

Abstract
The cholinesterases, acetylcholinesterase (AChE), and butyrylcholinesterase (BChE) (pseudocholinesterase), are abundant in the nervous system and in other tissues. The role of AChE in terminating transmitter action in the peripheral and central nervous system is well understood. However, both knowledge of the function(s) of the cholinesterases in serum, and of their metabolic and endocrine regulation under normal and pathological conditions, is limited. This study investigates AChE and BChE in sera of dystrophin-deficient mdx mutant mice, an animal model for the human Duchenne muscular dystrophy (DMD) and in control healthy mice. The data show systematic and differential variations in the concentrations of both enzymes in the sera, and specific changes dictated by alteration of hormonal balance in both healthy and dystrophic mice. While AChE in mdx-sera is elevated, BChE is markedly diminished, resulting in an overall cholinesterase decrease compared to sera of healthy controls. The androgen testosterone (T) is a negative modulator of BChE, but not of AChE, in male mouse sera. T-removal elevated both BChE activity and the BChE/AChE ratio in mdx male sera to values resembling those in healthy control male mice. Mechanisms of regulation of the circulating cholinesterases and their impairment in the dystrophic mice are suggested, and clinical implications for diagnosis and treatment are considered.
Keywords: acetylcholinesterase, butyrylcholinesterase, mouse serum, mdx, muscular dystrophy, testosterone, orchidectomy, H-P-G axis
Introduction
The presence of cholinesterase activity in serum was already described by Dale in 1914 (Dale, 1914). It was later found that this activity was due to the two acetylcholine (ACh)-hydrolyzing enzymes (Mendel and Rudney, 1943), acetylcholinesterase (AChE), and butyrylcholinesterase (pseudocholinesterase, BChE), that could be distinguished by their substrate specificity (Mendel and Rudney, 1943) and differential sensitivity to selective inhibitors (Austin and Berry, 1953). By now, both enzymes have been well-characterized in terms of structure and function (Massoulié, 2002; Massoulié et al., 2008). AChE is abundant in the nervous system and muscles, where it is clearly responsible for controlling the duration of cholinergic transmission (Katz and Miledi, 1973). Additional functions have been ascribed to both enzymes in normal development of the nervous system and in altered physiological or pathological conditions due to stress, inflammation, and neurodegenerative diseases, (e.g., Alzheimer's disease) and their treatment (for reviews, see e.g., Layer and Willbold, 1995; Darvesh et al., 2003, 2010; Ballard et al., 2005; Dori and Soreq, 2006; Meshorer and Soreq, 2006; Massoulié et al., 2008; Vogel-Höpker et al., 2012). Moreover, the soluble form of BChE in mammalian serum may serve as a safeguard against the diffusion of ACh into the bloodstream and/or against orally ingested toxic compounds, since it reacts with a broad range of substrates (Lockridge and Masson, 2000; Masson and Lockridge, 2010). In fact, serum BChE has been used as an indicator of hepatic, renal, and thyroid diseases and as a marker for pesticide toxicity. It is therefore important to understand how the levels and distribution of the ChEs are regulated. Such knowledge may have therapeutic consequences for the treatment of diseases modulated by ChE levels. However, little attention has been paid to the metabolic, and particularly to the hormonal, regulation of BChE or of AChE. Several lines of evidence suggest that such regulation may exist. Thus, rat serum BChE was shown to be under endocrine regulation by gonadal steroids and by growth hormone (GH) (Illsley and Lamartiniere, 1981; Lamartiniere, 1986). Furthermore, systematic changes in AChE/BChE ratios have been established during normal development. Thus, whereas AChE activity in rat muscle increased concomitantly with decrease in BChE, resulting in an inversion in ratio of the amounts of the two enzymes in adult muscle (Berman et al., 1987), an opposite change was observed in mouse serum, leading to a high BChE/AChE ratio post-puberty (Oliver et al., 1992). However, this was not the case for the serum of the mdx dystrophin-deficient mutant mouse, an animal model for Duchenne muscular dystrophy (DMD). In the mdx mouse, as in DMD patients, a point mutation in the dystrophin gene results in synthesis of a non-functional truncated protein, so that full-length dystrophin is absent from skeletal and cardiac muscles, as well as from certain neural cell populations in the brain (Sicinski et al., 1989; for a review see Muntoni et al., 2003), with devastating, and eventually fatal, consequences in humans. In mdx sera, AChE activity did not decline with the onset of puberty as in normal mice sera, but stayed at a high level (Oliver et al., 1992), which might indicate impaired hormonal regulation. In the present study we found not only that AChE activity was elevated but that BChE was drastically diminished in the mdx sera. We went on to show that in normal mouse serum BChE levels are subject to endocrine regulation by the androgen, testosterone (T) in male mice, whereas AChE levels are not influenced. We further examined whether the systematic differences in the levels of both ChEs in mdx sera were due to impaired endocrine regulation, and if hormonal manipulation can restore the AChE/BChE ratio to that in normal mouse serum.
Materials and methods
Animals
Male mdx and control (wt) mice from the same mouse strain (C57BL/10ScSn-Dmdx and C57BL10Sn, Jackson Laboratory, Bar Harbor, ME) were bred in our animal facility. Animals were maintained and treated according to an approved protocol of the Ethics Committee (IACUC) of the Hebrew University Medical School. The Hebrew University is an AAALAC internationally accredited institute. All mice were maintained under a controlled photoperiod (lights on 07:00–19:00). Food and water were provided ad libitum. Body weights were monitored.
Surgical manipulations
Bilateral orchidectomy
Exteriorized testes and epididymis of 19–21 week mice were removed under anaesthesia (0.45 mg ketamine/0.05 mg xylazine per ml saline solution, 0.9 ml/mouse, i.p.). Briefly, the spermatic cord was clamped with a hemostat. Then, the distal portions of the vas deferens and spermatic artery were severed. Once homeostasis had been achieved (~5 min), the spermatic cord was unclamped, and the incision was closed. All control animals underwent sham surgery.
Testosterone implants
This procedure was kindly provided by Dr. Robert A. Steiner (U. Washington, Seattle, WA). Immediately after orchidectomy (or sham surgery), testosterone capsules (T, 20–25 mg/pellet), were implanted subcutaneously (about halfway down the mouse's back) via a small incision at the base of the neck. Then the skin was sutured and the animal was allowed to wake up. The T pellet was designed to achieve normal physiological levels. The dose was chosen based on previous research, which established that it would produce significant androgenic actions at target tissues, i.e., spermatogenesis in gonadotropin-deficient mice (Singh et al., 1995; Lindzey et al., 1998). All untreated animals received empty (sham) capsules.
