

Abstract
Gene expression in eukaryotes can be regulated by controlling the efficiency of transcript elongation by RNA polymerase II. The composition of the elongation complex is, however, poorly understood. Previous work has identified DNA sequences which block RNA polymerase II transcription and factors which stimulate RNA chain elongation. Here, I have purified elongation complexes arrested at discrete template locations. Complexes were rapidly and efficiently precipitated from in vitro transcription reactions using a monoclonal antibody that binds RNA. The isolated complexes remained transcriptionally active. This technique enables the facile manipulation of transcription elongation complexes. Using this approach, I show that transcription initiation factor α is not associated with a RNA polymerase II elongation complex. Since others have shown that α associates stoichiometrically with DNA, RNA polymerase II, and other required factors in an initiation complex, this work suggests that α departs from the transcription complex after nucleotides are required but before extensive RNA chain synthesis. In this regard α resembles the bacterial promoter-recognition factor σ.
Many eukaryotic genes are regulated by controlling the efficiency of complete primary transcript synthesis (reviewed in Spencer and Groudine, 1990). Intragenic sites that block chain elongation by RNA polymerase II have been mapped (Maderious and Chen-Kiang, 1984; Bentley and Groudine, 1986; Reines et al., 1987; Kao et al., 1987; Skarnes et al., 1988; Resnekov and Aloni, 1989; Resnekov et al., 1989) and factors involved in transcript elongation have been identified (Sekimizu et al., 1976; Reinberg and Roeder, 1987b; Rappaport et al., 1987; Reines et al., 1989; Sluder et al., 1989; Price et al., 1989; Flores et al., 1989). The final target for the action of these transcription elongation factors is the RNA polymerase II elongation complex. How these proteins function to alter transcript synthesis is unclear. Indeed, the structure and composition of the RNA polymerase II elongation complex is poorly understood.
Promoter-dependent transcription by RNA polymerase II requires multiple initiation factors. Many of these factors have been purified and studied in vitro (reviewed in Sawadogo and Sentenac, 1990). They assemble, with RNA polymerase II and template DNA, into a nucleoprotein complex prior to transcript polymerization (Buratowski et al., 1989; Conaway and Conaway, 1990a; Maldonado et al., 1990); however, little information is available regarding the fate of these factors subsequent to the initiation of RNA synthesis. It has been suggested that human transcription initiation factors TFIIB and TFIIE/F are released from the template under transcription conditions (van Dyke et al., 1989); however, it is not known if transcribing RNA polymerase II is associated with any initiation factors after it departs the promoter region.
In bacteria, the sigma subunit of RNA polymerase is required for promoter recognition. σ binds to the core polymerase and is an integral component of the initiation complex and early elongation complex, but is not associated with the transcribing enzyme (Hansen and McClure, 1980; Carpousis and Gralla, 1985; Straney and Crothers, 1985; Krummel and Chamberlin, 1989).
Two RNA polymerase II initiation factors, TFIID and βγ, display σ-like structural or functional properties (Hahn et al., 1989; Horikoshi et al., 1990; Conaway and Conaway, 1990b). A number of eukaryotic transcription initiation factors can bind to purified RNA polymerase II (Zheng et al., 1987; Sopta et al., 1985; Reinberg and Roeder, 1987a; Flores et al., 1988; Burton et al., 1988). Therefore, it seemed possible that an initiation factor(s) may be associated with RNA polymerase II, either directly or indirectly, during RNA chain elongation. One function of an elongation complex-associated factor might be to alter the properties of the elongating RNA polymerase. Since elongation and termination by RNA polymerase II can be influenced by the nature of the promoter from which transcription originates (Neuman de Vegvar et al., 1986; Hernandez and Lucito, 1988; Bentley and Groudine, 1988; Miller et al., 1989; Spencer et al., 1990; Spencer and Groudine, 1990), it is plausible that initiation factors play an important role in the transcription elongation and termination reactions of RNA polymerase II. In fact, transcription initiation factors have been shown to affect transcript elongation by RNA polymerase II (Flores et al., 1989; Price et al., 1989). An elongation factor, SII, can enter the transcription cycle after initiation and alter the rate of RNA chain elongation (Sekimizu et al., 1976; Reinberg and Roeder, 1987a; Rappaport et al., 1987; Reines et al., 1989; Sluder et al., 1989). To understand how transcription elongation is regulated and how elongation factors modify the elongation complex, we need to identify the elements of the RNA polymerase II elongation complex.
Five fractions from rat liver, α, βγ, σ, ε, and τ, have been identified as general transcription initiation factors required for accurate RNA polymerase II transcription from the adenovirus major late promoter (Conaway and Conaway, 1989a, 1989b, 1990a, 1990b; Conaway et al., 1987, 1990). α and βγ have been purified to apparent homogeneity; they resemble human general transcription factors BTFIII and TFIIF, respectively, which have been shown to bind to RNA polymerase II (Zheng et al., 1987; Flores et al., 1989). Both join a DNA-bound preinitiation complex which contains RNA polymerase II. Therefore, these proteins are candidates for initiation factors which may associate with transcribing RNA polymerase II after the enzyme leaves the promoter.
The RNA polymerase II elongation complex must contain the DNA template, nascent RNA, and one or more subunits of RNA polymerase II; however, a direct study of the composition of the elongation complex with respect to the RNA polymerase II initiation factors is lacking. Separations employing gel filtration and electrophoresis have been designed to isolate RNA polymerase II elongation complexes from in vitro transcription reactions (Ackerman et al., 1983; Coppola and Luse, 1984; Bengal et al., 1989). These methods are slow, and gel filtration often results in the unacceptable dilution of proteins. Nonspecific, or partially assembled, nucleoprotein complexes may not be well resolved from functional elongation complexes using these methods and the efficacy of such separations has not always been examined thoroughly.
I describe here the use of an anti-RNA monoclonal antibody to explore the structure and function of an RNA polymerase II elongation complex. These data show that initiation factor α, which associates with the promoter, RNA polymerase II, and other accessory factors in an initiation complex, is not associated with an RNA polymerase II elongation complex. Thus, α appears to be released from the complex after nucleotides are required but before RNA chains of 145 nt1 are polymerized.
