

Abstract
Oligodendrocyte progenitor cells (OPCs) in the postnatal mouse corpus callosum (CC) and motor cortex (Ctx) reportedly generate only oligodendrocytes (OLs), whereas those in the piriform cortex may also generate neurons. OPCs have also been subdivided based on their expression of voltage-gated ion channels, ability to respond to neuronal activity, and proliferative state. To determine whether OPCs in the piriform cortex have inherently different physiological properties from those in the CC and Ctx, we studied acute brain slices from postnatal transgenic mice in which GFP expression identifies OL lineage cells. We whole-cell patch clamped GFP-expressing (GFP+) cells within the CC, Ctx, and anterior piriform cortex (aPC) and used prelabeling with 5-ethynyl-2′-deoxyuridine (EdU) to assess cell proliferation. After recording, slices were immunolabeled and OPCs were defined by strong expression of NG2. NG2+ OPCs in the white and gray matter proliferated and coexpressed PDGFRα and voltage-gated Na+ channels (INa). Approximately 70% of OPCs were capable of generating regenerative depolarizations. In addition to OLIG2+ NG2+ INa+ OPCs and OLIG2+ NG2neg INaneg OLs, we identified cells with low levels of NG2 limited to the soma or the base of some processes. These cells had a significantly reduced INa and a reduced ability to incorporate EdU when compared with OPCs and probably correspond to early differentiating OLs. By combining EdU labeling and lineage tracing using Pdgfrα–CreERT2 : R26R–YFP transgenic mice, we double labeled OPCs and traced their fate in the postnatal brain. These OPCs generated OLs but did not generate neurons in the aPC or elsewhere at any time that we examined.
Introduction
Oligodendrocytes (OLs) are produced by proliferating precursors, oligodendrocyte progenitor cells (OPCs), that express PDGFRα and NG2 proteoglycan. OPCs receive depolarizing synaptic input from unmyelinated glutamatergic and GABAergic neurons (Bergles et al., 2000; Lin and Bergles, 2004; Kukley et al., 2007, 2008, 2010; Ziskin et al., 2007), and some OPCs can fire regenerative action potential-like depolarizations (Chittajallu et al., 2004; Káradóttir et al., 2008; Ge et al., 2009). The function of this signaling is uncertain, but in vitro neurotransmitters alter OPC migration and differentiation (Yuan et al., 1998; Gudz et al., 2006; Tong et al., 2009). Furthermore, axonal stimulation promotes OPC proliferation and OL differentiation (Li et al., 2010), whereas blocking action potentials retards myelination (Barres and Raff, 1993; Demerens et al., 1996). More recently, vesicular release of glutamate was demonstrated to promote myelin protein translation at axon–OL contacts (Wake et al., 2011), supporting the idea that synaptic communication modulates myelination.
Whether all OPCs are the same is controversial. On the one hand, it has been suggested that all OPCs express voltage-gated sodium (INa) and potassium channels, which are downregulated when OPCs differentiate to form OLs (De Biase et al., 2010; Kukley et al., 2010). On the other hand, NG2-expressing (NG2+) OPCs in the rat cerebellar white matter fall into two classes: one that receives synaptic input, expresses INa, and can generate action potentials and another that lacks these attributes (Káradóttir et al., 2008). Voltage-gated ion channel expression and spiking behavior also reportedly differ between white and gray matter OPCs (Chittajallu et al., 2004). Furthermore, perinatal OPCs have been shown to produce OLs and astrocytes in vivo, whereas postnatal OPCs generate only OLs, except in the piriform cortex in which they have been reported to also produce some neurons (Dayer et al., 2005; Tamura et al., 2007; Rivers et al., 2008; Guo et al., 2010; Zhu et al., 2011).
Despite reports of neurogenesis in the piriform cortex (Bernier et al., 2002; Pekcec et al., 2006; Shapiro et al., 2007; Arisi et al., 2012), the source of the new neurons is contentious, with the main candidates being neural stem cells and OPCs. In many brain regions, OPCs express doublecortin (DCX) (Tamura et al., 2007; Guo et al., 2010; Ehninger et al., 2011), a marker of migratory neuroblasts, but those in the piriform cortex also express the neuroblast marker polysialylated neural cell adhesion molecule (PSA-NCAM) and the neural precursor markers Sox2 and Pax6 (Seki and Arai, 1999; Hayashi et al., 2001; Nacher et al., 2002; Pekcec et al., 2006; Shapiro et al., 2007; Bullmann et al., 2010; Guo et al., 2010), suggesting that they might be a functionally distinct cell population.