Collection of blood and separation of plasma
Submandibular vein withdrawal for ChE measurements
Blood samples for the measurement of circulating ChE concentrations were collected via heparinized capillaries from punctured submandibular vein of lightly sedated mice (ketamine hydrochloride/xylazine, ~0.1 ml/mouse, i.p.). Blood was drawn during the period of 10:00–12:00 am from control and castrated animals of age ~21 weeks. It should be noted that prior to blood collection, all mice were acclimated for 2 weeks under a new controlled photoperiod (lights on 09:00–21:00).
Orbital sinus withdrawal for creatine kinase measurements
Creatine kinase (CK) levels in circulation were used as a convenient index to confirm the genotype of the animals (see below). Animals were anesthetized (chloral hydrate solution, 0.05 g/ml, 0.1 ml/10 g body weight, i.p.) and 0.5 ml of blood was drawn from the orbital sinus via heparinized capillaries.
Sera
After blood collection, samples were allowed to clot for 30 min at room temperature (RT), and then centrifuged (Eppendorf 5415 at 1250 rpm, 15 min, RT). The separated plasma was stored at −80°C. Sera from male rhesus macaque were a generous gift from Dr. Tony Plant (U. Penn., Pittsburgh, PA) and bovine sera were kindly donated by Dr. Joel Zeron (SION A.I. Center & Breeding Ltd., Shikmim, Israel).
Determination of ChE activities in sera
Spectrophotometric method
ChE activity was measured in microplates by the colorimetric method of Ellman et al. (1961) in 1.0 mM 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) in 0.1 M Tris, pH 7.6, containing 0.5 mM acetylthiocholine iodide (ATCh) or butyrylthiocholine iodide (BTCh) (Sigma, St. Louis, MO). Selective inhibitors were used to distinguish between AChE and BChE as described below. The increase in absorbance at 412 nm was followed for 12–15 min at 31–34°C using a PowerWavex340 Model microplate reader (Bio-Tek Instruments, Inc, Winooski, VT). Activities were expressed as ΔOD units per minute.
Radiometric method
ChE activity was assayed by a radiometric method, monitoring the 3H-acetate generated during acetylcholine hydrolysis (Johnson and Russell, 1975). This method is of higher sensitivity than the Ellman's technique (see above) thus allowing for better resolution when measurement of variations in the low AChE activities of mouse sera was required. Briefly, samples were incubated in 0.1 ml of 1 mM of 3H-acetylcholine iodide (3H-ACh, PerkinElmer Life Sciences) in 0.1 M NaCl and 0.01 M Tris buffer (pH 7.4). The reaction was stopped as required by addition of 0.1 ml 1 M chloroacetic acid/0.5 M NaOH/2 M NaCl, followed by addition of 4 ml scintillation fluid composed of 90% QuickSafe N (Zinsser Analytic, Frankfurt, Germany) and 10% isoamyl alcohol. The reaction product, [3H]-acetate, was extracted into the organic phase and counted in a Tri-CARB 2900TR liquid scintillation counter (Packard BioScience, Meriden, CT). Selective inhibitors were used to distinguish AChE and BChE activities (see below).
Selective ChE inhibitors
Tetra-isopropyl-pyrophosphoramide (0.1 mM, iso-OMPA, Sigma) and 1,5-bis(4-allyldimethylammoniumphenyl)pentan-3-one dibromide (0.01 mM, BW284c51, Burroughs Wellcome Co.) were used to selectively inhibit BChE and AChE activities, respectively (Austin and Berry, 1953). Samples were pre-incubated with either inhibitor for 20 min at RT and then assayed for ChE activity in the presence of the inhibitors using either the colorimetric or radiometric method.
Determination of creatine kinase (CK) activity
Circulating CK values, expressed as units of enzyme activity per liter of serum sample (U/L), were used as an index to identify the genotype of the animals (mdx or wt). CK values in blood samples drawn from orbital sinuses were determined using a standard enzymatic method and an automated clinical analyzer.
Statistical analyses
Data were collected for samples from individual animals in all experiments. Mean values were obtained by averaging measurements obtained for a group of 4–10 animals for a particular experimental day (as indicated in each experiment). The significance of differences between groups was determined by One-Way ANOVA (parametric with Tukey's multiple comparison post-test) for all groups, or by Kruskal–Wallis (non-parametric) tests, depending whether or not the data showed normal Gaussian distribution. Comparison between two groups was by non-paired t-test with unequal variances (our usual case). All tests were available in the GraphPad Prism4 (GraphPad Inc.), Statistica10 (StatSoft) and Microsoft Office Excel Professional Software. The difference between groups was considered significant when (p < 0.05).
Results
Post-pubertal effects of dystrophy on body weight
Although the mdx mouse was reproductively competent (thus, T was present and functional), its postnatal growth was hindered compared to that of normal controls. The mdx mice displayed lower and more variable body weight compared to the controls, with the phenomenon being more pronounced in mdx females than in males (not shown). However, the body weight stabilized by 11 weeks of age at levels not significantly different from those of the control mice (Figure 1). The oscillatory shape of the curve was consistent with the finding that mdx mouse muscles undergo cycles of degeneration and regeneration until most muscles stabilize by 10 weeks (Settles et al., 1996). The mice we used were therefore aged 19–22 weeks, at which age their body weights are stable, and the majority of muscle fibers no longer display transient degeneration (Karpati et al., 1988).
Figure 1.

Postnatal changes in body weight in male mice. The effect of strain on body weight is shown. Several litters of C57BL/10J (wt,  ) and mdx (○) male mice were weighed over 3–21 weeks. Each data point is the mean ± SEM (n = 6–34). The dashed line at ~5 weeks marks initiation of puberty in the wt. *p < 0.05 between wt and mdx of the same age, showing that mdx mice gain less weight than wt during postnatal development but reach normal level by 11 weeks.
) and mdx (○) male mice were weighed over 3–21 weeks. Each data point is the mean ± SEM (n = 6–34). The dashed line at ~5 weeks marks initiation of puberty in the wt. *p < 0.05 between wt and mdx of the same age, showing that mdx mice gain less weight than wt during postnatal development but reach normal level by 11 weeks.