EXPERIMENTAL PROCEDURES
Materials and Methods
Enzymes and Proteins
RNA polymerase II was purified from rat liver as described (Conaway et al., 1987) except the cytosol fraction (800 × g supernatant) was used as starting material for phosphocellulose (P11, Whatman), DEAE-cellulose (DE-52, Whatman), and DEAE-Sephadex A25 (Sigma) chromatography. Rat liver transcription initiation factors α and B′ (partially purified βγ) were prepared as described (Conaway et al., 1987) except the DE52 step was omitted from the B′ purification.
Anti-RNA-producing hybridoma D44 was established previously (Eilat et al., 1982) and ascites fluid from mice innoculated with D44 hybridoma cells was kindly provided by Dr. Dan Eilat (Hadassah University Hospital, Jerusalem, Israel). D44 IgG was isolated from ascites fluid by protein A-Sepharose chromatography.
SII was purified from calf thymus as described (Rappaport et al., 1987). An elongation factor that enables RNA polymerase II to elongate past site Ia was prepared from bovine brain by chromatography of a cytosolic extract on phosphocellulose as described (rat liver fraction B; Conaway et al., 1987).2 This fraction from rat liver also contains transcription read-through activity. For convenience, this activity will be tentatively referred to as SII.
Protein concentration was determined with protein assay dye reagent (Bio-Rad) according to the manufacturer’s directions using BSA (Sigma, Fraction V) as a standard.
DNA Templates
The plasmid pDNAdML contains the core adenovirus major later promoter (AdMLP; −50 to +10) inserted into pUC18 at the KpnI and XbaI sites (Conaway and Conaway, 1988). When linearized with NdeI, this template yields a 250-nt runoff transcript. The plasmid pAdTerm-2 contains a segment of the human histone H3.3 gene in which transcription by RNA polymerase II is arrested at three discrete sites (II, Ib, and Ia; Reines et al., 1989). The histone gene fragment was inserted into the unique AccI site of pDNAdML and yields RNA transcripts of 145, 185, and 205 when polymerase initiates transcription from the AdMLP and stops at sites II, Ib, and Ia, respectively. When cleaved with NdeI this plasmid yields a runoff transcript of 530 nt. The plasmid pAdTerm-2ΔXB was generated from pAdTerm-2 by removal of the 110-bp fragment extending from the XbaI to BstEII site.
Other Materials
Actinomycin D and yeast RNA were purchased from Sigma. Acetylated BSA was from Promega. Formalin-fixed Staphylococcus aureus (Immunoprecipitin) and NdeI were purchased from Bethesda Research Laboratories/Life Technologies. FPLC-purified NTPs, T4 polynucleotide kinase, PvuII, and PstI were purchased from Pharmacia LKB Biotechnology Inc. [α-32P]CTP and [γ-32P]ATP were purchased from Amersham Corp. and ICN, respectively.
Immunoprecipitation of Purified Nucleic Acids with D44 IgG
10-μl aliquots of the indicated amounts of D44 IgG in phosphate-buffered saline (20 mM sodium phosphate, pH 7.0, 150 mM NaCl) were mixed with 9,000 cpm (<6 ng) of 32P-labeled RNA or 60,000 cpm (approximately 20 ng) of 32P-labeled DNA in 60 μl of reaction buffer (20 mM Tris, 3 mM HEPES, pH 7.9, 62 mM KCl,2.2% polyvinyl alcohol, 3% (v/v) glycerol, 2 mM dithiothreitol, 0.5 mM EDTA, and 0.3 mg/ml acetylated BSA). Reactions were incubated for 30 min on ice. 20 μl of fixed S. aureus (washed three times in an equal volume of reaction buffer) were added and incubation was continued for 15 min on ice. Reactions were centrifuged for 1 min at 16,000 × g in a Fisher model 235C microcentrifuge. Supernatants were precipitated with 5% (w/v) trichloroacetic acid and filtered on Whatman GF/C filters (Chamberlin et al., 1979). Nucleic acids were eluted from the S. aureus pellets after resuspension in 60 μl of reaction buffer by the addition of an equal volume of SDS buffer (0.2 M Tris-HCl, pH 7.5, 25 mM EDTA, 0.3 M NaCl, 2% (w/v) SDS). Fixed S. aureus was removed by centrifugation at 16,000 × g for 1 min. This eluate was trichloroacetic acid-precipitated as above and quantitated by scintillation counting. 32P-labeled RNA was synthesized by Escherichia coli RNA polymerase (Gonzalez et al., 1977) from the plasmid pKK34–121 which contains an E. coli 5 S gene (Thayer and Brosius, 1985; transcripts shown in Fig. 1B, lane M). Template DNA was not removed from the reaction. 32P-labeled DNA was prepared by phosphorylating PstI-cleaved pAdTerm-2ΔXB with [γ-32P]ATP (4500 Ci/mmol) and T4 polynucleotide kinase (Maniatis et al., 1982). Labeled nucleic acids were purified by phenol/chloroform extraction and ethanol precipitation.
Fig. 1. Immunoprecipitation of nucleic acids with monoclonal antibody D44.

A, preference of D44 for RNA over DNA. Deproteinized [32P]RNA (<6 ng; ■) or 32P-labeled double-stranded DNA (20 ng; ●) were incubated with the indicated concentrations of D44 IgG. IgG-bound nucleic acids were precipitated with formalin-fixed S. aureus and quantitated by liquid scintillation counting. B, D44-dependent precipitation of 32P-labeled runoff (RO) transcript. [32P]RNA was synthesized in a reconstituted rat liver transcription reaction. 400 ng of D44 IgG (+) or buffer (−) were added and immune complexes were precipitated with fixed S. aureus. Soluble (S) and precipitable (P) fractions were analyzed by electrophoresis and autoradiography. The migration position of RNAs synthesized from pKK 34–121 (from bottom to top: 260, 380, 420, and 540 nt, lane M) are indicated by arrowheads at the left.