We report that OPCs in the mouse corpus callosum (CC), motor cortex (Ctx), and anterior piriform cortex (aPC) have similar membrane properties: they express INa but do not generate bona fide action potentials. By combining 5-ethynyl-2′-deoxyuridine (EdU) administration with transgenic lineage tracing, we demonstrate that OPCs within the postnatal forebrain proliferate and generate OLs but do not generate neurons at any time that we examined after postnatal day 25 (P25).
Materials and Methods
Transgenic mice.
Pdgfrα–H2BGFP knock-in mice (Hamilton et al., 2003), referred to as Pdgfrα–GFP mice, were purchased from The Jackson Laboratory (line B6.129S4–Pdgfrαtm11(EGFP)Sor/J). These mice have a histone–GFP fusion gene knocked into the Pdgfrα locus, resulting in nuclear labeling of PDGFRα-expressing cells, including OPCs. Pdgfrα–GFP mice were genotyped by PCR using the following primers: Ra-lox, 5′-ACGAAGTTATTAGGTCCCTCGAC-3′; AR4, 5′-CCCTTGTGGTCATGCCAAAC-3′; and AR5, 5′-GCTTTTGCCTCCATTACACTGG-3′, which amplified a 451 bp wild-type and/or a 242 bp mutant DNA fragment.
Homozygous Pdgfrα–CreERT2 bacterial artificial chromosome (BAC) transgenic mice (Rivers et al., 2008) were crossed with Cre-sensitive reporter mice [homozygous R26R–YFP transgenic mice (Srinivas et al., 2001) or heterozygous Tau–lox–STOP–lox–mGFP–IRES–NLS–LacZ–pA (Tau–mGFP) mice (Hippenmeyer et al., 2005)] to generate double-heterozygous offspring for analysis. Genotyping for Cre and R26R–YFP was performed by PCR amplification of genomic DNA as described previously (Rivers et al., 2008). PCR genotyping for mGFP (encoding a membrane-tethered version of GFP) was with GFP-specific primers: 5′-CCCTGAAGTTCATCTGCACCAC-3′ and 5′-TTCTCGTTGGGGTCTTTGCTC-3′.
Tamoxifen administration.
Tamoxifen (Sigma) was dissolved to 40 mg/ml in corn oil by sonicating at 21°C for ≥35 min. It was administered to young adult (P45) Pdgfrα–CreERT2 : R26R–YFP or Pdgfrα–CreERT2 : Tau–mGFP double-heterozygous mice, of either sex, by oral gavage on 4 consecutive days (each dose was 300 mg/kg tamoxifen body weight).
Electrophysiology.
Coronal brain slices (225 μm thick) were prepared from P9 (P7–P10) and P33 (P32–P34) mice, of either sex, that were prelabeled with EdU as described below. Slices were prepared in a solution containing 1 mm Na-kynurenate (Sigma-Aldrich) to block glutamate receptors (this was omitted from solutions for experiments) and superfused at 33 ± 1°C with bicarbonate-buffered solution, as described previously (Tripathi et al., 2011). Within a slice, a single cell expressing GFP under Pdgfrα transcriptional control (see above) from the CC, Ctx, or aPC was whole-cell voltage clamped to determine whether it expressed or lacked INa and filled with Alexa Fluor-568 dye. Whole-cell recordings were made from GFP+ cells (OPCs had brighter GFP fluorescence than OLs; see Results). After recording, the patch electrode was removed from the cell, leaving the soma intact. The slice was then immersion fixed in 4% (w/v) paraformaldehyde (PFA) in PBS at 21°C for a minimum of 1 h (maximum of 6 h). These cells were also processed to detect GFP and NG2, with or without EdU, as described below. Some slices were briefly fixed for 20 min at 21°C to allow immunolabeling with anti-PDGFRα (this antibody is fixation sensitive).