Serum ChE activities in normal and dystrophic mice
Mouse serum contains BChE and AChE, both of which hydrolyze ATCh. Total ChE activities in sera of male mdx mice were at least 15% (p < 0.01) below those in normal mice sera (Figure 2A), indicating reduced levels of AChE and/or BChE in the mdx sera. AChE activity, which was found to be very low in sera of normal mice, was significantly elevated in the mdx sera (Figure 2A, detailed below). In contrast, BChE levels, assayed on BTCh, were found to decrease in male mdx-sera to 64–67% of the levels in wt sera (Figure 2B, p < 0.002). Similar results were observed when BChE was assayed on ATCh, with AChE activity being selectively inhibited by BW284c51 (Figure 2A). Since AChE levels in normal mouse serum are very low, the more sensitive radiometric assay was used to confirm the results obtained using the colorimetric Ellman procedure (Figures 2C and D), showing that AChE activity in mdx-sera indeed increased by 50–80% over the level in control wt sera (p < 0.05), confirming an earlier report (Oliver et al., 1992). BChE activity was shown, also by the radiometric assay, to decrease in mdx-sera to 61–76% of wt values (Figure 2D, p < 0.001). Since BChE is the major ChE form in mouse serum (>70%), the large drop in BChE was responsible for the overall reduction in total ChE activity observed in mdx sera. However, the decrease in BChE levels, taken together with the increase in AChE levels, in mdx sera (as shown in Figures 2C and D), reduced the BChE/AChE ratio from 2.3 ± 0.2 in the wt-sera to 1.3 ± 0.5 in mdx-sera. Similar results were found for adult female mdx-mice and are summarized elsewhere (Anglister et al., 2008).
Figure 2.

Circulating ChE activities in wt and mdx mice. ChE activities in the sera of adult male wt (C57Bl/10J)  and mdx □ mice were assayed by: (A,B) Measurement using the Ellman colorimetric method, with ATCh as substrate and the selective inhibitors, iso-OMPA, for measurement of AChE (striped columns), or BW284c51, for measurement of BChE (plain columns) (A); BChE activity was also measured using BTCh (B); (C,D) The radiometric method with 3H-ACh as substrate and with iso-OMPA for AChE (C), or with BW284c51 for BChE (D). (E) CK levels in the sera of wt and mdx mice. Activity values represent the mean ± SEM, of samples from 6 to 8 mice per group for ChEs and of 12 animals per group for CK. Activities in mdx sera differed from wt significantly: The BChE level in mdx sera was reduced compared to wt (*p < 0.01 in B and < 0.05 in D), while AChE in mdx sera was elevated compared with wt (*p < 0.05 in A,C). As expected, CK levels in mdx sera were elevated, compared to those in the wt sera (E, *p < 0.001).
and mdx □ mice were assayed by: (A,B) Measurement using the Ellman colorimetric method, with ATCh as substrate and the selective inhibitors, iso-OMPA, for measurement of AChE (striped columns), or BW284c51, for measurement of BChE (plain columns) (A); BChE activity was also measured using BTCh (B); (C,D) The radiometric method with 3H-ACh as substrate and with iso-OMPA for AChE (C), or with BW284c51 for BChE (D). (E) CK levels in the sera of wt and mdx mice. Activity values represent the mean ± SEM, of samples from 6 to 8 mice per group for ChEs and of 12 animals per group for CK. Activities in mdx sera differed from wt significantly: The BChE level in mdx sera was reduced compared to wt (*p < 0.01 in B and < 0.05 in D), while AChE in mdx sera was elevated compared with wt (*p < 0.05 in A,C). As expected, CK levels in mdx sera were elevated, compared to those in the wt sera (E, *p < 0.001).
As an internal control, CK levels in sera were measured to validate the genotype of the animals (mdx compared to wt). Consistent with the 3–14-fold elevation reported by Glesby et al. (1988), the male mdx mice used in our studies showed variable, but consistently increased, CK levels in sera, viz., 9.9 ± 1.4 (× 103 U/L, mean ± SEM) in mdx mice, compared to 1.3 ± 0.3 in controls (Figure 2E). These differences were highly significant (p < 0.001).
Endocrine regulation of normal mouse serum BChE activity
To determine whether the abnormal BChE/AChE ratio in mdx sera is due to hormonal influences, we first investigated regulation by the androgen, T, of ChEs in the sera of adult male wt mice.
Sera from orchidectomized 21 week adult male wt mice were analyzed for ChE activities (Figure 3). Gonadectomy raised BChE activities in adult male mice serum by 20–37% (p < 0.001), whether measured on BTCh, or on ACh in the presence of a selective AChE inhibitor (Figure 3A and B, respectively)
Figure 3.
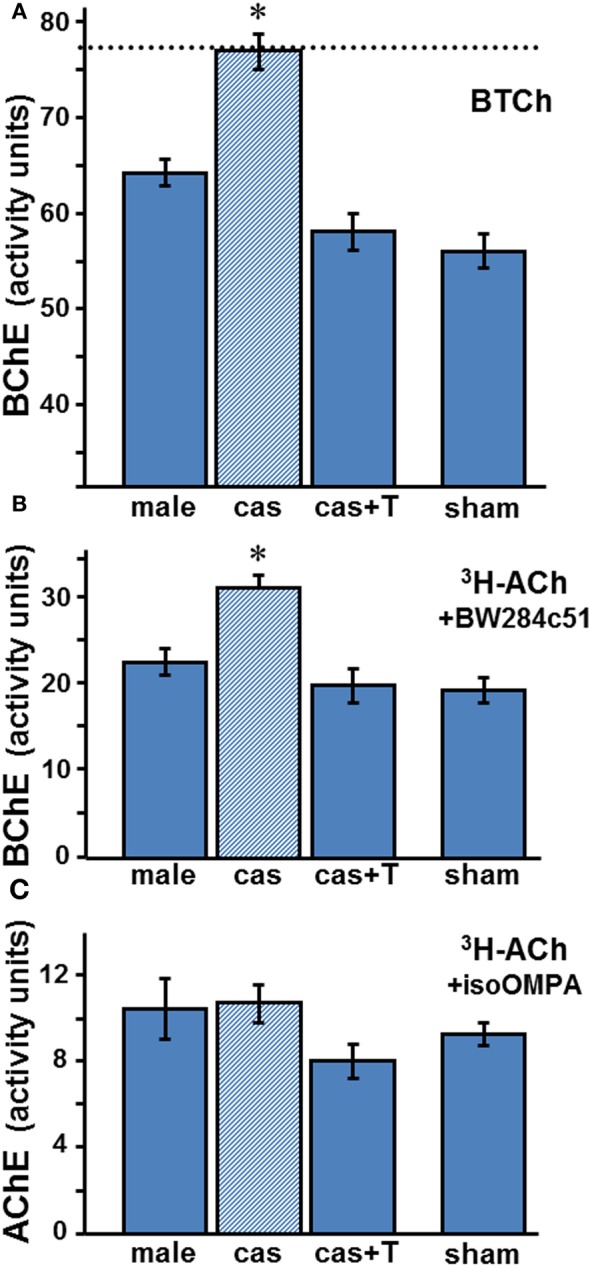
Effect of orchidectomy and T-replacement appears to be on BChE, not on AChE. Activities in wt sera of BChE (A,B) and AChE (C) were determined colorimetrically (A) or radiometrically (B,C) with appropriate substrates and selective inhibitors as described in the legend to Figure 2. Values are the mean ± SEM for 6–8 samples per group (sham group, n = 3). BChE levels in gonadectomized animals (dashed line in A) differ significantly from those in intact, T-treated and sham animals: Data were analyzed by One-Way ANOVA, showing statistical significance in A (p « 0.0002) and in B (p « 0.0001), with Tukey's post-hoc revealing significant differences (*) between castrated male (cas, shaded column) and the other three groups: intact male (male) (p < 0.0003), castrated with T-replacement (cas + T) (p < 0.0002) or sham (p < 0.0002). All other comparisons showed insignificance (p > 0.05): male compared to cas + T (p = 0.086), or to sham (p = 0.07), and cas + T compared to sham (p = 0.92). Similarly, post-hoc test in B showed significant difference between cas and the other groups: male (p < 0.001), cas + T (p < 0.0004), and sham (p < 0.001), which were very similar. No significant changes were observed in AChE activity (C).