Reconstituted Rat Liver Transcription Reactions
AdMLP-directed RNA polymerase II transcription was reconstituted in vitro with partially purified initiation factors and RNA polymerase II as described by Conaway et al. (1987) (modified by Reines et al., 1989). 100–200 ng of DNA template were incubated with rat liver RNA polymerase II (0.5 μg) and fraction D (2 μg) in 20 μl of 20 mM HEPES, pH 7.9, 20 mM Tris-HCl, pH 7.9, 2% (w/v) polyvinyl alcohol, 0.4 mg/ml acetylated BSA (Promega), 0.15 M KCl, 2 mM DTT, and 7% (v/v) glycerol for 30 min at 28 °C. 33 μl of a solution containing fraction B′ (1 μg) and α (TSK phenyl 5-PW fraction; 3 ng unless stated otherwise) in the same buffer without KCl, were added and incubation continued for another 20 min. MgCl2, ATP, UTP, and [α-32P]CTP (>400 Ci/mmol) were added in 6 μl to final concentrations of 7 mM, 20 μM, 20 μM, and approximately 0.6 μM, respectively. In the absence of GTP, ternary complexes containing a 14-nt transcript were synthesized since the first G residue appears in the transcript at position +15. Unless stated otherwise, heparin (10 μg/ml) was added to ternary complexes before any additional nucleotides. Runoff transcripts were generated by providing 800 μM more of each NTP and continuing incubation for 15 min at 28 °C. Nucleic acids were isolated and processed for electrophoresis.
Synthesis of RNA Polymerase II Elongation Complexes
RNA polymerase II elongation complexes were formed on NdeI-cleaved pAdTerm-2 DNA as described by Reines et al. (1989). To form elongation complexes arrested at sites II, Ib, and Ia, heparin-treated ternary complexes were made 620 and 200 μM in unlabeled CTP and GTP, respectively, and incubated for 30 min at 28 °C. To form elongation complexes arrested exclusively at site Ia, heparin-treated ternary complexes were made 820 μM each in ATP and UTP, and 800 μM each in GTP and CTP, and incubated at 28 °C for 15 min.
Immunoprecipitation of RNA and Elongation Complexes from in Vitro Transcription Reactions
RNA or elongation complexes were precipitated directly from transcription reactions. Addition of the indicated amount of D44 IgG was followed by an incubation of 5–30 min on ice. (Maximum binding of D44 (at 6.5 μg/ml) was reached before 5 min.2) 10–20 μl of washed, formalin-fixed S. aureus were incubated with complexes on ice for 5 min. The mixture was centrifuged and RNA was eluted from S. aureus with SDS buffer as described above. For primer extension analysis, unlabeled RNA was extracted with phenol/chloroform and precipitated with ethanol. For electrophoresis and autoradiography labeled RNA was treated with 5 μg of proteinase K at 20 °C for 5 min and ethanol-precipitated.
Primer Extension Assay of Soluble α Transcription Activity after Precipitation of Elongation Complexes
Elongation complexes arrested at sites II, Ib, and Ia were formed as described above except ternary complexes were synthesized with unlabeled CTP (1 μM) and heparin was omitted. Control reactions were identical except they did not contain pAdTerm-2 DNA. Elongation complexes were precipitated with 0.4 μg of D44 IgG and fixed S. aureus as described above. The supernatant was removed and added to a reaction mixture containing 100 ng of supercoiled pDNAdML DNA that had been incubated with fraction D and RNA polymerase II for 30 min at 28 °C as described above. Fraction B′ (1 μg) was added and incubated for 20 min at 28 °C. MgCl2 was adjusted to 7 mM; ATP, UTP, CTP, and GTP were adjusted to 800 μM each. After an additional 15 min at 28 °C an equal volume of SDS buffer was added with 20 μg of yeast RNA. Nucleic acids were extracted with phenol and chloroform and precipitated with ethanol. Primer extension analysis was carried out as described (McKnight and Kingsbury, 1982). Nucleic acids were dissolved in 0.3 M sodium acetate with 25 fmol of a synthetic DNA primer (d(5′-GTTTTCCCAGT-CACGAC-3′); U.S. Biochemical Corp.) which was 5′-end-labeled with 32P to a specific activity of 2–3 × 106 cpm/pmol. After ethanol precipitation, nucleic acids were dissolved in 10 μl of 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1.25 M KCl, heated to 60 °C for 3 min, and hybridized at 45 °C for 1 h. 23 μl of extension buffer (20 mM Tris-HCl, pH 8.7, 10 mM MgCl2, 100 μg/ml actinomycin D, 5 mM dithiothreitol, 330 μM each of dATP, dGTP, dCTP, and TTP) were added and reactions were incubated with 180–200 units of Moloney murine leukemia virus reverse transcriptase (Bethesda Research Laboratories/Life Technologies) for 1 h at 37 °C. Nucleic acids were ethanol-precipitated and processed for electrophoresis.
Electrophoresis and Autoradiography
RNA was dissolved in 80% (v/v) formamide, 0.025% (w/v) xylene cyanole, and 0.025% (w/v) bromphenol blue in TBE (89 mM Tris, 89 mM boric acid, pH 8.0, 1 mM EDTA) and denatured at 95 °C for 3 min. DNA was dissolved in 80% (v/v) formamide, 0.025% (w/v) xylene cyanole, and 0.025% (w/v) bromphenol blue, 10 mM NaOH, and 1 mM EDTA. Samples were subjected to electrophoresis on 8% (primer extension experiments) or 5% ([32P]RNA) polyacrylamide gels in TBE. Gels were dried and exposed to X-Omat (Kodak) film at −80 °C with an intensifying screen. Quantitation of autoradiographic signals was performed with a Bio-Rad model 620 densitometer.
RESULTS
Characterization of the RNA Binding Properties of Monoclonal Antibody D44
A hybridoma (D44) which secretes RNA-reactive IgG3 has been identified previously (Eilat et al., 1982). To test the suitability of this reagent for immunoprecipitating RNA-associated elongation complexes, I characterized first the interaction of D44 with isolated RNA and DNA. It was important to show that D44 recognized RNA in preference to DNA such that transcription complexes would be selected via their RNA, and not DNA, moiety.