Patch electrodes had a resistance between 5 and 8 MΩ when filled with a solution containing the following (in mm): 130 potassium gluconate, 4 NaCl, 0.5 CaCl2, 10 HEPES, 10 BAPTA, 4 MgATP, and 0.5 Na2GTP, with Alexa Fluor-568 dye (Invitrogen), pH set to 7.3 with KOH. Voltage-gated sodium and potassium currents were elicited with a series of voltage steps (from −110 to 10 mV, in 20 mV increments) from a holding potential of −70 mV. Tetrodotoxin (TTX) at 1 μm (Tocris Bioscience) was applied in the superfusion solution to block sodium current. The peak net inward current (reflecting the difference between the inward INa and the other voltage-dependent outward currents) was quantified after subtracting the capacity current and an assumed ohmic leak current, scaled from the response to hyperpolarizing pulses. Voltage responses to current injection were recorded from the resting potential of the cell (applying 20 pA steps for 200 ms). An Axopatch 200B amplifier (Molecular Devices) was used for voltage and current clamp, and electrode junction potentials were compensated. Data were sampled at 50 kHz and filtered at 10 kHz using pClamp 9.2, and offline analysis was performed using Clampfit 9.2 (Molecular Devices).
Tissue preparation and immunolabeling.
Mice were perfusion fixed with 4% PFA in PBS. Brains were removed and sliced into 2 mm coronal slices using a rodent brain matrix (Agar Scientific). Brain slices were immersion fixed in 4% PFA in PBS at 4°C overnight (except for PDGFRα immunolabeling in which tissue was immersion fixed for 45 min at 21°C) and cryoprotected in 20% (w/v) sucrose in PBS at 4°C for 6 h, before embedding in Optimal Cutting Temperature medium (Tissue-Tek). Frozen tissue was stored at −80°C. Cryosections (30 μm thick) were collected and immunohistochemistry was performed on floating sections as described previously (Young et al., 2007).
The following primary antibodies were used: rabbit anti-GFP serum (1:1000; Synaptic Systems), rat anti-GFP IgG2a (1:3000; Nacalai Tesque), rat monoclonal anti-PDGFRα (1:400; BD Biosciences Pharmingen), rabbit anti-NG2 (1:500; Millipore), rabbit anti-OLIG2 (1:1000; Millipore), mouse anti-neuron-specific nuclear protein (NeuN) (1:500; Millipore), mouse anti-glial fibrillary acidic protein (1:2000; Sigma), rabbit anti-somatostatin (SST) (1:100; Peninsular Laboratories), rabbit anti-neuropeptide Y (NPY) (1:1000; Insight), rabbit anti-parvalbumin (1:1000; Millipore Bioscience Research Reagents), mouse anti-calbindin (1:1000; Swant), rabbit anti-calretinin (1:1000; Swant), mouse anti-PSA-NCAM IgM (1:500; Millipore Bioscience Research Reagents), mouse anti-reelin (1:2000; a gift from A. Goffinet, University of Louvain, Louvain, Belgium), rabbit anti-Iba1 (1:1000; Synaptic Systems), guinea-pig anti-DCX (1:1000; Millipore), and mouse monoclonal CC1 (1:200; against adenomatous polyposis antigen; Calbiochem). Secondary antibodies were goat anti-rat (1:500), donkey anti-rabbit (1:1000), goat anti-mouse IgM or goat anti-mouse IgG (heavy and light chains) (1:1000) conjugated with Alexa Fluor-405, Alexa Fluor-488, Alexa Fluor-568, or Alexa Fluor-647 (Invitrogen), and goat anti-guinea pig Cy3 (1:500; Millipore Bioscience Research Reagents). Cell nuclei were visualized by post-staining with Hoechst 33258 (1:1000; Sigma). When immunolabeling with CC1, Tris-buffered saline (TBS) blocking solution [TBS containing 0.5% (v/v) Triton X-100 and 10% fetal calf serum] was used in place of PBS blocking solution, and all washes were performed in TBS.
EdU labeling and detection.
EdU (Invitrogen) is a thymidine analog that is incorporated into the DNA of cells as they undergo DNA replication. EdU was administered to mice either as a series of subcutaneous injections (for mice aged P10 or younger) or via the drinking water (for mice aged P20 or older). For subcutaneous injections, EdU was dissolved at 1 mg/ml in PBS, and each pup was injected with 50 μl at 7:30 A.M., 10:30 A.M., 1:30 P.M., 4:30 P.M., and 7:30 P.M. for 5 consecutive days. Alternatively, EdU was dissolved in the drinking water (0.3 mg/ml) that was available ad libitum and replaced every 2–3 d. Optimization of this in vivo EdU labeling method and a more detailed comparison of in vivo BrdU and EdU labeling and detection methodologies will be detailed in a later publication (K. M. Young, K. Psachoulia, and W. D. Richardson, unpublished observations).
