runx2 was expressed in hypertrophic chondrocytes, Sox9 and Runx3 were excluded from these populations in all genotypes (data not shown). We quantified the number of Sox9, Runx2, and Runx3 expressing cells in the proliferating zone as percentages of immunoreactive cells over total number of cells per fixed surface area. We found that, when Runx1 activity was abrogated by 50% or more, the number of Sox9, Runx2 and Runx3 cells was significantly decreased in the proliferating zone.
Inhibited Runx1 activity results in delayed fracture healing
To examine whether Runx1 plays a role during bone repair we assessed the ability of Runx1+/+, Runx1L148A/+, Runx1+/− and Runx1L148A/− mice to heal their bones following a femoral fracture. Radiographic evaluation of the mid-diaphyseal fractures over a time course of 21 days post-fracture showed callus formation in fractured bones from all genotypes. At day 14, while radiolucency persisted in calluses of Runx1L148A/− mice, the other genotypes showed fully mineralized bridging. By day 21, all genotypes had achieved union (Supplementary Figure 2A).
MicroCT imaging showed decreased callus size with an unmineralized void at the center of calluses in the Runx1L148A/− mice compared to mice of other genotypes at 14 days post-fracture (Supplementary Figure 2B). By day 21 all mineralized calluses were smaller than at day 14, indicating the onset of fracture callus remodeling (Supplementary Figure 2B). Quantitative analyses of these images confirmed that total callus size was significantly smaller in Runx1L148A/− mice at day 14 compared to the wild type control (Supplementary Figure 2C), and mineralized callus size was also reduced (Supplementary Figure 2D). Total callus size was significantly decreased in all genotypes at day 21 compared to day 14 (Supplementary Figure 2C). However, no significant differences were observed between day 14 and day 21 in mineralized callus size of Runx1L148A/− mice (Supplementary Figure 2D). The data indicate that global loss of Runx1 activity is concomitant with delayed fracture repair of long bones.
Incremental loss of Runx1 function results in altered cartilaginous callus formation during bone fracture healing
We complemented our imaging results with histological and histomorphometric analyses of safranin-O/fast green-stained fractured calluses of all genotypes at days 7, 14, and 21 post-operation. Total callus size was smaller in Runx1+/− and Runx1L148A/− compared to Runx1+/+ and Runx1L148A/+ mice at day 7 post-fracture (Fig. 3A). At 14 days post-fracture, while the majority of the cartilaginous callus from Runx1+/+, Runx1L148A/+ and Runx1+/− fractured bone was replaced with mineralized bone, residual cartilage remained in calluses from Runx1L148A/− mice. While callus sizes were similar among all genotypes at day 21 postfracture, residual cartilage persisted in the mineralized callus of Runx1L148A/− mice.
Figure 3. Runx1 controls the stromal and cartilaginous callus formation at the early stage of bone fracture healing.
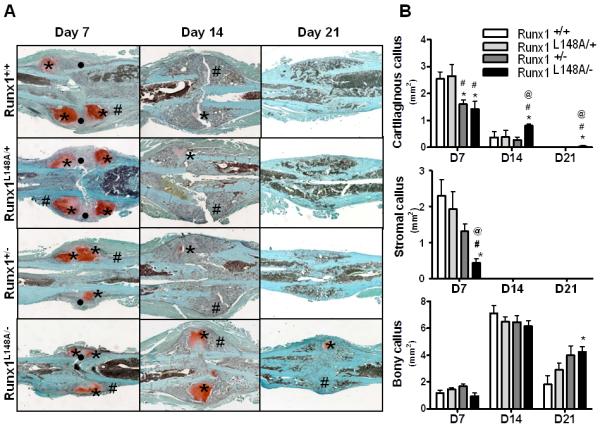
Seven to 9 week old mice were used for the femoral fracture experiments as described in the materials and methods section. (A) For histological analysis, the fracture calluses were stained with Safonin O (Red: cartilage) and Fast Green (Green: mineralized tissue) to identify the stromal (•), cartilaginous (*), and mineralized (#) callus formation. (B) Histomorphometric assessment was also performed to measure the size of the stromal, cartilaginous, and bony callus at day 7, 14, and 21 post fracture. n=4-7 mice per group. *p<0.05 vs Runx1 +/+; #p<0.05 vs Runx1 L148A/+; and @p<0.05 vs Runx1 +/−.
Histomorpometric analyses showed that cartilage content in the early (Day7) fracture calluses was significantly decreased in Runx1+/− and Runx1L148A/− mice compared to that of Runx1+/+ and Runx1L148/+ mice (Fig. 3B). Conversely, a significant increase in cartilage content of the calluses from Runx1L148A/− mice was seen at days 14 and 21 compared to all other genotypes. Stromal tissue contents were also significantly decreased in Runx1L148A/− mice at day 7. The bony callus content remained unchanged between all genotypes at days 7 and 14. However, at day 21 post fracture, significant amounts of residual bony callus persisted in Runx1L148A/− mice compared to Runx1+/+ mice. Together, the data indicate that loss of Runx1 function results in delayed cartilaginous callus formation and remodeling, leading to impaired fracture healing.