A fixed amount of deproteinized RNA or DNA was mixed with various amounts of D44 IgG. Immune complexes (anti-body-RNA complexes) were separated from unbound nucleic acid by adsorption to formalin-fixed S. aureus which contains surface bound protein A. This titration demonstrated that, at relatively low concentrations of IgG (9–35 nM), binding of D44 was highly selective for RNA over DNA (Fig. 1A). In the experiments described below, D44 concentrations in this range were used. Significant binding of D44 to DNA was detected only at high antibody concentrations. In addition, DNA in these reactions was present at a 4-fold higher concentration than the RNA in an equivalent reaction. Quantities even as low as 6 ng of RNA can be precipitated quantitatively using this antibody. This confirms and extends previous work which employed a competition, solid-phase binding assay to define the binding properties of D44 (Eilat et al., 1982). The nucleotide sequence of the RNAs studied here are largely those of the E. coli 5S rRNA (Thayer and Brosius, 1985); however, RNA transcribed from numerous templates of differing nucleotide sequence also bind D44 (Figs. 2–4).2
Fig. 2. Immunoprecipitation of RNA polymerase II elongation complexes.

A, D44-precipitated elongation complexes arrested at histone sites II and Ib remain NTP-responsive. Elongation complexes containing [32P]RNA were assembled at sites II, Ib, and Ia using limiting amounts of ATP and UTP as described under “Materials and Methods.” D44 (400 ng) and fixed S. aureus were added and the mixture was centrifuged. The precipitable fraction was resuspended and divided in half. One half (−) was prepared for electrophoresis. The other half was made 7 mM in MgCl2, 800 μM each in the four NTPs, and incubated for 30 min at 28 °C before preparation for electrophoresis. Reference RNAs are indicated as described in Fig. 1B, legend. Runoff (RO) and RNAs with 3′ termini at sites Ia. Ib. and II are indicated to the right. B, D44-precipitated elongation complexes arrested at site Ia remain SII-responsive. Elongation complexes containing [32P]RNA were assembled at site Ia, precipitated with D44 (400 ng), and resuspended in buffer. One sample was challenged with 12 ng of pure calf thymus SII (CT) or 3.3 μg of an Sll-containing fraction from bovine brain (Br) in the presence of 800 μM each of the four NTPs and 7 mM MgCl2. The identity of each RNA is indicated as above.
Fig. 4. Sedimentation of RNA polymerase II elongation complexes requires D44 IgG.

Elongation complexes containing [32P]RNA were formed at sites II, Ib, and Ia (lanes 1–4) or exclusively at site Ia (lanes 5–8). They were incubated with buffer (−) or D44 IgG (400 ng; +), fixed S. aureus and separated into soluble (S) and precipitable fractions (P). RNA was isolated from each fraction and subjected to electrophoresis and autoradiography. RNA identities are indicated as described above.
Specific initiation of transcription has been reconstituted in vitro using purified rat liver transcription initiation factors and RNA polymerase II (Conaway and Conaway, 1988, 1989a, 1989b, 1990a, 1990b; Conaway et al., 1987, 1990). The use of this purified, reconstituted transcription system has facilitated the analysis of transcription elongation by RNA polymerase II (Reines et al., 1989). To determine if D44 could mediate the direct isolation of RNA from a reconstituted transcription reaction, runoff RNA was synthesized in vitro and subjected to immunoprecipitation. Efficient, antibody-dependent precipitation of intact runoff transcript was observed (Fig. 1b).
Immunopurified Elongation Complexes Remain Functional
The ability of D44 to mediate the precipitation of elongation complex-associated RNA was tested next. That an elongation complex was precipitated in addition to labeled, nascent RNA, was assayed by allowing the D44-isolated material to renew RNA chain elongation.
Elongation complexes were synthesized using partially purified rat liver transcription factors, RNA polymerase II, and a template containing previously characterized transcription arrest sites (II, Ib, and Ia) from a human histone gene (H3.3; Reines et al., 1987, 1989). In the first set of experiments (Fig. 2A) elongation complexes were formed at sites II, Ib, and Ia by restricting the levels of ATP and UTP to 20 μM each. Complexes were selected with D44 and fixed S. aureus. After selection with D44, RNA polymerase II was efficiently chased from sites II and Ib when a high concentration of NTPs was added, indicating that immunopurified elongation complexes remained fully functional. In agreement with previous results (Reines et al., 1989), only a fraction of the RNA polymerases can elongate through site Ia even in the presence of high levels of NTPs.
Next, I tested whether or not elongation complexes arrested at site Ia remained SII-responsive after D44 selection. Elongation complexes assembled exclusively at site Ia were immunopurified with D44 and challenged with elongation factor SII. Polymerase arrested at site Ia was capable of extending RNA chains to runoff length in the presence of pure calf thymus SII or partially purified bovine brain SII (Fig. 2B). Thus, D44-purified elongation complexes remained both nucleotide-responsive (at sites II and Ib) and SII-responsive (site Ia). Remarkably, neither RNA-bound IgG, nor the associated S. aureus, inhibited elongation by RNA polymerase II in these assays.
Immunoprecipitation of elongation complexes provided for a rapid change in the solute composition of the reaction mixture. This was evident from the fact that efficient elongation by immunopurified complexes was dependent upon adding back both NTPs and MgCl2, which has been depleted during immunopurification. In the absence of added MgCl2 or NTPs, immunopurified elongation complexes failed to elongate RNA chains even when incubated for 1 h in the presence of bovine brain SII (Fig. 3). A control reaction containing α-amanitin (1 μg/ml) confirmed that the activity of RNA polymerase II was essential for transcript elongation.
Fig. 3. Reactivation of D44-purified elongation complexes requires the addition of NTPs and MgCl2.

Elongation complexes containing [32P]RNA were assembled at site Ia, D44-precipitated, and resuspended in reaction buffer with 3.3 μg of an SII-containing fraction from bovine brain (P.6). MgCl2 (7 mM) and all four NTPs (800 μM) were added as indicated and incubation continued for 30 or 60 min at 28 °C. One reaction also received α -amanitin (1 μg/ml; aman). RNA was isolated and analyzed by electrophoresis and autoradiography. RNA identities are indicated as described above.