After fixation, tissue slices were washed once with PBS and then incubated for 20 min at 21°C in 0.8% (v/v) Triton X-100 in PBS on an orbital shaker. The slices were again washed in PBS before EdU detection with the Click-iT EdU Alexa Fluor-647 Imaging kit (Invitrogen). Each slice was immersed in ∼120 μl of Click-iT developing mixture and incubated for 45 min at 21°C in the dark, according to the instructions of the manufacturer. Slices were washed once with PBS before immunolabeling for NG2 (see above).
For floating cryosections, the EdU labeling was developed immediately after immunolabeling, because some antibodies failed if the order was reversed. Floating cryosections were incubated at 21°C for 15 min in PBS with 0.5% (v/v) Triton X-100, transferred to the EdU developing mixture, incubated in the dark at 21°C for 40 min, washed three times in PBS, post-stained with Hoechst 33258 (1:1000; Sigma) to visualize cell nuclei, and mounted under coverslips in fluorescence mounting medium (Dako). Unlike BrdU detection, EdU detection does not require antigen retrieval protocols.
Microscopy and cell counts.
All images were collected on an Ultraview confocal microscope (PerkinElmer Life and Analytical Sciences) as Z-stacks with 1 μm spacing, using standard excitation and emission filters for DAPI, FITC (Alexa Fluor-488), TRITC (Alexa Fluor-568), and Far Red (Alexa Fluor-647). For quantification, eight or more low-magnification (20× objective) fields were collected within each anatomical region of interest, for each coronal brain section, including all layers of the medial Ctx, layer II of the aPC, and the CC (region between the lateral edges of the lateral ventricles). Cells were counted manually, viewing each image in Adobe Photoshop. For post-electrophysiological confocal imaging of individual cells, the Alexa Fluor-568 dye-filled cell was located within the section and scored for EdU and NG2 expression, blind to the electrophysiology data for that cell. Fluorescence intensities and cell body diameters were measured in photomicrographs using NIH Image J software (http://rsbweb.nih.gov/ij/).
Statistics.
Data are presented as mean ± SEM. P values were obtained from Student's two-tailed t tests or ANOVA, as specified. Cell-cycle parameters were estimated from EdU labeling as described previously (Psachoulia et al., 2009).
Results
Using Pdgfrα–GFP transgenic mice to identify OL lineage cells in the postnatal brain
To investigate whether oligodendrogenic OPCs in the Ctx and CC of the postnatal mouse brain have electrical properties similar to the potentially neurogenic OPCs in the aPC, putative OPCs were identified by their intrinsic fluorescence in brain slices from Pdgfrα–GFP transgenic mice (Hamilton et al., 2003). Because PDGFRα is expressed by OPCs but not by differentiated OLs, we would expect OPCs to be specifically labeled in these mice. Pdgfrα–GFP transgenic mice were used previously to identify embryonic OPCs (Hamilton et al., 2003), but the specificity of the GFP expression was not characterized postnatally. Therefore, we coimmunolabeled P9 and P33 coronal brain sections for GFP and the OL lineage marker OLIG2 (Fig. 1a–c). In the Ctx, CC, and aPC, >96% of all GFP+ cells also expressed OLIG2 (Fig. 1g). Because NG2 immunolabeling is intense in OPCs but is lost as they differentiate into OLs, we immunolabeled additional sections for GFP and NG2 (Fig. 1d–f) to determine what proportion of the GFP+ cells corresponds to OPCs. At P9, ∼93% of GFP antibody-labeled cells also labeled for NG2. However, by P33, only ∼70% of all GFP+ cells in the Ctx, ∼30% of those in the CC, and ∼60% in the aPC also expressed NG2, suggesting that GFP persists in more mature oligodendroglial cells (OLIG2+ NG2neg) (Fig. 1g). Therefore, in the postnatal brain of Pdgfrα–GFP transgenic mice, GFP labeling identifies cells of the OL lineage (OLIG2+) but does not distinguish between OPCs and recently differentiated OLs.
Figure 1.