Runx1 regulates chondrogenic transcription factors
Because of the changes in the soft callus composition in Runx1L148A/−, we performed immunohistochemical analyses to examine the cellular representation of other chondrogenic transcription factors using specific antibodies for Sox9, Runx2 and Runx3. Images of immunostained calluses of all genotypes are presented in supplementary Figure 3. Immunohistochemical data was quantified by counting the number of Sox9, Runx2, and Runx3 positive cells per fixed surface area in both the stroma and cartilage of fracture calluses. No significant differences were observed in the percentages of Sox9, Runx2 and Runx3 in the stroma of calluses between Runx1+/+ and Runx1L148A/+ calluses (Fig. 4). The percentages of Sox9, Runx2 and Runx3 positive cells were significantly decreased in the stroma of Runx1+/− compared to Runx1+/+ calluses by 39%, 38% and 51% respectively. This decrease was further accentuated in the stroma of Runx1L148A/− calluses by 81%, 92%, and 93% in comparison to Runx1+/+, respectively. The number of Sox9 positive cells was slightly but significantly decreased in the cartilaginous calluses of Runx1L148A/+ compared to Runx1+/+. No significant changes in the percentages of Runx2 or Runx3 positive cells were observed between Runx1L148A/+ and Runx1+/+ calluses. The percentages of Sox9, Runx2 and Runx3 positive cells were all significantly decreased in cartilaginous calluses of Runx1+/− compared to Runx1+/+ mice by 29%, 36% and 47% respectively and that of Runx1 L148A/− compared to Runx1+/+ mice by 25%, 39% and 38% respectively. Together, the data suggest that Runx1 enhanced the differentiation of mesenchymal progenitor cell into chondrocytes in the fracture callus, possibly through modulation of Sox9, Runx2, and Runx3.
Figure 4. Runx1 affects chondrogenic transcription factors in early fracture callus formation.
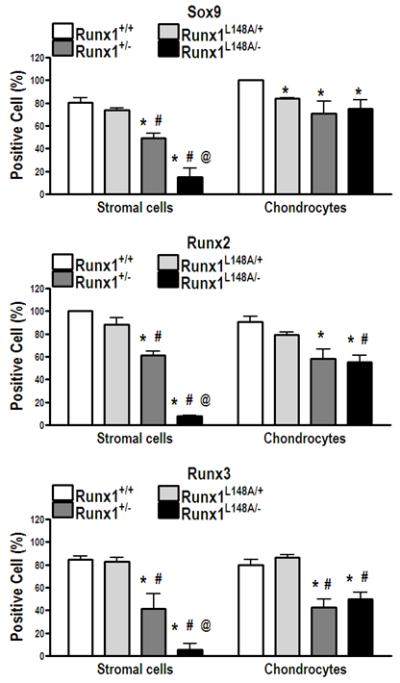
Calluses at day 7 postfracture were used to measure Sox9, Runx2, and Runx3 positive cells within both stroma and cartilaginous callus following immunohistochemical staining. Data are presented as a ratio of immunoreactive cells over total number of either stromal cells or chondrocytes per fixed surface area. n=4-7 mice per group. *p<0.05 vs Runx1 +/+; #p<0.05 vs Runx1 L148A/+; and @p<0.05 vs Runx1 +/−.
We also performed Q-PCR analyses of Runx1, 2, and 3 and cartilage phenotypic marker genes Sox9, type II collagen and type X collagen using RNA extracted from fracture calluses at day 7 post-fracture. Runx1 transcripts were reduced in Runx1+/− and Runx1L148A/− mice. Neither Runx2 nor Runx3 expression was affected by the loss of Runx1 activities. However, gene expression of the chondrocyte phenotypic markers Sox9 and type II collagen was inhibited in the calluses of Runx1+/− mice, and to a greater degree in Runx1L148/− mice. Type X collagen expression was only significantly decreased in Runx1L148/− mice (data not shown).
Impaired cartilaginous callus formation following tissue-specific deletion of Runx1
The periosteum contains Prx1-positive mesenchymal stem cells that can differentiate into chondrocytes during cartilaginous callus formation (5). Runx1F/F mice were crossed with Prx1-Cre transgenic mice and femoral fractures performed in the resulting Prx1-Cre;Runx1F/F mutants. Mice with 100% Runx1 activity (Runx1F/+) and partial deletion of Runx1 function (Prx1-Cre;Runx1F/+) were used as controls. Figure 5A shows that the area of the cartilaginous tissue and bony tissue within the fracture calluses were comparable between Prx1-Cre;Runx1F/+ and Runx1F./+. Conversely, the fracture callus of Prx1-Cre;Runx1F/F mice was much smaller than those of Prx1-Cre;Runx1F/+ and Runx1F/+ littermate controls, with limited cartilage formation. Histomophormetric analyses showed that Prx1-Cre; Runx1F/F calluses contained significantly reduced amounts of both cartilaginous and bony tissues compared to Prx1-Cre;Runx1F/+ and Runx1F/+ (Fig. 5B). We further extracted RNA from fracture calluses of Prx1-Cre;Runx1F/F and Runx1F/+ at day 7 postfracture and examined mRNA levels of Runx1 as well as Runx2, Runx3, Sox9, Sox5 and Sox6 (Fig. 5C). We observed a significant reduction of Runx1 expression by 81% in fracture callus of Prx1-Cre; Runx1F/F mice, and significant decreases in Sox9, Runx2, and Runx3 expression by 42%, 29%, and 39% respectively. Levels of Sox5 and Sox6 expression were also decreased by 23% and 43% respectively (data not shown). These results suggest that Prx1-mediated deletion of Runx1 resulted in decreased cartilaginous callus formation in the early stages of fracture healing.