These elongation complexes contained at least: 1) the 3-kbp DNA template; 2) a nascent transcript of 145–205 nt; and 3) RNA polymerase II. An experiment was designed to prove that sedimentation of elongation complexes was dependent upon D44 IgG and that the large elongation complex was not inherently insoluble. Elongation complexes arrested at sites II, Ib, and Ia were subjected to the precipitation procedure in the presence or absence of D44 IgG. Sedimentation of these RNA polymerase II elongation complexes were absolutely dependent upon D44 IgG (Fig. 4). From these and other data,2 it can be concluded that neither DNA, RNA, nor any individual rat liver transcription factor sediments under these conditions.
In summary, isolation of RNA polymerase II elongation complexes was dependent upon an anti-RNA antibody and RNA synthesis. RNA chain elongation by the isolated complexes was indistinguishable from that of native complexes.
Transcription Initiation Factor α Is Not Associated with Immunopurified RNA Polymerase II Elongation Complexes
The possible association of rat liver α with RNA polymerase II elongation complexes was studied. If α, which binds to the RNA polymerase II initiation complex (Conaway and Conaway, 1990), departs from the complex after nucleotide addition, it should be found in the soluble phase of a reaction from which elongation complexes have been extracted. This was observed when supernatant fractions, following removal of elongation complexes, were assayed for α activity in a second transcription reaction (Fig. 5; +DNA lanes).
Fig. 5. Primer extension assay of soluble α transcription activity.
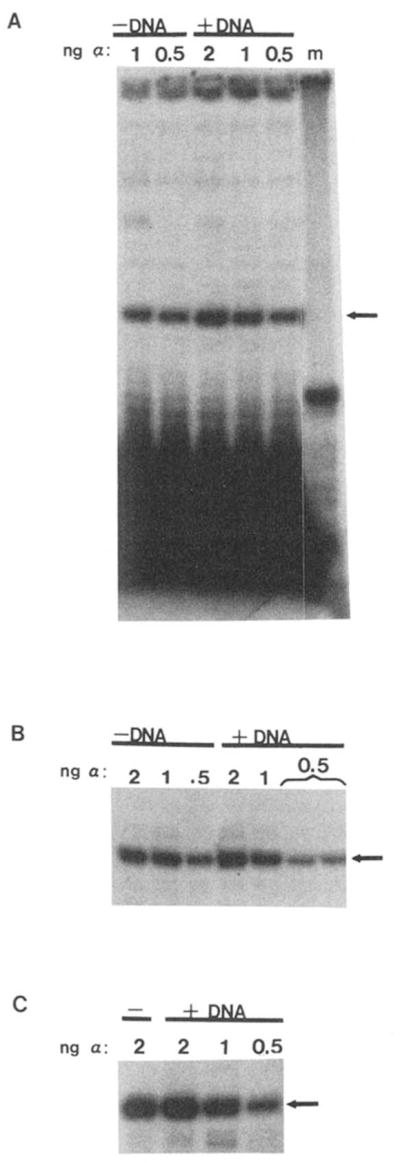
Elongation complex assembly reactions (+DNA) were performed as described under “Materials and Methods” using 0.5, 1, or 2 ng of α. In control reactions, pAdTerm-2 template DNA was omitted (−DNA). D44 IgG (400 ng) and fixed S. aureus were added, and soluble and precipitable fractions were separated by centrifugation. The soluble fractions were added to a second template (pDNAdML) which had been incubated with partially purified rat liver RNA polymerase II and fraction D. Fraction B′, MgCl2, and NTPs were added and accurate transcription in this reaction was measured using a primer-extension assay. A, B, and C are the results from three independent experiments. A shows the entire gel including the 17-nt primer at the bottom and the origin at top. B and C show only the 80-nt extension product (arrow). Lane m contains a 32P-labeled 50-nt DNA oligonucleotide as a size reference. Note that in B one 0.5-ng sample was applied to two lanes.
All of the α activity used to assemble elongation complexes was recovered after elongation complexes had been removed by D44 precipitation. This is apparent from controls where pAdTerm-2 DNA template was omitted from elongation complex assembly reactions (Fig. 5A, compare −DNA to +DNA lanes). Since, in the absence of DNA, transcription complexes could not form and RNA could not be synthesized, these mock reactions served as an index of the amount of α added to each reaction. The recovered α transcription activity was quantitated by densitometric scanning of autoradiographs. No significant difference was detected between control (−DNA) and experimental (+DNA) data points (Student’s t test; p < 0.05).
To substantiate these results, limiting amounts of α were used for complex synthesis such that α found in the soluble phase of an immunoprecipitation reaction would not simply reflect a saturation of α binding sites. 0.5–2 ng of α gave a linear response when assayed in a reconstituted runoff transcription reaction2 and in this template-selective primer extension assay (Fig. 5) and was consistent with previous measurements (Conaway et al., 1987). Control experiments demonstrated that the major extension product representing accurate transcription was: 1) dependent upon added α, 2) dependent upon the adenovirus major late promoter sequences in pAdTerm-2, and 3) abolished in the presence of 1 μg/ml α-amanitin.2
DISCUSSION
This report describes a novel way both to isolate and concentrate rapidly RNA polymerase II elongation complexes. Using a monoclonal antibody that binds tightly to RNA, I have been able to isolate a population of template-associated RNA polymerase II molecules. These enzyme molecules are, by definition, the active species of RNA polymerase II since they were selected through an RNA moiety. This is significant since highly purified and even homogeneous preparations of bacterial and eukaryotic RNA polymerases are mixtures of active and inactive molecules (Chamberlin et al., 1979; Kadesch and Chamberlin, 1982).