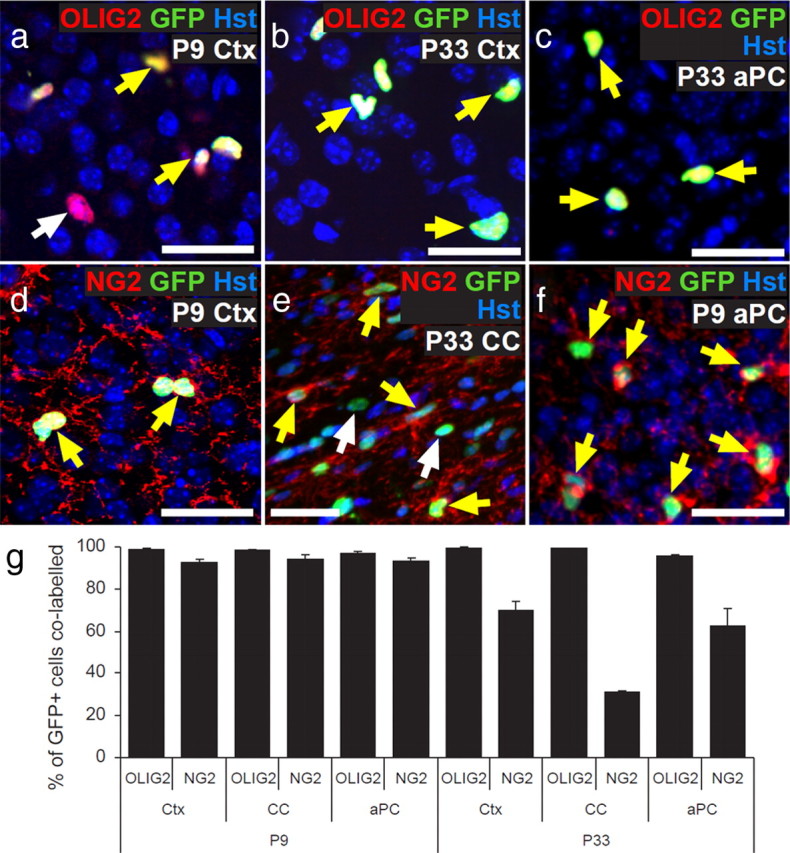
Pdgfrα–GFP mice label oligodendroglial cells in the postnatal brain. a–f, Cryosections from P9 to P33 Pdgfrα–GFP mice were immunolabeled to detect GFP (green) with either OLIG2 (red; a–c) or NG2 (red; d–f). Double-labeled cells are indicated by a yellow arrow and single-labeled cells by a white arrow. Sections were also stained with Hoechst 33258 (Hst; blue) to detect cell nuclei. g, The proportions of GFP+ cells that coexpressed each marker were quantified for the Ctx, CC, and aPC. The proportion of GFP+ cells that coexpressed NG2 fell significantly from P9 to P33 (ANOVA; p < 0.01). Scale bars, 20 μm.
Expression of voltage-gated sodium channels defines two populations of OL lineage cells in the postnatal mouse brain
To investigate the electrophysiological properties of OL lineage cells, we whole-cell patch clamped cells in the CC, Ctx, and aPC. Identified by their GFP expression, OL lineage cells in the brain of postnatal Pdgfrα–GFP mice could be divided into two broad classes of cell. One class (termed INa+) produced a transient inward current after depolarization beyond −30 mV (Fig. 2a–c), which was blocked by TTX (Fig. 3a), indicating that it was mediated by INa. This transient inward current was followed by activation of a more sustained outward, presumably K+ current (IK) (Fig. 2a–c). In contrast, the other cell type (termed INaneg) showed an essentially time-independent current–voltage (I–V) relationship at negative potentials but often an outward (presumably K+) voltage-gated current on depolarization (Fig. 2d–f). When TTX was applied to this cell class, the I–V relationship remained unchanged (Fig. 3b), excluding the possibility that the K+ conductance was masking a voltage-gated Na+ current in these cells (n = 3). Similar classes of cell, with and without INa, were also found in GFP+ cells in the cerebellum (data not shown).
Figure 2.