T-replacement in adult wt males, performed concomitantly with castration, abolished the increase caused by castration, restoring BChE activity to the levels observed in the sera of intact males (Figures 3A and B). Thus, a high basal level of BChE activity is found in the sera of agonadal males (in the absence of T), and both endogenous and exogenous androgen down-regulate the enzyme.
Whereas castration of the male wt markedly raised the BChE level in the serum, that of AChE (measured with ACh in the presence of iso-OMPA) did not change significantly (Figure 3C; p = 0.41). The ranges of values recorded radiometrically, with ACh and the respective selective inhibitors of BChE and AChE, in sera of male adult wt mice under the different treatment regimes, are shown in Table 1. The data show clearly that BChE activities were increased from a low level (1830–2350 arbitrary assay units) to a high level (2770–3870 units) after orchidectomy, and reverted to the base level after T-implant. In contrast, the AChE activities observed in adult male mice sera fell in a similar range (530–1460 arbitrary units), whether the mice were intact or castrated, with or without T-implant. These results indicate a specific and selective negative androgen modulation of BChE, but not of AChE. Furthermore, the large increase in BChE after castration resulted in an increased ratio of BChE to AChE activities from 2.3 ± 0.2 in sera of intact wt males to 3.0 ± 0.1 in castrated wt males (Table 1).
Table 1.
Effect of gonadectomy on serum BChE and AChE levels in adult male wt mice.
| wild-type | n | BChE | AChE | BChE:AChE |
|---|---|---|---|---|
| No surgery | 5 | 1830–2350 | 790–1010 | 2.3 ± 0.2 |
| Castrate | 8 | 2770–3870 | 730–1460 | 3.0 ± 0.1 |
| Castrate + T | 5 | 1570–2550 | 530–980 | 2.5 ± 0.2 |
| Sham | 3 | 1660–2130 | 870–1040 | 2.1 ± 0.1 |
Effects of orchidectomy and T-replacement on serum BChE levels in adult mdx mice
We went on to examine whether orchidectomy raises BChE levels also in the sera of mdx mice, and whether, as a consequence, the high agonadal baseline ChE levels seen in wt mice sera can be reached. The data presented in Table 2 and Figure 4 reveal low levels of BChE in the sera of control adult mdx males (32.2–48.6 assay units). Although levels increase substantially after gonadectomy (48.7–61.9 assay units), by an average of 35%, the BChE deficiency observed in untreated mdx mice is sustained (Figure 4): thus, the agonadal “baseline” level in mdx mice was significantly below the baseline level in wt agonadal mice (29%, p < 0.002). It was slightly lower than the level in control wt mice (14%, p = 0.06) or in sham-operated wt mice. As in wt mice, T-replacement reversed the effect of orchidectomy on BChE activity. Thus, after orchidectomy, the BChE in mdx circulation may almost reach the gonadal baseline level of BChE activity in the serum of intact male wt mice, but not the agonadal baseline level in the serum of castrated wt mice.
Table 2.
Effect of gonadectomy in elevating serum BChE activity in normal and dystrophic mice.
| wt | mdx | p | |
|---|---|---|---|
| No surgery | 56.1–68.2 (8) | 32.2–48.6 (5) | <0.002 |
| Castrate | 68.0–83.3 (6) | 48.7–61.9 (4) | <0.002 |
| Castrate + T | 50.1–62.3 (6) | 34.0–46.2 (5) | <0.001 |
| Sham | 51.6–57.8 (3) | 40.8–44.8 (3) | <0.006 |
Activities relate to the same animals and assay conditions as for the data displayed in Figure 4.
Figure 4.

T-regulation of BChE activity in male mdx sera. BChE activities were measured colorimetrically with BTCh as substrate in sera of intact, castrated, castrated with T-replacement, and sham operated wt
and mdx □ mice, (as in Figure 3A). Values, mean ± SEM for samples taken from 4 to 8 mice per group (sham group, n = 3). *p < 0.001 between BChE levels in castrated wt and in intact, T-treated, and sham mice sera (of either wt or mdx strains). **p < 0.05 (detailed in 3) between BChE levels in castrated mdx and in intact, T-treated, and sham mdx mice sera. #Significant difference between BChE level in mdx and wt in all the experimental groups (p-values are listed in 2). The dashed black and red lines indicate BChE levels in agonadal and gonadal wt sera, respectively.
T-regulation of serum AChE activities in mdx mice
We went on to examine the effects of orchidectomy and T-replacement on AChE levels in the sera of mdx mice. Specifically, we checked whether orchidectomy of mdx mice, with or without T-implant, affected serum levels of AChE. As documented in Table 3, the mean AChE activity in the sera of intact mdx mice was 1260 ± 120 units, as compared to 790 ± 70 units in the castrated mice. Despite the great variability in AChE levels, castration appears to reduce AChE activity by >30%, p < 0.02). Interestingly, the mean value for agonadal mdx mice was not different from that for gonadal wt mice (p = 0.2).
Table 3.
Changes in BChE and AChE activities in the sera of adult wt and mdx adult male mice before and after orchidectomy, and after T-implant.
| wt | mdx | ||||
|---|---|---|---|---|---|
| Male | Male | Castrate | Castrate + T | Sham | |
| Total ChEs | 3700 ± 100** | 3000 ± 90 | 3340 ± 220 | 2880 ± 120 | 2940 ± 270 |
| BChE | 2140 ± 90 | 1440 ± 70 | 1970 ± 110* | 1540 ± 130 | 1350 ± 90 |
| AChE | 910 ± 40 | 1260 ± 120# | 790 ± 70 | 920 ± 140 | 1070 ± 140 |
| BChE/AChE | 2.3 ± 0.2 | 1.2 ± 0.5 | 2.5 ± 0.4 | 1.7 ± 0.3 | 1.3 ± 0.2 |
Data shown refer to the same animals as those used in Figure 4.