Figure 5. Runx1 expression in periosteum-derived mesenchymal stem cells is necessary for cartilaginous callus formation during fracture healing.
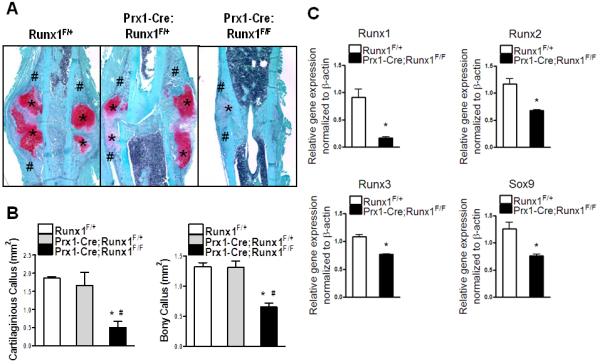
(A) The calluses of Runx1F/+, Prx1-Cre; Runx1F/+, and Prx1-Cre; Runx1F/F mice at day 7 postfracture were stained with Safonin O (Red: cartilage) and Fast Green (Green: mineralized tissue) to identify the cartilaginous (*) and bony (#) callus formation. (B) Histomorphometric assessment was performed to measure the size of the cartilaginous and bony callus. n=4-5 mice per group (C) Gene expression of Runx1, Runx2, Runx3, and Sox9 within the fracture calluses of Runx1F/+ and Prx1-Cre; Runx1F/F was measured by real-time PCR and normalized to β-actin. n=4 mice per group with a duplicate format of real-time PCR. *p<0.05 vs Runx1F/+; #p<0.05 vs Prx1-Cre; Runx1F/+.
Runx1 regulates mesenchymal progenitor cell differentiation into chondrocytes
To examine whether Runx1 is required for mesenchymal cell differentiation into the cartilaginous phenotype, we performed in vitro deletion of Runx1 from Runx1F/F derived micromass cultures using adeno-Cre viruses or adeno-GFP controls. Runx1 protein expression was efficiently inhibited in the adeno-Cre infected cultures at all time points compared to GFP-infected controls (Fig. 6A). Expression of Runx2 and Sox9 proteins was significantly inhibited at day 4 following Runx1 deletion (by 59% and 63% respectively) then it was significantly upregulated at 14 days (by 3.7 and 7.2 folds respectively) compared to controls. Runx3 protein levels were only significantly inhibited in micromass lacking Runx1 after 4 days in culture (by 44%). Loss of Runx1 function also resulted in decreased chondrogenesis and chondrocyte differentiation compared to GFP infected controls as evidenced by decreased alcian blue staining at day 4 and fewer nodules at later stages (Fig. 6B). The data indicate that Runx1 regulates chondrogenesis of a mesenchymal progenitor cell population in vitro, independently from its potential role in hematopoiesis.
Figure 6. Runx1 regulates the chondrogenic differentiation of the early mesenchymal progenitor cells in vitro.
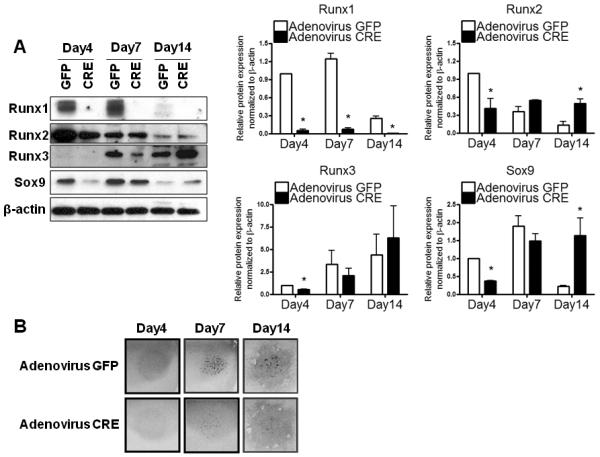
Pure mesenchymal stem cell populations isolated from E12.5 limb buds of Runx1F/F were infected with adeno-Cre virus and then cultured as micromass. Adeno-GFP virus was used as a control. (A) Nuclear fractions isolated from micromass culture were used for protein analyses of Runx1, Runx2, Runx3, Sox9, and β-actin at day 4, 7, and 14. Three independent experiments were performed to measure protein levels of Runx1, Runx2, Runx3, and Sox9. Their band intensities were quantified by NIH ImageJ. Values are expressed as means ± SE, normalized to β-actin. *p<0.05 vs adeno-GFP infected controls at the same time point. (B) Micromass cultures were stained with Alcian blue at the time points indicated above.