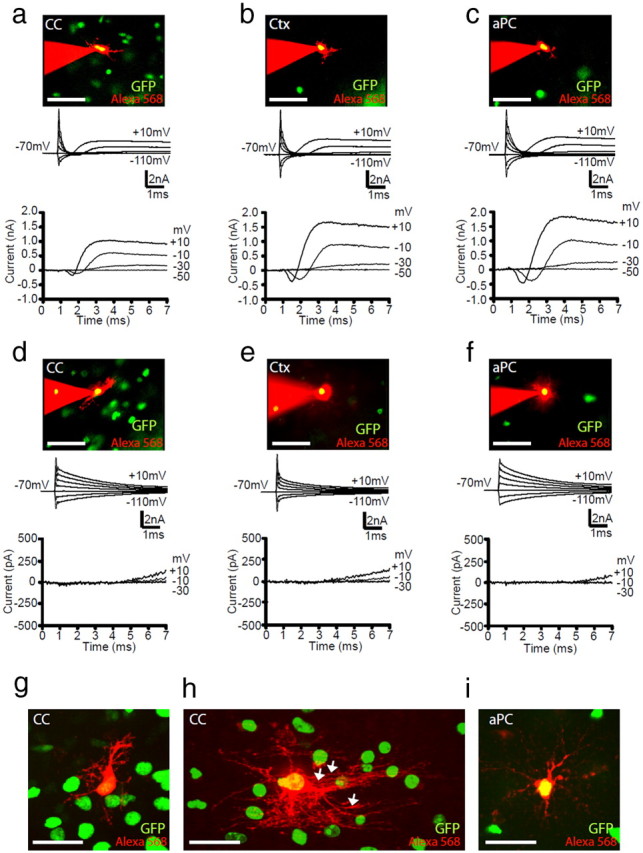
Two types of GFP+ OL lineage cells in the postnatal mouse brain. a–f, Cells expressing GFP driven by the Pdgfrα promoter (green) were whole-cell patch clamped and filled with Alexa Fluor-568 dye (red) (top image in columns a–f). Current responses (second panel of a–f) were recorded for GFP+ cells with and without INa (a–c and d–f, respectively), to voltage steps in 20 mV increments from −70 mV. Voltage-gated currents were isolated by subtracting the linearly scaled capacity transient and ohmic leak current evoked by a hyperpolarizing pulse (bottom sets of traces). Representative data, at either P9 (a–d) or P33 (e, f) from GFP+ cells in the CC (a, d), Ctx (b, e), and aPC (c, f) are shown. g–i, GFP was mainly expressed by cells with typical OPC morphology in white and gray matter [e.g., dye-filled GFP+ cells in the CC (g) and aPC (i)], but GFP could also be occasionally detected in cells that were more advanced in their differentiation with more OL-like morphology, including myelinating internodes (e.g., dye-filled GFP+ cell in the CC; h). White arrows indicate myelin internodes. Scale bars: a–f, 50 μm; g–i, 30 μm.
Figure 3.
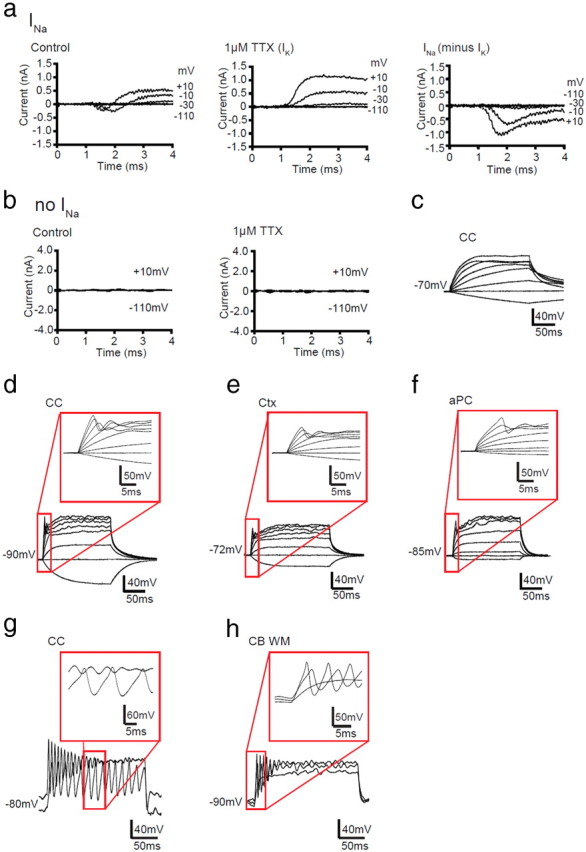
INa+ GFP+ cells generate action potential-like events in response to current injection. a, Left, Capacity transient and ohmic leak-subtracted responses of a GFP+ cell with INa to voltage steps in 20 mV increments from −70 mV. Middle, TTX at 1 μm blocked the INa. Right, Subtracting the voltage-gated IK isolated in TTX from the total voltage-gated current gave the voltage-gated Na+ current. b, Left, Capacity transient and ohmic leak-subtracted responses of a GFP+ cell without INa to voltage steps in increments of 20 mV (to −110 to +10 mV) from −70 mV. Right, TTX did not change the response of the cell to voltage steps. Traces representative of three GFP+ cells without INa are shown. c–f, Responses of white and gray matter GFP+ cells to depolarizing current injection (−20, 0, 20, 60, 100, 160, 220, and 260 pA of current was injected). c, Current-clamp recording of a GFP+ cell without INa, at P9, in the CC; similar responses were seen in all other brain areas examined. d–f, Current-clamp recordings for GFP+ cells with INa, at P9, in the CC (d), Ctx (e), and aPC (f). g, h, Current-clamp recordings from GFP+ INa cells, which produced repetitive action potential-like waves in the CC at P33 (40, 140 pA of current was injected) (g) and cerebellar white matter (CB WM) at P9 (40, 80, 120 pA of current was injected) (h). Red boxes indicate the region of the trace that has been enlarged.