A variety of approaches to isolate elongation complexes from reconstituted transcription reactions were considered. One approach was to incorporate nucleotide analogs that contain affinity ligands (e.g. thio-UTP or biotinylated UTP) into RNA chains (Melvin and Keir, 1977; Langer et al., 1981). RNA, and in the case of paused polymerase enzymes, elongation complexes, could then be extracted from solution with solid phase-bound reagents (e.g. mercury or avidin). However, substitution of base analogs for naturally occurring nucleotides can be problematic. For example, RNA polymerase II cannot use biotinylated UTP as a substrate (Langer et al., 1981).2
The immunopurification technique described here is preferable to existing methods for elongation complex isolation. Compared to column chromatography it is both more rapid and more efficient. While gel filtration often results in sample dilution, immunoprecipitation effectively concentrates the sample. This should allow one to identify various proteins present in the complex, particularly those which may be of low abundance. Furthermore, D44 is reactive with a variety of RNAs of differing sequence.2 This should permit the analysis of many different RNA-containing nucleoprotein complexes without the need to incorporate derivatized nucleotides into RNA chains.
D44-mediated sedimentation of elongation complexes resulted in the cosedimentation of 5% of the input DNA.2 This serves as an estimation of the maximum number of templates utilized since some DNA precipitation could be accounted for by cross-reactivity of the DNA with D44 (Fig. 1A). This is consistent with previous findings that relatively few templates are utilized in in vitro transcription reactions (Weil et al., 1979; Manley et al., 1980; Ackerman et al., 1983; Coppola and Luse, 1984; Conaway et al., 1987; Lue and Romberg, 1987; Wampler et al., 1990) and underscores the value of this approach in selecting components which participate in transcription over those that are inactive.
The elongation complexes examined here result from a cessation of transcription by RNA polymerase II at naturally occurring genetic elements. Polymerase arrested at one of these sites (Ia) can respond to the stimulatory action of elongation factor SII (Reines et al., 1989). This block to elongation may be used in vivo to regulate histone H3.3 expression.3 Whether complexes arrested at these sites are representative of RNA polymerase II elongation complexes in general, or are somehow unique, will have to await a comparison between complexes located at these and other sites. In any case, the study of discrete elongation complexes arrested at the H3.3 gene sites has enabled me to begin to analyze the protein composition of a discrete, and in the case of site Ia, homogeneous, collection of elongation complexes.
A complex set of initiation factors are involved in transcription by RNA polymerase II (reviewed in Sawadogo and Sentenac, 1990). One factor, TFIID, is involved in nucleating assembly of the initiation complex on promoter DNA. Most other factors do not, however, bind DNA directly. How these factors function in initiation is less clear. Separation and purification of factors has resulted in the construction of models which describe the assembly of the initiation complex (Fire et al., 1984; Reinberg et al., 1987; van Dyke et al., 1988; Buratowski et al., 1989; Conaway and Conaway, 1990a; Sawadogo and Sentenac, 1990). Most of this work has focused on the steps leading up to initiation; little information is available regarding the fate of these factors subsequent to nucleotide addition. In contrast, the role of the E. coli accessory factor, σ, as well as other factors that participate in transcript elongation in bacteria, are relatively well understood (reviewed in Yager and von Hippel, 1987; Helmann and Chamberlin, 1988). E. coli RNA polymerase undergoes a number of structural and functional transitions which lead to the departure of σ factor from the core enzyme after approximately 10 residues have been polymerized (Hansen and McClure, 1980; Carpousis and Gralla, 1985; Straney and Crothers, 1985; Krummel and Chamberlin, 1989). σ release accompanies a profound change in the stability of the initial transcription complex after which RNA polymerase can become committed to extensive chain elongation. It is noteworthy therefore, that an RNA polymerase II transcription complex that has polymerized 10 nt is more stable than one which has incorporated only seven (Coppola and Luse, 1984). By analogy to the bacterial elongation complex, the ability of RNA polymerase II to clear the promoter could be related to the departure of a factor from the initiation complex. This step could in turn be governed by regulatory transcription factors.
Some RNA polymerase II initiation factors resemble bacterial σ in that they: 1) bind purified RNA polymerase II (RAP proteins, BTFIII, TFIIE/F; Sopta et al., 1985; Reinberg and Roeder, 1987a, 1987b; Zheng et al., 1987; Flores et al., 1988, 1989); 2) regulate the interaction between DNA and RNA polymerase II (βγ Conaway and Conaway, 1990b); or 3) share sequence homology with σ (TFIID; Hahn et al., 1989; Horikoshi et al., 1990), although the significance of this latter relationship has been questioned (Hahn et al., 1989). Our understanding of the role of these factors in each phase of the transcription cycle is limited. Thus, it is of interest to investigate any association the transcription initiation factors may have with the RNA polymerase II elongation complex.
Using the detergent Sarkosyl, it has been suggested that at least one RNA polymerase II initiation factor remains template-associated following transcript synthesis (Hawley and Roeder, 1985). Other studies have suggested that the entire initiation complex is disassembled after each round of initiation (Tolunay et al., 1984; Kadonaga, 1990). Yet none of these experiments were designed to measure a physical association of initiation factors with elongation complexes.
By immunoprecipitation of elongation complexes I have been able to isolate an intermediate in the transcription cycle (the engaged elongation complex) and show that α does not cosediment with these complexes. This approach permits the analysis of the fate of initiation factors subsequent to nucleotide addition but prior to the completion of transcript synthesis; i.e before RNA polymerase II leaves the template. Although α, RNA polymerase II, template DNA, and transcription factors βγ, δ, ε and τ, assemble into a transcriptionally competent nucleoprotein complex (Conaway and Conaway, 1990a), these data indicate that, at some point after NTPs are required, α departs from the RNA polymerase II complex (Buratowski et al., 1989; Conaway and Conaway, 1990a; Maldonado et al., 1990). It has been suggested that α is the rat counterpart to the human factor BTFIII which binds RNA polymerase II (Sawadogo and Sentenac, 1990). This raises the possibility that α binds to RNA polymerase II. If so, α would behave much like bacterial σ factors which bind to RNA polymerase and whose departure from core RNA polymerase denotes a transition from initiation to elongation.
The possibility that α was displaced from the elongation complex during centrifugation of the elongation complexes cannot be dismissed. Since immunopurified elongation complexes are fully active, it is clear however, that α is not required by promoter-initiated RNA polymerase for transcript elongation, although it is required for chain initiation. In this context, it should be noted that RNA polymerase III transcription initiation factors TFIIIA and TPIIIC can be removed from assembled RNA polymerase III initiation complexes with 0.5 M NaCl or heparin, and are dispensable subsequently for accurate transcript synthesis by RNA polymerase III (Kassavetis et al., 1990). These factors have been considered assembly factors (Paule, 1990). α is an RNA chain initiation factor that is required for the assembly of a complete initiation complex but does not require magnesium or nucleotides to do so (Conaway and Conaway, 1990). Whether α departs from the initiation complex after RNA synthesis starts or after a distinct nucleotide-dependent event remains to be studied.