BChE in castrated mdx sera is significantly elevated compared with mdx male (p < 0.02), castrated with T implant (p < 0.05) and sham sera (<0.01).
BChE in wt male sera is significantly higher than in mdx male (p < 0.001), castrated with T implant (p < 0.006) and sham sera (p < 0.001).
AChE in mdx male serum is significantly higher than in castrated mdx (p < 0.02) and in wt male sera (p < 0.05).
T-implant into agonadal mdx mice resulted in a mean serum AChE activity of 930 ± 180, not significantly different from the value for agonadal mdx mice lacking the T-implant (p = 0.40). This value is also very similar to the level in the gonadal wt mice (p = 0.89), but lower than the average value for gonadal mdx mice, although a statistically significant difference could not be demonstrated due to the large standard errors within the two groups, resulting in p = 0.11. Furthermore, the values obtained for the small group of “sham” mdx mice analyzed, were intermediate, not being significantly different from either the values for sera of intact mdx and intact wt mice (p = 0.34 and 0.39, respectively). These limitations prevent us from drawing definite conclusions concerning the effect of orchidectomy on serum AChE in mdx mice, although orchidectomy decreases AChE levels. However, the large increase in BChE levels, coupled with the apparent decrease in AChE levels, in the agondal mdx mice, results in an increase in the BChE/AChE activity ratio from 1.2 ± 0.5 in the male mdx-sera to 2.5 ± 0.4 after castration (Table 3), a ratio similar to that in wt-sera (2.3 ± 0.2, Tables 1 and 3).
Species-specific and developmental variations in circulating ChEs
Fetal calf serum is known to be a good source of AChE (Ralston et al., 1985). It occurred to us that bovine serum might serve as a good system for testing postnatal down-regulation of serum AChE, and for examining, in parallel, the regulation of BChE, assuming BChE to be a serum component common to all species. We therefore compared ChE activities in fetal bovine sera and in post-pubertal (adult) bull sera. Consistent with the literature, we observed a high level of AChE activity, >96% of total ChE, in fetal bovine sera. Interestingly, AChE activity was >10-fold lower in adult bull sera (Table 4). However, whereas in adult mouse serum, BChE was the major ChE component, as in several other species (Ecobichon and Corneau, 1973; Chatonnet and Lockridge, 1989), in bovine sera it was the minor ChE component, with very low activity (Table 4). Although BChE activity in fetal and adult bovine sera was similar, its fraction of the total ChE activity rose from fetal to adult (~4–25%) due to the big drop in AChE activity. Because of the striking difference in ChE composition of mouse and bovine sera, we decided to examine the ChE composition of the sera of rhesus (RHS) monkeys, which are closely related to humans and for which many endocrinological studies have been performed, including examination of the effects of androgens. As seen in Table 4, in adult RHS serum, as in mouse serum, BChE is the major component (89%).
Table 4.
Species-specific variations in circulating ChE activities.
| Bovine | Macaque | Mouse | |||
|---|---|---|---|---|---|
| Fetal | Adult | Rhesus | wt (C57BL) | mdx | |
| Total ChE (no inhibitor) | 5020 | 470 | 2240 | 3500 ± 210 | 3260 ± 270 |
| BChE (+BW) | 190 | 100 | 1990 | 2140 ± 90 | 1650 ± 220 |
| AChE (+iso-OMPA) | 4830 | 370 | 250 | 910 ± 40 | 1260 ± 130 |
| BuChE/AChE | 0.04 | 0.27 | 3.9 ± 0.4 | 2.3 ± 0.2 | 1.3 ± 0.5 |
| AChE U/mL* | 1.2–1.7 | 0.11 | 0.081 | 0.24 | 0.35 |
All sera were from male animals. ChE activities were determined by radiometric assay with ACh and the respective inhibitors, and presented as units in 125 μL serum. Macaque and bovine values were obtained from pooled sera of three animals each. The values observed for bovine sera were confirmed by Drs. B. P. Doctor and H. Saxena (Walter Reed Army Institute of Research, Silver Spring, MD, pers. communication).
AChE U/mL values represent activities calculated in enzyme units (U = μmoles ACh hydrolyzed/min).
Discussion
In the present study we explored the regulation of serum ChEs in normal and dystrophic mice. Several findings were made: (1) While there is more BChE than AChE activity in mouse serum, as in the sera of several other mammals (reviewed by Chatonnet and Lockridge, 1989), the value of the BChE/AChE ratio for the sera of mdx mice is about half of the ratio for the sera of wt mice; (2) While AChE levels increased slightly in the mdx sera (Oliver et al., 1992), BChE levels decreased by >30%, resulting in an overall decrease in ChE activity of ~20% in mdx as compared to wt sera; (3) Although orchidectomy elevated levels of BChE activity in the sera of both strains, levels in mdx sera remained lower than in wt sera; (4) T-replacement reversed the effect of orchidectomy on BChE activity in both wt and mdx sera; (5) T-modulation was specific to serum BChE, not significantly affecting levels of AChE; (6) Orchidectomy did not increase AChE activities in either strain. These findings are consistent with involvement of the hypothalamic-hypophysial-gonadal (H-P-G) axis in the impairment of BChE activity in mdx sera.
The regulation of mouse serum ChEs, either AChE or BChE, via the (H-P-G) axis, has not been previously explored. Our results demonstrate that testosterone (T) down-regulates the expression of BChE in sera of mice, confirming the early observations on male rat sera that the total ChE content was reduced by circulating T (or increased by estrogen in female), not as a result of a direct interaction of the steroid with the enzyme but via higher level regulation (Everett and Sawyer, 1946; Sawyer and Everett, 1946, 1947; Illsley and Lamartiniere, 1981).
How can T modulate levels of serum BChE?
The target of testosterone (or estrogen) that produces modulation of serum BChE levels is not known. It may be in the hypothalamus, in the pituitary, or perhaps in a target organ that produces serum BChE, such as the liver (Leeuwin, 1965), together with pituitary factors. Experiments on hypophysectomized rats have demonstrated that pituitary mediation is required for both androgen and estrogen action (Everett and Sawyer, 1946; Illsley and Lamartiniere, 1981): while sex steroids do modulate ChE activity from a basal level, either due to suppression by androgen in males, or due to stimulation by estrogen in females, their action is abolished by hypophysectomy. It has also been shown in rats that hypophysectomy (Illsley and Lamartiniere, 1981; Edwards and Brimijoin, 1983) or lesion of the arcuate nucleus in the hypothalamus of males (Lamartiniere, 1986) reduces levels of GH and increases levels of ChE in the serum. The effects on serum ChEs were shown to be reversed by exogenous administration of GH after either type of surgery. Thus, both T and GH appear to be negative modulators of rat serum BChE in male rats. Moreover, they can act synergistically, since their levels are inter-related: the hypothalamus regulates the release of pituitary hormones (e.g., GH) and gonadotropins that, in turn, drive the gonads to produce T and estrogen, viz., the H-P-G axis. In addition, T itself influences GH secretion, and GH in turn reduces serum BChE levels; thus, T can also reinforce down-regulation of BChE via GH.