Owing to the rapid kinetics of INa, it partly overlaps with the transient capacity current, so the net inward current was quantified after subtracting the linearly scaled capacity transient and ohmic leak current that was evoked by a hyperpolarizing pulse (Fig. 2, bottom traces). The resulting peak inward current represents a combination of INa and IK. To assess how the overlap of these currents alters the apparent magnitude of the voltage-gated Na+ current, TTX was applied to some cells to isolate IK (Fig. 3a). Subtraction of IK in these cells from the total inward current revealed a 2.5-fold increase in the amplitude of the net inward current (n = 3) (Fig. 3a).
INa cells show regenerative action potential-like waves but do not express neuronal markers
It has been reported that some OPCs can fire action potentials in response to depolarization (Chittajallu et al., 2004; Káradóttir et al., 2008; Ge et al., 2009). In our present study, depolarizing current injection resulted in passive membrane responses in INaneg GFP+ cells (Fig. 3c). INa+ GFP+ cells exhibited a range of action potential-like waves in response to depolarizing current pulses (Fig. 3d–h). Early in development (P9), the majority of INa+ GFP+ cells (69 of 87) in both gray and white matter showed a single regenerative depolarization (Fig. 3d–f). Single action potential-like events were also detected in 21 of 37 INa+ GFP+ cells in the white and gray matter later in development (P33). A minority of INa-expressing cells (3 of 124) exhibited repetitive action potential-like waves on depolarization (Fig. 3g,h). Similar regenerative responses have been reported for OPCs in white (Chittajallu et al., 2004; De Biase et al., 2010) and gray (Chittajallu et al., 2004; Ge et al., 2009; De Biase et al., 2010) matter regions of the postnatal mouse brain.
Although INa+ GFP+ cells could generate action potential-like events, they were not like typical neuronal action potentials or those described for INa+ OPCs in the rat cerebellar white matter (Káradóttir et al., 2008). The action potential-like events had a higher threshold for activation (−21 ± 1.5 mV) than action potentials in neurons, their amplitudes increased somewhat with larger current injections (Fig. 3d–f) unlike neuronal action potentials, and the timescale of the spikes (4.3 ± 0.7 ms, measuring the duration at half of the amplitude) was longer than those for most neurons (for examples, see Fig. 3d–h). This probably reflects the lower ratio of INa current magnitude to cell input resistance in mouse OPCs (mean peak inward current of <300 pA; see Fig. 5k) than was found previously for rat OPCs (mean peak inward current of ∼1000 pA; Káradóttir et al., 2008) and suggests that mouse INa+ cells do not have a sufficient density of INa to fire bona fide action potentials.
Figure 5.