Runx1 directly binds regulatory sequences on the Sox 5, 6, and 9 promoters
Given the effects observed on chondrogenesis we examined the direct occupancy of the promoter regions of the three major chondrogenic factors Sox9, Sox5 and Sox6 (39). We first identified two 100% matching Runx responsive elements between nuclotides -1705 and -1700 and between nucleotides -1047 and -1042 on the Sox9 promoter sequence. Similarly, the Sox5 and Sox6 promoters harbor Runx1 response elements (between nucleotides -1824 and -1818 for Sox5) and (between nucleotides -2923 and -2918 as well as between nucleotides -1123 and -1119 for Sox6) (Fig. 7A).
Figure 7. Runx1 directly binds regulatory sequences on the Sox 5, 6, and 9 promoters.
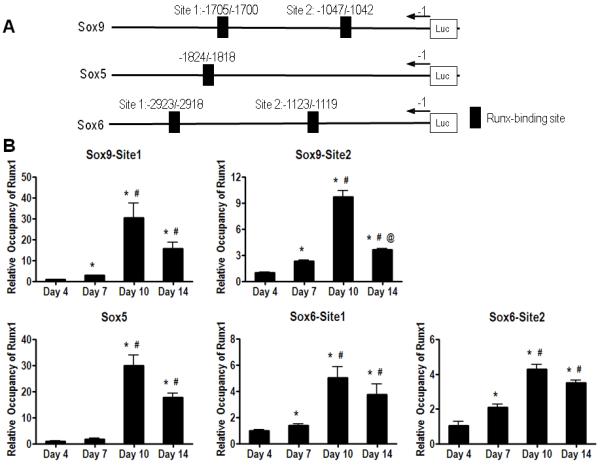
(A) 100% matched Runx1 binding sites of Sox9, Sox5, and Sox6 promoters are presented. (B) Chromatin extracted from micromass culture at days 4, 7, 10, and 14 was used for ChIP analyses using anti-Runx1 antibody or a negative control IgG antibody. Immunoprecipitated and input DNA were analyzed by real time-PCR using primers, which embrace Runx1 responsive elements of each Sox9, Sox5, and Sox6 genes. Values are expressed as means ± SE from three independent experiments. *p < 0.05 vs day 4; #p<0.05 vs day 7; and @p<0.05 vs day 10.
Figure 7B shows that Runx1 binds directly to all response elements on Sox9, 5 & 6 promoters as evidenced by ChIP assay analyses using chromatin from limb-bud micromass cultured for 4, 7, 10 and 14 days. A single PCR product for each of the Sox9, Sox5, and Sox6 promoter regions was obtained in input regardless of the stage of chondrdocyte differentiation (data not shown). Conversely, no or minimal PCR amplification was observed in chromatin extracts that were immunoprecipitated with a negative IgG control antibody (data not shown). Quantification of these bindings (Fig. 7B) showed that binding of endogenous Runx1 to both sites on the Sox9 promoter was increased with chondrocyte differentiation between days 4 and 10 then decreased by day 14. Similarly, all Runx binding sites on the Sox5 and Sox6 promoters were gradually occupied with Runx1 protein between days 4 and 14 with a maximum of residence in 10-day micromass cultures. The data indicate that Runx1 directly bound regulatory sequences of the promoters of Sox9, Sox5, and Sox6 genes.
Discussion
The observation that Runx1 is expressed in skeletal elements of developing mice (23-26) and the periosteum of adult mice (24) prompted us to examine its potential roles in skeletal tissue. Additionally, while the function of Runx1 during skeletal development remains poorly understood, its role during skeletal repair was completely unknown. Thus, to circumvent the embryonic lethality of the Runx1 global knock-out mice (16), we generated surviving mice with global incremental loss of Runx1 function and found that mutants with the lowest level of Runx1 activity (Runx1L148A/−) were runted. This runted phenotype was observed in Runx1L148A/− mice but not in mice happloinsufficient for Runx1, indicating that more than 50% loss of Runx1 activity was required to observe a growth plate phenotype. These findings are consistent with Yoshida and colleagues’ findings showing that Runx2−/− mice and Runx1+/−; Runx2−/− mice exhibited a similar degree of delayed chondrocyte differentiation (40), indicating that happloinsufficiency for Runx1 may not be sufficient to impair chondrogenesis; even when Runx2 is also deleted. Our data clearly indicate that a threshold of Runx1 is necessary to mediate proper growth.
The Runx1L148A/− mice displayed extended proliferating and hypertrophic zones within their growth plates. Similar phenotypes were previously observed in neonatal pups lacking the bone-related isoform of Runx2, which exhibited elongation of both the proliferating and hypertrophic zones (41). Subsequent delay in chondrocyte differentiation and terminal maturation also occurred as a consequence of absence or decrease of Runx3 (36,40). Mice haploinsufficient for Sox9 exhibited extension of their hypertrophic zone while their proliferating zone was not affected (42). Since we demonstrated that incremental loss of Runx1 function resulted in decreased number of Sox9, Runx2, and Runx3 positive cells in the proliferating zone of the Runx1L148A/− growth plates, we speculate that the delayed endochondral ossification in the Runx1L148A/− mice was likely due to the inhibition of chondrocyte differentiation and subsequently that of terminal maturation.