Where can T and GH exert their influences on serum BChE?
Serum BChE in adults is apparently produced in the liver (Silver, 1974). Indeed, GH has been shown to modulate several sex-differentiated hepatic enzymes, including serum ChE (Lamartiniere, 1981, 1986). Since hypophysectomy abolishes rat liver estrogen receptors and GH restores them, GH may also restore responsiveness of the liver to circulating sex steroids, and result in androgen or estrogen modulation of serum ChE. A similar mechanism could apply to GH regulation of other potential target organ for serum BChE production. In considering the liver as the source for serum BChE, it should be taken into account that, the liver secretes BChE monomers and dimers (Perelman et al., 1990), not the G4 tetramer which is the major isoform in serum (Lockridge et al., 1979). Furthermore, we have preliminary evidence that T, castration, and T-replacement after castration, alter predominantly the level of G4 BChE in mouse serum. Thus, it appears that the BChE isoforms produced/secreted by the liver are not the isoform present in serum and modulated by the hormones. This unresolved issue, as well as the influence of T and/or GH on BChE isoforms in serum, liver, and other tissues need to be revisited and further investigated.
Is H-P-G axis (and GH) control of serum BChE impaired in mdx mice?
It became apparent in this study that the mdx mice, that lacked dystrophin, had less BChE in their sera (both before and after orchidectomy), relative to wt controls. Since T and GH have both been found to be negative modulators of BChE, it is possible that the intact mdx mice secrete higher levels of T and/or GH than the wt mice. Consistent with this suggestion, mdx mice exhibit hypertrophy of somatotroph cells in the pituitary, and slightly elevated pituitary GH in adult females (Anderson et al., 1994). The reduced levels of BChE we show in castrated mdx mice compared to castrated wt are also likely to be due to gender-specific effects of GH at the hypothalamic-pituitary level, and not to a direct effect of T, since castration has eliminated T from the system. Further studies will be needed to determine whether the perturbation is at the pituitary or hypothalamic level.
Further correlation of GH levels with dystrophin deficiency comes from studies on DMD patients that reported mitigation of the severity of the dystrophy in GH-deficient patients (Zatz et al., 1981a,b), or after treatment with a GH inhibitor (Zatz et al., 1986). Another report provided some evidence for an altered hypothalamic-hypophyseal axis (Zaccaria et al., 1989). This is consistent with the observation referred to above, of high GH levels in mdx sera (Anderson et al., 1994). In addition, insulin growth factor-1 (IGF-1), which is secreted in correlation with GH, is elevated in mdx plasma (De Luca et al., 1999). Altogether, dystrophin deficiency is likely to be associated with impaired H-P-G-regulation.
Removal of gonadal control normalizes the BChE/AChE ratio in male mdx sera
Consistent with Oliver et al. (1992), it became apparent in this study that mdx mice had more AChE in their sera (prior to orchidectomy), than wt controls. Elevated levels of serum AChE were also reported for dystrophic chickens (Lyles et al., 1980) and for other animal dystrophies (Skau, 1985), as well as for fetal calf serum (Ralston et al., 1985). Oliver et al. (1992) proposed that the increased AChE levels in the sera of the dystrophic mice reflect increased release of muscle AChE relative to release in wt controls. Our finding that post-pubertal castration of adult mdx males normalizes their serum ChE levels by elevating BChE and, perhaps reducing AChE, and thus normalizing the BChE:AChE ratio, suggests that the mdx sera BChE levels are not just a reflection of muscle degradation but are under control of the gonads and of the aforementioned higher centers. Moreover, in addition to T, there is likely to be an additional factor involved in the impaired down-regulation of AChE and up-regulation of BChE in mdx sera. This seems likely since although the BChE level increased after castration, it did not reach the agonadal wt level, and T-replacement did not reverse the effect on mdx serum AChE. Before one can extrapolate from our results in mice to human patients, one has to prove that gonadal regulation of BChE occurs in higher primates.
Postnatal maturation of serum BChE/AChE ratio
Because, in mdx sera, AChE failed to decrease upon maturation and BChE was low in the adult, we considered the possibility that mdx mice are locked in a pre-adult stage and retain some embryonic regulatory properties. It was shown in mouse sera that in the embryo AChE predominates, while in the adult BChE is the major ChE species (Oliver et al., 1992). Surprisingly, we report here strong postnatal down-regulation of AChE in bovine serum, with only small amounts of BChE in both fetal and adult serum relative to other species (Table 4). However, we found BChE to be the major ChE component in sera of adult normal wt mice and rhesus monkeys (Table 4), as has been reported for many animals (Ecobichon and Corneau, 1973; Chatonnet and Lockridge, 1989). Indeed, while human 12th week fetal serum may contain 40% AChE (Hahn et al., 1993), adult human plasma contains >99% BChE (Brimijoin and Hammond, 1988). Higher primates, such as the rhesus macaque, should provide an appropriate model for further studies on the regulation of both AChE and BChE in human serum.
While in the present study we have shown that the AChE in mouse serum is not regulated by circulating T, it should be noted as mentioned, that AChE decreased ~2-fold in the serum of normal mice, prior to puberty. During that time, a progressive increase in AChE coincident with a reduction in BChE occurred in muscle resulting in inversion in the amounts of the two enzymes in adult muscle (Berman et al., 1987). Thus it may still be that the decrease in serum AChE and increase in muscle AChE are due to one or more postnatal triggers, e.g., humoral factor(s) discharged before the episodic T release at puberty (~5 weeks). In this context, it should be noted that BChE precedes AChE expression in embryonic neural development of birds (Layer, 1991; Layer and Willbold, 1994) and mammals (Koenigsberger et al., 1998).
One needs to bear in mind, considering the various roles attributed to ChEs in development, during normal function and in pathological conditions, that modulation of their levels may both reflect and influence normal processes or existing impairments. In the context of the present study, the abnormal BChE/AChE ratio in the serum of dystrophic mice, as compared to normal, may be developed as a tool for easy detection of dystrophy, provided similar observations are made for DMD or for other human dystrophies.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by MDA and ISF to Lili Anglister. We thank Dr. Ifat Uzi (Hebrew University) for expert veterinary advice on surgical procedures, and Prof. Israel Silman (Weizmann Institute) for his useful comments on the manuscript.