Acknowledgments
I thank Dr. Dan Eilat for the D44 antibody, Drs. R. and J. Conaway for the TSK-phenyl α, and Drs. C. Kane and L. SivaRaman for calf thymus SII. I also thank Drs. J. Conaway, R. Conaway, and L. Coluccio for continuing and helpful discussion and colleagueship and Drs. Coluccio, Conaway, and J. Boss for a critical reading of the manuscript.
Footnotes
This work was supported by Biomedical Research Support Grant S07-RR05364 from the National Institutes of Health and Grant IRG-182 from the American Cancer Society.
The abbreviations used are: nt, nucleotide; bp, base pair(s); BSA, bovine serum albumin; HEPES, 4-(2-hydroxyethyl)-l-piperazineeth-anesulfonic acid; kbp, kilobase pair(s); SDS, sodium dodecyl sulfate; FPLC, fast protein liquid chromatography.
D. Reines, data not shown.
D. Wells, manuscript in preparation.
References
- Ackerman S, Bunick D, Zandomeni R, Weinmann R. Nucleic Acids Res. 1983;11:6041–6064. doi: 10.1093/nar/11.17.6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengal E, Goldring A, Aloni Y. J Biol Chem. 1989;264:18926–18932. [PubMed] [Google Scholar]
- Bentley DL, Groudine M. Nature. 1986;321:702–706. doi: 10.1038/321702a0. [DOI] [PubMed] [Google Scholar]
- Bentley DL, Groudine M. Cell. 1988;53:245–256. doi: 10.1016/0092-8674(88)90386-8. [DOI] [PubMed] [Google Scholar]
- Buratowski S, Hahn S, Guarente L, Sharp PA. Cell. 1989;56:549–561. doi: 10.1016/0092-8674(89)90578-3. [DOI] [PubMed] [Google Scholar]
- Burton Z, Killeen M, Sopta M, Ortolan L, Greenblatt J. Mol Cell Biol. 1988;8:1602–1613. doi: 10.1128/mcb.8.4.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpousis A, Gralla J. J Mol Biol. 1985;183:165–177. doi: 10.1016/0022-2836(85)90210-4. [DOI] [PubMed] [Google Scholar]
- Chamberlin MJ, Nierman W, Wiggs J, Neff N. J Biol Chem. 1979;254:10061–10069. [PubMed] [Google Scholar]
- Conaway R, Conaway JW. J Biol Chem. 1988;263:2962–2968. [PubMed] [Google Scholar]
- Conaway JW, Conaway R. J Biol Chem. 1989a;264:2357–2362. [PubMed] [Google Scholar]
- Conaway R, Conaway JW. Proc Natl Acad Sci U S A. 1989b;86:7356–7360. doi: 10.1073/pnas.86.19.7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway R, Conaway JW. J Biol Chem. 1990a;265:7559–7563. [PubMed] [Google Scholar]
- Conaway JW, Conaway R. Science. 1990b;248:1550–1553. doi: 10.1126/science.2193400. [DOI] [PubMed] [Google Scholar]
- Conaway JW, Bond M, Conaway R. J Biol Chem. 1987;262:8293–8297. [PubMed] [Google Scholar]
- Conaway JW, Reines D, Conaway R. J Biol Chem. 1990;265:7552–7558. [PMC free article] [PubMed] [Google Scholar]
- Coppola JA, Luse DS. J Mol Biol. 1984;178:415–437. doi: 10.1016/0022-2836(84)90151-7. [DOI] [PubMed] [Google Scholar]
- Eilat D, Hochberg M, Fischel R, Laskov R. Proc Natl Acad Sci U S A. 1982;79:3818–3822. doi: 10.1073/pnas.79.12.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Samuels M, Sharp PA. J Biol Chem. 1984;259:2509–2516. [PubMed] [Google Scholar]
- Flores O, Maldonado E, Burton Z, Greenblatt J, Reinberg D. J Biol Chem. 1988;263:10812–10816. [PubMed] [Google Scholar]
- Flores O, Maldonado E, Reinberg D. J Biol Chem. 1989;264:8913–8921. [PubMed] [Google Scholar]
- Gonzalez N, Wiggs J, Chamberlin MJ. Arch Biochem Biophys. 1977;182:404–408. doi: 10.1016/0003-9861(77)90521-5. [DOI] [PubMed] [Google Scholar]
- Hahn S, Buratowski S, Sharp PA, Guarente L. Cell. 1989;58:1173–1181. doi: 10.1016/0092-8674(89)90515-1. [DOI] [PubMed] [Google Scholar]
- Hansen UM, McClure WR. J Biol Chem. 1980;255:9564–9570. [PubMed] [Google Scholar]
- Hawley DK, Roeder RG. J Biol Chem. 1985;260:8163–8172. [PubMed] [Google Scholar]
- Helmann JD, Chamberlin MJ. Annu Rev Biochem. 1988;57:839–872. doi: 10.1146/annurev.bi.57.070188.004203. [DOI] [PubMed] [Google Scholar]
- Hernandez N, Lucito R. EMBO J. 1988;7:3125–3134. doi: 10.1002/j.1460-2075.1988.tb03179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi M, Yamamoto T, Ohkuma Y, Weil PA, Roeder RG. Cell. 1990;61:1171–1178. doi: 10.1016/0092-8674(90)90681-4. [DOI] [PubMed] [Google Scholar]
- Kadesch TR, Chamberlin MJ. J Biol Chem. 1982;257:5286–5295. [PubMed] [Google Scholar]
- Kadonaga JT. J Biol Chem. 1990;265:2624–2631. [PubMed] [Google Scholar]
- Kao SY, Calman AF, Luciw PA, Peterlin BM. Nature. 1987;330:489–493. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- Kassavetis GA, Bruan BR, Nguyen LH, Geiduschek EP. Cell. 1990;60:235–245. doi: 10.1016/0092-8674(90)90739-2. [DOI] [PubMed] [Google Scholar]