We and others have generated tissue specific Runx1 deletions in chondrocytes using the collagen type II-Cre transgenic mice (19) (data not shown). No skeletal phenotype was observed in these conditional knock-outs, indicating that Runx1 may be important at a stage of mesenchymal cell differentiation into the chondrogenic phenotype that is prior to type II collagen expression. Interestingly, when Runx1 was specifically deleted in limb mesenchyme using Prx1-Cre, only a slight and transient inhibition of sternal and xiphoidal mineralization was observed (19). Limb specific deletion of Runx1 and global loss of Runx2 double mutants (DKO: Prx1-Cre;Runx1F/F; Runx2−/−) also displayed a defective rib cage due to the absence of sternal structures, but no limb phenotype was observed (19). When Liakhovistskaia et al. (20) generated mice that express Runx1 only in endothelial and hematopoietic cells they found that sternebrae, the mineralization centers in the sternum, were less well developed at E17.5 - E18.5 (the mice died at birth), but noted no limb defects. Conditional deletion of Runx1 in all blood cells using Vav1-Cre also failed to produce a growth phenotype (43). It is possible that the difference in limb phenotypes between the Prx1-Cre;Runx1F/F conditional knock-outs the Runx1L148A/− mice could only be reconciled if the mesenchymal and hematopoietic cell lineages both require Runx1 for normal limb development. Because of the heterogeneity of the cell populations that contribute to limb development, we speculate that both Prx1 positive and negative cells contributes to the overall development of the limb in vivo, which could explain why we didn’t see a limb phenotype in the Runx1 conditional knock-outs. The 90% loss of Runx1 function in our micromass model further supports a role for Runx1 in regulating mesenchymal chondroprogenitor cell differentiation in vitro.
When we challenged adult Runx1L148A/− and Prx1-Cre;Runx1F/F mice for femoral fractures, both genetic models displayed impaired cartilaginous callus formation during bone repair. This delay in fracture healing may be due to inhibited recruitment of mesenchymal progenitor cells at the fracture site and a subsequent delay in chondrocyte differentiation. Interestingly, the absence of a limb phenotype in the fetus of Prx1-Cre;Runx1F/F contrasts with the obvious defect in fracture callus formation observed in adult mice. This is not the first report in which a phenotype is revealed by injury or stress. For example, mice with deletion of the bone morphogenetic protein BMP-2 by Prx1-Cre did not display severe limb abnormalities, yet when fractured, cartilaginous callus formation was abrogated (8). One of the reservoirs of Prx1-positive cells in adult mice is the periosteum, and these Prx1 positive periosteal progenitors are recruited to the fracture site for cartilaginous callus formation during fracture repair (5). Thus, our data imply that unlike embryonic skeletal development, intact Prx1-positive periosteal progenitor cells are necessary for cartilaginous callus formation during fracture healing. We speculate that loss of Runx1 function in Prx1-positive periosteal cells may lead to decreased commitment of these progenitor cells to the chondrogenic lineage.
Runx1L148A/− mice exhibited significantly decreased expression of Sox9, Runx2, and Runx3 positive stromal cells and chondrocytes within the fracture calluses. Fracture calluses of Prx1-Cre; Runx1F/F mutants also showed significantly reduced mRNA expression of Sox9, Sox5 and Sox6, which is reflective of decreased mesenchymal progenitor cell commitment to the early chondrogenic phenotype. Furthermore, Runx2 and Runx3 transcripts were inhibited in these calluses. This finding could explain the decreased cartilaginous callus formation observed. Our in vitro limb bud cultures in which Runx1 was abrogated demonstrated that Sox9, Runx2 and Runx3 gene expression was only inhibited in the early stages of chondrocyte differentiation. These results further suggest that Runx1 function in cartilage is primarily associated with the early stages of chondrogenesis. Kimura et al. (19) showed that dose-dependent abrogation of Runx1 and Runx2 resulted in a strong although highly localized cartilage phenotype in the rib cage, and concluded that Runx1 is essential for the differentiation of mesenchymal cells into chondrocytes through its regulation of Sox5 and Sox6. They reported that Runx1 did not regulate Sox9. Our ChIP data, however, indicate that the chondrogenic transcription factors Sox9, Sox5 and Sox6 are all direct targets of Runx1 during chondrogenesis in vitro. We show that while relatively lower occupancy of Sox5, 6, and 9 promoters by Runx1 is observed at day 4, Sox9 protein levels were relatively higher at the same time point. Based on the data, we speculate that since Runx binding sites on the promoter of Sox9 are potential targets for Runx2 and Runx3 as well as Runx1, endogenous Runx2 and Runx3 may competitively bind the Runx binding sites on these promoters. Future studies will delineate the temporal regulation of Sox genes by Runx1 and potentially the other members of the Runx family of transcription factors.
Based on our results, we conclude that while Runx1 alone in the Prx1 positive mesenchyme may not be required for the normal development of the limb, during fracture healing Runx1 controls early chondrocyte differentiation possibly through temporal regulation of the Sox genes as well as Runx2 and Runx3.
Supplementary Material
Supplementary Figure 1. Runx1 regulates the expression of chondrogenic transcription factors in growth plates during endochondral ossification. (A) The protein expressions of Sox9, Runx2, and Runx3 were detected in growth plates of tibias from all genotypes at 4-week-old age by immunohistochemistry. (B) Data were presented as a ratio of immunoreactive cells over total number of cells per w *p<0.05 vs Runx1 +/+; #p<0.05 vs Runx1 L148A/+; and @p<0.05 vs Runx1 +/−.