References
- Anderson J. E., Liu L., Kardami E., Murphy L. J. (1994). The pituitary-muscle axis in mdx dystrophic mice. J. Neurol. Sci. 123, 80–87 [DOI] [PubMed] [Google Scholar]
- Anglister L., Etlin A., Finkel E., Durrant A. R., Lev-Tov A. (2008). Cholinesterases in development and disease. Chem. Biol. Interact. 175, 92–100 10.1016/j.cbi.2008.04.046 [DOI] [PubMed] [Google Scholar]
- Austin L., Berry W. K. (1953). Two selective inhibitors of cholinesterase. Biochem. J. 54, 695–700 [PMC free article] [PubMed] [Google Scholar]
- Ballard C. G., Greig N. H., Guillozet-Bongaarts A. L., Enz A., Darvesh S. (2005). Cholinesterases: roles in the brain during health and disease. Curr. Alzheimer Res. 2, 307–318 [DOI] [PubMed] [Google Scholar]
- Berman H. A., Decker M. M., Jo S. (1987). Reciprocal regulation of acetylcholiesterase and butyrlcholinesterase in mammalian skeletal muscle. Dev. Biol. 120, 154–161 10.1016/0012-1606(87)90113-8 [DOI] [PubMed] [Google Scholar]
- Brimijoin S., Hammond P. (1988). Butyrylcholinesterase in human brain and acetylcholinesterase in human plasma: trace enzymes measured by two-site immunoassay. J. Neurochem. 51, 1227–1231 [DOI] [PubMed] [Google Scholar]
- Chatonnet A., Lockridge O. (1989). Comparison of butyrylcholinesterase and acetylcholinesterase. Biochem. J. 260, 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale H. H. (1914). The action of certain esters of choline, and their relation to muscarine. J. Pharmacol. Exp. Ther. 6, 147–190 [Google Scholar]
- Darvesh S., Hopkins D. A., Geula C. (2003). The biology of butyrylcholinesterase. Nat. Rev. 4, 131–138 10.1038/nrn1035 [DOI] [PubMed] [Google Scholar]
- Darvesh S., Reid G. A., Martin E. (2010). Biochemical and histochemical comparison of cholinesterases in normal and Alzheimer brain tissues. Curr. Alzheimer Res. 5, 386–400 [DOI] [PubMed] [Google Scholar]
- De Luca A., Pierno S., Camerino C., Cocchi D., Camerino D. C. (1999). Higher content of insulin-like growth factor-1 in dystrophic mdx mouse: potential role in the spontaneous regeneration through an electrophysiological investigation of muscle function. Neuromuscul. Disord. 9, 11–18 [DOI] [PubMed] [Google Scholar]
- Dori A., Soreq H. (2006). ARP, the cleavable C-terminal peptide of “readthrough” acetylcholinesterase, promotes neuronal development and plasticity. J. Mol. Neurosci. 28, 247–255 10.1385/JMN:28:3:247 [DOI] [PubMed] [Google Scholar]
- Ecobichon D. J., Corneau A. M. (1973). Pseudocholinesterase of mammalian plasma: physiochemical properties and organophosphate inhibition in eleven species. Toxicol. Appl. Pharmacol. 24, 92–100 [DOI] [PubMed] [Google Scholar]
- Edwards J. A., Brimijoin S. (1983). Effects of hypophysectomy on acetylcholinesterase and butyrylcholinesterase in the rat. Biochem. Pharmacol. 32, 1183–1189 [DOI] [PubMed] [Google Scholar]
- Ellman G. L., Courtney K. D., Andres V., Featherstone K. D. (1961). A new and rapid colorimetric determination of acetylcho-linesterase. Biochem. Pharmacol. 7, 88–95 [DOI] [PubMed] [Google Scholar]
- Everett J. W., Sawyer C. H. (1946). Effects of castration and treatment with sex steroids on the synthesis of serum cholinesterase in the rat. Endocrinology 39, 323–343 [DOI] [PubMed] [Google Scholar]
- Glesby M. J., Rosenmann E., Nylen E. G., Wrogemann K. (1988). Serum, CK, calcium, magnesium, and oxidative phosporylation in mdx mouse muscular dystrophy. Muscle Nerve 11, 852–856 10.1002/mus.880110809 [DOI] [PubMed] [Google Scholar]
- Hahn T., Desoye G., Lang I., Skofitsch G. (1993). Location and activities of acetylcholinesterase and butyrylcholinesterase in the rat and human placenta. Anat. Embryol. (Berl.) 188, 435–440 [DOI] [PubMed] [Google Scholar]
- Illsley N. P., Lamartiniere C. A. (1981). Endocrine regulation of rat serum cholinesterase activity. Endocrinology 108, 1737–1743 10.1210/endo-108-5-1737 [DOI] [PubMed] [Google Scholar]
- Johnson C. D., Russell R. L. (1975). A rapid, simple radiometric assay for cholinesterase, suitable for multiple determinations. Anal. Biochem. 64, 229–238 10.1016/0003-2697(75)90423-6 [DOI] [PubMed] [Google Scholar]
- Karpati G., Carpenter S., Prescott S. (1988). Small-caliber skeletal muscle fibers do not suffer necrosis in mdx mouse dystrophy. Muscle Nerve 11, 795–803 10.1002/mus.880110802 [DOI] [PubMed] [Google Scholar]
- Katz B., Miledi R. (1973). The binding of acetylcholine to receptors and its removal from the synaptic cleft. J. Physiol. 231, 549–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsberger C., Hammond P. I., Brimijoin S. (1998). Developmental expression of acetyl- and butyrylcholinesterase in the rat: enzyme and mRNA levels in embryonic dorsal root ganglia. Brain Res. 787, 248–258 10.1016/S0006-8993(97)01507-2 [DOI] [PubMed] [Google Scholar]
- Lamartiniere C. A. (1981). The hypothalamic-hypophyseal-gonadal regulation of hepatic glutathione S-transferase in the rat. Biochem. J. 198, 211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamartiniere C. A. (1986). Growth hormone modulates serum choline-sterase. Endocrinology 118, 1252–1254 10.1210/endo-118-3-1252 [DOI] [PubMed] [Google Scholar]
- Layer P. G. (1991). Cholinesterases during development of the avian nervous system. Cell. Mol. Neurobiol. 11, 7–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer P. G., Willbold E. (1994). Cholinesterases in avian neurogenesis. Int. Rev. Cytol. 151, 139–181 [DOI] [PubMed] [Google Scholar]
- Layer P. G., Willbold E. (1995). Novel functions of cholinesterases in development, physiology and disease. Prog. Histochem. Cytochem. 29, 1–94 [DOI] [PubMed] [Google Scholar]