- Krummel B, Chamberlin MJ. Biochemistry. 1989;28:7829–7842. doi: 10.1021/bi00445a045. [DOI] [PubMed] [Google Scholar]
- Langer L, Waldrop A, Ward D. Proc Natl Acad Sci U SA. 1981;7:6633–6637. doi: 10.1073/pnas.78.11.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue NF, Kornberg RD. Proc Natl Acad Sci U S A. 1987;84:8839–8843. doi: 10.1073/pnas.84.24.8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maderious A, Chen-Kiang S. Proc Natl Acad Sci U S A. 1984;81:5931–5935. doi: 10.1073/pnas.81.19.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado E, Ha I, Cortes P, Weis L, Reinberg D. Mol Cell Biol. 1990;10:6335–6347. doi: 10.1128/mcb.10.12.6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1982. [Google Scholar]
- Manley JL, Fire A, Cano A, Sharp PA, Gefter ML. Proc Natl Acad Sci U S A. 1980;77:3855–3859. doi: 10.1073/pnas.77.7.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight SL, Kingsbury R. Science. 1982;217:316–324. doi: 10.1126/science.6283634. [DOI] [PubMed] [Google Scholar]
- Melvin W, Keir H. Biochemical J. 1977;168:595–597. doi: 10.1042/bj1680595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller H, Asselin C, Dufort D, Yang JQ, Gupta K, Marcu KB, Nepveu A. Mol Cell Biol. 1989;9:5340–5349. doi: 10.1128/mcb.9.12.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman de Vegvar HE, Lund E, Dahlberg J. Cell. 1986;47:259–266. doi: 10.1016/0092-8674(86)90448-4. [DOI] [PubMed] [Google Scholar]
- Paule MR. Nature. 1990;344:819–820. doi: 10.1038/344819a0. [DOI] [PubMed] [Google Scholar]
- Price DH, Sluder AE, Greenleaf AL. Mol Cell Biol. 1989;9:1465–1475. doi: 10.1128/mcb.9.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappaport J, Reinberg D, Zandomeni R, Weinmann R. J Biol Chem. 1987;262:5227–5232. [PubMed] [Google Scholar]
- Reinberg D, Roeder RG. J Biol Chem. 1987a;262:3310–3321. [PubMed] [Google Scholar]
- Reinberg D, Roeder RG. J Biol Chem. 1987b;262:3331–3337. [PubMed] [Google Scholar]
- Reines D, Wells D, Chamberlin MJ, Kane CM. J Mol Biol. 1987;196:299–312. doi: 10.1016/0022-2836(87)90691-7. [DOI] [PubMed] [Google Scholar]
- Reines D, Chamberlin MJ, Kane CM. J Biol Chem. 1989;264:10799–10809. [PubMed] [Google Scholar]
- Resnekov O, Aloni Y. Proc Natl Acad Sci U S A. 1989;86:12–16. doi: 10.1073/pnas.86.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnekov O, Kessler M„, Aloni Y. J Biol Chem. 1989;264:9953–9959. [PubMed] [Google Scholar]
- Sawadogo M, Sentenac A. Annu Rev Biochem. 1990;59:711–754. doi: 10.1146/annurev.bi.59.070190.003431. [DOI] [PubMed] [Google Scholar]
- Sekimizu K, Kobayashi N, Mizuno D, Natori S. Biochemistry. 1976;15:5064–5070. doi: 10.1021/bi00668a018. [DOI] [PubMed] [Google Scholar]
- Skarnes WC, Tessier DC, Acheson NH. J Mol Biol. 1988;203:153–171. doi: 10.1016/0022-2836(88)90099-x. [DOI] [PubMed] [Google Scholar]
- Sluder AE, Greenleaf AL, Price DH. J Biol Chem. 1989;264:8963–8969. [PubMed] [Google Scholar]
- Sopta M, Carthew R, Greenblatt J. J Biol Chem. 1985;260:10353–10360. [PubMed] [Google Scholar]
- Spencer C, Groudine M. Oncogene. 1990;5:777–785. [PubMed] [Google Scholar]
- Spencer C, LeStrange R, Novak U, Hayward W, Groudine M. Genes & Dev. 1990;4:75–88. doi: 10.1101/gad.4.1.75. [DOI] [PubMed] [Google Scholar]
- Straney DC, Crothers DM. Cell. 1985;43:449–459. doi: 10.1016/0092-8674(85)90175-8. [DOI] [PubMed] [Google Scholar]
- Thayer GC, Brosius J. Mol Gen Genet. 1985;199:55–58. doi: 10.1007/BF00327509. [DOI] [PubMed] [Google Scholar]
- Tolunay HE, Yang L, Anderson WF, Safer B. Proc Natl Acad Sci U S A. 1984;81:5916–5920. doi: 10.1073/pnas.81.19.5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyke MW, Roeder RG, Sawadogo M. Science. 1988;241:1335–1338. doi: 10.1126/science.3413495. [DOI] [PubMed] [Google Scholar]
- van Dyke MW, Sawadogo M, Roeder RG. Mol Cell Biol. 1989;9:342–344. doi: 10.1128/mcb.9.1.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wampler SL, Tyree CM, Kadonaga JT. J Biol Chem. 1990;265:21223–21231. [PubMed] [Google Scholar]
- Weil AP, Luse DS, Segall J, Roeder RG. Cell. 1979;18:469–484. doi: 10.1016/0092-8674(79)90065-5. [DOI] [PubMed] [Google Scholar]
- Yager T, von Hippel P. In: Eschericia coli and Salmonella Typhimurium: Cellular and Molecular Biology. Neidhart F, editor. American Society for Microbiology; Wash. D. C: 1987. pp. 1241–1270. [Google Scholar]
- Zheng XM, Moncollin V, Egly J, Chambon P. Cell. 1987;50:361–368. doi: 10.1016/0092-8674(87)90490-9. [DOI] [PubMed] [Google Scholar]