Supplementary Figure 2. The incremental loss of Runx1 impairs the mineralization and remodeling of callus during bone fracture healing. (A) All fracture callus sites from all genotypes mice were radiographed by x-ray at day 0, 7, 14, and 21 post fracture. (B) Mineralized fracture calluses of all genotypes at day 14 and 21 postfracture were graphically re-constructed by μCT. Total callus size and mineralized callus size were also measured from the fracture callus of all genotypes at day 14 and 21 post-fracture. n=4-7 mice per group. •p < 0.05 vs its own day 14 data. *p<0.05 vs Runx1 +/+; #p<0.05 vs Runx1 L148A/+; and @p<0.05 vs Runx1 +/−.
Supplementary Figure 3. Runx1 affects chondrogenic transcription factors during the early fracture callus formation. Calluses at day 7 postfracture were used for immunohistochemical analyses. The protein expressions of Sox9, Runx2, and Runx3 were detected in stromal cells (•) and chondrocytes (*) within the fracture calluses. n=4-7 mice per group.
Acknowledgments
We would like to acknowledge NIAMS and NCI for the support to conduct these experiments (NIH-RO1: AR052674-01 to Drissi MH and CA89419 to Speck NA). The University of Connecticut Health Center financed a part of this work. The authors are grateful to Robert Kosher and Caroline Dealy for sharing the Prx1-Cre transgenic mice.
DS, NS and HD participated in the study design. DS, LT, CM and MS performed the experiments. DS, LT, CM and MS collected the data. All authors participated in data interpretation and analyses as well as manuscript preparation. All the authors approved the final version of the manuscript.
Footnotes
Funding Source: NIH awards RO1: AR052674-01 (HD) and CA89419 (NAS)
Disclosures
All the authors state that they have no conflicts of interest
Author’s email address: Do Y. Soung: soung@uchc.edu, Laleh Talebian:laleh.talebian@dartmouth.edu, Christina J. Matheny:Christina.J.Matheny.Adv05@Alum.Dartmouth.ORG, Rosa Guzzo: guzzo@uchc.edu, Maren E. Speck: Maren.E.Speck@hitchcock.org, Jay R. Lieberman, JLieberman@uchc.edu, Nancy A. Speck: nancyas@exchange.upenn.edu, Hicham Drissi: drissi@uchc.edu
References
- 1.Shapiro F. Bone development and its relation to fracture repair. The role of mesenchymal osteoblasts and surface osteoblasts. Eur Cell Mater. 2008;15:53–76. doi: 10.22203/ecm.v015a05. [DOI] [PubMed] [Google Scholar]
- 2.Einhorn TA. The cell and molecular biology of fracture healing. Clin Orthop Relat Res. 1998;(355 Suppl):S7–21. doi: 10.1097/00003086-199810001-00003. [DOI] [PubMed] [Google Scholar]
- 3.Ushiku C, Adams DJ, Jiang X, Wang L, Rowe DW. Long bone fracture repair in mice harboring GFP reporters for cells within the osteoblastic lineage. J Orthop Res. 28(10):1338–47. doi: 10.1002/jor.21105. [DOI] [PubMed] [Google Scholar]
- 4.Colnot C. Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J Bone Miner Res. 2009;24(2):274–82. doi: 10.1359/jbmr.081003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawanami A, Matsushita T, Chan YY, Murakami S. Mice expressing GFP and CreER in osteochondro progenitor cells in the periosteum. Biochem Biophys Res Commun. 2009;386(3):477–82. doi: 10.1016/j.bbrc.2009.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu C, Miclau T, Hu D, Hansen E, Tsui K, Puttlitz C, Marcucio RS. Cellular basis for age-related changes in fracture repair. J Orthop Res. 2005;23(6):1300–7. doi: 10.1016/j.orthres.2005.04.003.1100230610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naik AA, Xie C, Zuscik MJ, Kingsley P, Schwarz EM, Awad H, Guldberg R, Drissi H, Puzas JE, Boyce B, Zhang X, O’Keefe RJ. Reduced COX-2 expression in aged mice is associated with impaired fracture healing. J Bone Miner Res. 2009;24(2):251–64. doi: 10.1359/jbmr.081002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsuji K, Bandyopadhyay A, Harfe BD, Cox K, Kakar S, Gerstenfeld L, Einhorn T, Tabin CJ, Rosen V. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet. 2006;38(12):1424–9. doi: 10.1038/ng1916. [DOI] [PubMed] [Google Scholar]
- 9.Kagoshima H, Shigesada K, Satake M, Ito Y, Miyoshi H, Ohki M, Pepling M, Gergen JP. The Runt-domain identifies a new family of heteromeric DNA-binding transcriptional regulatory proteins. Trends Genet. 1993;9:338–341. doi: 10.1016/0168-9525(93)90026-e. [DOI] [PubMed] [Google Scholar]
- 10.Daga A, Karlovich CA, Dumstrei K, Banerjee U. Patterning of cells in the Drosophila eye by Lozenge, which shares homologous domains with AML1. Genes Dev. 1996;10:1194–1205. doi: 10.1101/gad.10.10.1194. [DOI] [PubMed] [Google Scholar]