- Leeuwin R. S. (1965). Effect of androgenic and anabolic compounds on pseudocholinesterase activity in the liver and serum of the rat. Acta Endocrinol. 50, 391–402 [DOI] [PubMed] [Google Scholar]
- Lindzey J., Wetsel W. C., Couse J. F., Stoker T., Cooper R., Korach K. S. (1998). Effects of castration and chronic steroid treatments on hypothalamic gonadotropin-releasing hormone content and pituitary gonadotropins in male wild-type and estrogen receptor-α knockout mice. Endocrinology 139, 4092–4101 10.1210/en.139.10.4092 [DOI] [PubMed] [Google Scholar]
- Lockridge O., Masson P. (2000). Pesticides and susceptible populations: people with butyrylcholinesterase genetic variants may be at risk. Neurotoxicology 21, 113–126 [PubMed] [Google Scholar]
- Lockridge O., Eckerson H. W., La Du B. N. (1979). Interchain disulfide bonds and subunit organization in human serum cholinesterase. J. Biol. Chem. 254, 8324–8330 [PubMed] [Google Scholar]
- Lyles J. M., Barnard E. A., Silman I. (1980). Changes in the levels and forms of cholinesterases in the blood plasma of normal and dystrophic chickens. J. Neurochem. 34, 979–988 [DOI] [PubMed] [Google Scholar]
- Masson P., Lockridge O. (2010). Butyrylcholinesterase for protection from organophosphorus poisons: catalytic complexities and hysteretic behavior. Arch. Biochem. Biophys. 494, 107–120 10.1016/j.abb.2009.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoulié J. (2002). The origin of the molecular diversity and functional anchoring of cholinesterases. Neurosignals 11, 130–143 [DOI] [PubMed] [Google Scholar]
- Massoulié J., Perrier N., Noureddine H., Liang D., Bon S. (2008). Old and new questions about cholinesterases. Chem. Biol. Interact. 175, 30–44 10.1016/j.cbi.2008.04.039 [DOI] [PubMed] [Google Scholar]
- Mendel B., Rudney H. (1943). Cholinesterases. 3. Specific tests for true cholinesterase and pseudo-cholinesterase. Biochem. J. 37, 473–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E., Soreq H. (2006). Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends Neurosci. 29, 216–224 10.1016/j.tins.2006.02.005 [DOI] [PubMed] [Google Scholar]
- Muntoni F., Torelli S., Ferlini A. (2003). Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2, 731–740 [DOI] [PubMed] [Google Scholar]
- Oliver L. J., Chatel J.-M., Massoulie J., Vigny M., Vallette F. M. (1992). Molecular forms of acetylcholinesterase in dystrophic (mdx) mouse tissues. Neuromuscul. Disord. 2, 87–97 10.1016/0960-8966(92)90040-D [DOI] [PubMed] [Google Scholar]
- Perelman A., Abeijon C., Hirschberg C. B., Inestrosa N. C., Brandan E. (1990). Differential association and distribution of acetyl- and butyrylcholinesterases within rat liver subcellular organelles. J. Biol. Chem. 265, 214–220 [PubMed] [Google Scholar]
- Ralston J. S., Rush R. S., Doctor B. P., Wolfe A. D. (1985). Acetylcholinesterase from fetal bovine serum. Purification and characterization of soluble G4 enzyme. J. Biol. Chem. 260, 4312–4316 [PubMed] [Google Scholar]
- Sawyer C. H., Everett J. W. (1946). Effects of various hormonal conditions in the intact rat on the synthesis of serum cholinesterase. Endocrinology 39, 307 [DOI] [PubMed] [Google Scholar]
- Sawyer C. H., Everett J. W. (1947). Cholinesterase in rat tissues and the site of serum non-specific cholinesterase production. Am. J. Physiol. 148, 675 [DOI] [PubMed] [Google Scholar]
- Settles D. L., Cihak R. A., Erickson H. P. (1996). Tenasin-C expression in dystrophin-related muscular dystrophy. Muscle Nerve 19, 147–154 [DOI] [PubMed] [Google Scholar]
- Sicinski P., Geng Y., Ryder-Cook A. S., Barnard E. A., Darlison M. G., Barnard P. J. (1989). The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244, 1578–1580 10.1126/science.2662404 [DOI] [PubMed] [Google Scholar]
- Silver A. (1974). “Pseudocholinesterases,” in The Biology of Cholinesterases, Vol. 36, eds Neuberger A., Tatum E. L. (New York, NY: North-Holland Publishing Co; ), 441 [Google Scholar]
- Singh J., O'Neill C., Handelsman D. J. (1995). Induction of spermatogenesis by androgens in gonadotropin-deficient (hpg) mice. Endocrinology 136, 5311–5321 10.1210/en.136.12.5311 [DOI] [PubMed] [Google Scholar]
- Skau K. A. (1985). Acetylcholinesterase molecular forms in serum and erythrocytes of laboratory animals. Comp. Biochem. Physiol. C 80, 207–210 [DOI] [PubMed] [Google Scholar]
- Vogel-Höpker A., Sperling L. E., Layer P. G. (2012). Co-opting functions of cholinesterases in neural, limb and stem cell development. Protein Pept. Lett. 19, 155–164 [DOI] [PubMed] [Google Scholar]
- Zaccaria M., Angelini C., Dal Pont O., Pegorara E., Marchetti M., Scandellari C. (1989). “Duchenne muscular dystrophy: lack of response to arginine test and low somatomedin-C levels,” in Growth Abnormalities, Serono Symposia Publications, Vol. 56, eds Bierich J. R., Cacciari E., Raiti S. (New York, NY: Raven Press; ), 469–473 [Google Scholar]
- Zatz M., Betti R. T., Frota-Pessoa O. (1986). Treatment of Duchenne muscular dystrophy with growth hormone inhibitors. Am. J. Med. Genet. 24, 549–566 10.1002/ajmg.1320240322 [DOI] [PubMed] [Google Scholar]
- Zatz M., Betti R. T., Levy J. A. (1981a). Benign Duchenne muscular dystrophy in a patient with growth hormone deficiency. Am. J. Med. Genet. 10, 301–304 10.1002/ajmg.1320100313 [DOI] [PubMed] [Google Scholar]
- Zatz M., Toledo S., Frota-Pessoa O. (1981b). Suggestion for a possible mitigating treatment of Duchenne muscular dystrophy. Am. J. Med. Genet. 10, 305–398 10.1002/ajmg.1320100314 [DOI] [PubMed] [Google Scholar]