- 11.omori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755–64. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 12.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GWH, Beddington RSP, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for Cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–772. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 13.Levanon D, Bettoun D, Harris-Cerruti C, Woolf E, Negreanu V, Eilam R, Bernstein Y, Goldenberg D, Xiao C, Fliegauf M, Kremer E, Otto F, Brenner O, Lev-Tov A, Groner Y. The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. EMBO J. 2002;21(13):3454–63. doi: 10.1093/emboj/cdf370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoue K, Ozaki S, Shiga T, Ito K, Masuda T, Okado N, Iseda T, Kawaguchi S, Ogawa M, Bae SC, Yamashita N, Itohara S, Kudo N, Ito Y. Runx3 controls the axonal projection of proprioceptive dorsal root ganglion neurons. Nat Neurosci. 2002;5:946–954. doi: 10.1038/nn925. [DOI] [PubMed] [Google Scholar]
- 15.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321–30. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 16.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci U S A. 1996;93(8):3444–9. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inoue K, Shiga T, Ito Y. Runx transcription factors in neuronal development. Neural Dev. 2008;3:20. doi: 10.1186/1749-8104-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong WF, Kurokawa M, Satake M, Kohu K. Down-regulation of Runx1 expression by TCR signal involves an auto-regulatory mechanism and contributes to IL-2 production. J Biol Chem. doi: 10.1074/jbc.M110.166694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimura A, Inose H, Yano F, Fujita K, Ikeda T, Sato S, Iwasaki M, Jinno T, Ae K, Fukumoto S, Takeuchi Y, Itoh H, Imamura T, Kawaguchi H, Chung UI, Martin JF, Iseki S, Shinomiya K, Takeda S. Runx1 and Runx2 cooperate during sternal morphogenesis. Development. 137(7):1159–67. doi: 10.1242/dev.045005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liakhovitskaia A, Lana-Elola E, Stamateris E, Rice DP, van ’t Hof RJ, Medvinsky A. The essential requirement for Runx1 in the development of the sternum. Dev Biol. 2010;340(2):539–46. doi: 10.1016/j.ydbio.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 21.Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, Curley DP, Kutok JL, Akashi K, Williams IR, Speck NA, Gilliland DG. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106(2):494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho CY, Otterud B, Legare RD, Varvil T, Saxena R, DeHart DB, Kohler SE, Aster JC, Dowton SB, Li FP, Leppert M, Gilliland DG. Linkage of a familial platelet disorder with a propensity to develop myeloid malignancies to human chromosome 21q22.1-22.2. Blood. 1996;87(12):5218–24. [PubMed] [Google Scholar]
- 23.Smith N, Dong Y, Lian JB, Pratap J, Kingsley PD, van Wijnen AJ, Stein JL, Schwarz EM, O’Keefe RJ, Stein GS, Drissi MH. Overlapping expression of Runx1(Cbfa2) and Runx2(Cbfa1) transcription factors supports cooperative induction of skeletal development. J Cell Physiol. 2005;203(1):133–43. doi: 10.1002/jcp.20210. [DOI] [PubMed] [Google Scholar]
- 24.Lian JB, Balint E, Javed A, Drissi H, Vitti R, Quinlan EJ, Zhang L, Van Wijnen AJ, Stein JL, Speck N, Stein GS. Runx1/AML1 hematopoietic transcription factor contributes to skeletal development in vivo. J Cell Physiol. 2003;196(2):301–11. doi: 10.1002/jcp.10316. [DOI] [PubMed] [Google Scholar]
- 25.Levanon D, Brenner O, Negreanu V, Bettoun D, Woolf E, Eilam R, Lotem J, Gat U, Otto F, Speck N, Groner Y. Spatial and temporal expression pattern of Runx3 (Aml2) and Runx1 (Aml1) indicates non-redundant functions during mouse embryogenesis. Mech Dev. 2001;109(2):413–7. doi: 10.1016/s0925-4773(01)00537-8. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Belflower RM, Dong YF, Schwarz EM, O’Keefe RJ, Drissi H. Runx1/AML1/Cbfa2 mediates onset of mesenchymal cell differentiation toward chondrogenesis. J Bone Miner Res. 2005;20(9):1624–36. doi: 10.1359/JBMR.050516. [DOI] [PubMed] [Google Scholar]
- 27.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33(2):77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 28.Matheny CJ, Speck ME, Cushing PR, Zhou Y, Corpora T, Regan M, Newman M, Roudaia L, Speck CL, Gu TL, Griffey SM, Bushweller JH, Speck NA. Disease mutations in RUNX1 and RUNX2 create nonfunctional, dominant-negative, or hypomorphic alleles. EMBO J. 2007;26(4):1163–75. doi: 10.1038/sj.emboj.7601568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bravo J, Li Z, Speck NA, Warren AJ. The leukaemia-associated AML1 (Runx1)-CBFβ complex functions as a DNA-induced molecular clamp. Nat Struct Biol. 2001;8:371–377. doi: 10.1038/86264. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Yan J, Matheny CJ, Corpora T, Bravo J, Warren AJ, Bushweller JH, Speck NA. Energetic contribution of residues in the Runx1 Runt domain to DNA binding. J. Biol Chem. 2003;278:33088–33096. doi: 10.1074/jbc.M303973200. [DOI] [PubMed] [Google Scholar]
- 31.Berardi M, Sun C, Zehr M, Abildgaard F, Peng J, Speck NA, Bushweller JH. The Ig fold of the core binding factor α Runt domain is a member of a family of structurally and functionally related Ig fold DNA binding domains. Structure Fold Des. 1999;7:1247–1256. doi: 10.1016/s0969-2126(00)80058-1. [DOI] [PubMed] [Google Scholar]
- 32.Bonnarens F, Einhorn TA. Production of a standard closed fracture in laboratory animal bone. J Orthop Res. 1984;2(1):97–101. doi: 10.1002/jor.1100020115. [DOI] [PubMed] [Google Scholar]
- 33.Kaback LA, Soung do Y, Naik A, Smith N, Schwarz EM, O’Keefe RJ, Drissi H. Osterix/Sp7 regulates mesenchymal stem cell mediated endochondral ossification. J Cell Physiol. 2008;214(1):173–82. doi: 10.1002/jcp.21176. [DOI] [PubMed] [Google Scholar]
- 34.Kaback LA, Soung do Y, Naik A, Geneau G, Schwarz EM, Rosier RN, O’Keefe RJ, Drissi H. Teriparatide (1-34 human PTH) regulation of osterix during fracture repair. J Cell Biochem. 2008;105(1):219–26. doi: 10.1002/jcb.21816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Kong L, Carlson CS, Liu CJ. Cbfa1-dependent expression of an interferon-inducible p204 protein is required for chondrocyte differentiation. Cell Death Differ. 2008;15(11):1760–71. doi: 10.1038/cdd.2008.112. [DOI] [PubMed] [Google Scholar]
- 36.Soung do Y, Dong Y, Wang Y, Zuscik MJ, Schwarz EM, O’Keefe RJ, Drissi H. Runx3/AML2/Cbfa3 regulates early and late chondrocyte differentiation. J Bone Miner Res. 2007;22(8):1260–70. doi: 10.1359/jbmr.070502. [DOI] [PubMed] [Google Scholar]
- 37.Bradley EW, Drissi MH. WNT5A regulates chondrocyte differentiation through differential use of the CaN/NFAT and IKK/NF-kappaB pathways. Mol Endocrinol. 24(8):1581–93. doi: 10.1210/me.2010-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, Shiina M, Sato K, Kumasaka T, Yamamoto M, Ishii S. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFβ. Cell. 2001;104:755–767. doi: 10.1016/s0092-8674(01)00271-9. [DOI] [PubMed] [Google Scholar]
- 39.Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16(21):2813–28. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, Yamana K, Zanma A, Takada K, Ito Y, Komori T. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004;18(8):952–63. doi: 10.1101/gad.1174704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu JC, Lengner CJ, Gaur T, Lou Y, Hussain S, Jones MD, Borodic B, Colby JL, Steinman HA, van Wijnen AJ, Stein JL, Jones SN, Stein GS, Lian JB. Runx2 protein expression utilizes the Runx2 P1 promoter to establish osteoprogenitor cell number for normal bone formation. J Biol Chem. 2011;286(34):30057–70. doi: 10.1074/jbc.M111.241505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bi W, Huang W, Whitworth DJ, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci U S A. 2001;98(12):6698–703. doi: 10.1073/pnas.111092198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature. 2009;457(7231):887–91. doi: 10.1038/nature07619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang YY, Crute BE, Kelley JJ, Huang X, Yan J, Shi J, Hartman KL, Laue TM, Speck NA, Bushweller JH. Biophysical characterization of interactions between the core binding factor alpha and beta subunits and DNA. FEBS Lett. 2000;470(2):167–72. doi: 10.1016/s0014-5793(00)01312-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Runx1 regulates the expression of chondrogenic transcription factors in growth plates during endochondral ossification. (A) The protein expressions of Sox9, Runx2, and Runx3 were detected in growth plates of tibias from all genotypes at 4-week-old age by immunohistochemistry. (B) Data were presented as a ratio of immunoreactive cells over total number of cells per w *p<0.05 vs Runx1 +/+; #p<0.05 vs Runx1 L148A/+; and @p<0.05 vs Runx1 +/−.
Supplementary Figure 2. The incremental loss of Runx1 impairs the mineralization and remodeling of callus during bone fracture healing. (A) All fracture callus sites from all genotypes mice were radiographed by x-ray at day 0, 7, 14, and 21 post fracture. (B) Mineralized fracture calluses of all genotypes at day 14 and 21 postfracture were graphically re-constructed by μCT. Total callus size and mineralized callus size were also measured from the fracture callus of all genotypes at day 14 and 21 post-fracture. n=4-7 mice per group. •p < 0.05 vs its own day 14 data. *p<0.05 vs Runx1 +/+; #p<0.05 vs Runx1 L148A/+; and @p<0.05 vs Runx1 +/−.
Supplementary Figure 3. Runx1 affects chondrogenic transcription factors during the early fracture callus formation. Calluses at day 7 postfracture were used for immunohistochemical analyses. The protein expressions of Sox9, Runx2, and Runx3 were detected in stromal cells (•) and chondrocytes (*) within the fracture calluses. n=4-7 mice per group.