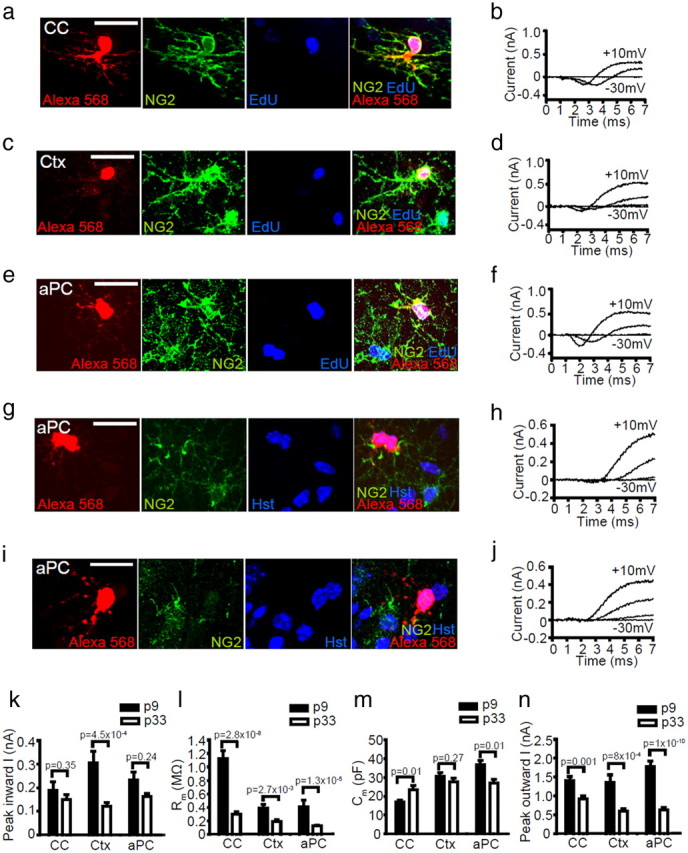
All brain NG2+ OPCs express INa and proliferate. a–j, Images of dye fills of whole-cell patch-clamped GFP+ cells in CC, Ctx, and aPC (red) and labeling for NG2 (green) and EdU [or Hoechst 33258 (Hst) in g, i] (blue) (left), together with current responses to depolarizing voltage steps in 20 mV increments from −70 mV (right). Cells fell into one of three main groups: a–f, NG2+ INa+ mitotic (EdU+) OPCs; g, h, NG2low INalow early differentiating OLs; and i, j, electrically passive OLs (NG2neg, INaneg). k–n, The average electrophysiological data for NG2+ OPCs (mean ± SEM) at P9 (CC, n = 20; Ctx, n = 15; aPC, n = 14 cells) and P33 (CC, n = 22; Ctx, n = 17; aPC, n = 41 cells) are shown, including the following: k, peak inward current on depolarization from −70 to +10 mV; l, membrane resistance at −70 mV; m, membrane capacitance; and n, peak outward current (on depolarization from −70 to +10 mV). Scale bars, 20 μm.
As INa+ GFP+ cells were detected in all brain regions, including the white matter of the CC, it seemed unlikely that the excitability of the INa+ GFP+ cells analyzed was a result of ectopic GFP expression by immature neurons or cortical interneurons. However, because some OPCs reportedly express immature neuronal markers, such as TOAD-64 (turned on after division-64), DCX, Tuj1, and HuC/D (Belachew et al., 2003; Chittajallu et al., 2004; Tamura et al., 2007), suggesting that they might turn into neurons in various brain regions (for review, see Richardson et al., 2011), we investigated this possibility by immunolabeling coronal brain sections from P9 and P33 Pdgfrα–GFP transgenic mice to detect GFP and a variety of neuronal markers (Fig. 4). At both ages, GFP+ cells in the Ctx were negative for the expression of parvalbumin (Fig. 4a,m), SST (Fig. 4b,m), NPY (Fig. 4c,m), calbindin (Fig. 4d,m), reelin (Fig. 4e,m), and PSA-NCAM (Fig. 4l,m). Although the vast majority of GFP+ cells were also negative for calretinin (Fig. 4f,m) and NeuN (Fig. 4i,m), a small number of cells that exhibited faint GFP expression were found to express calretinin (Fig. 4g,h,m) or NeuN (Fig. 4j,k,m) strongly and so are presumed to be neurons. In the Ctx, these presumptive neurons were located in the superficial cortex, concentrated near the brain midline. Even if the calretinin+ and NeuN+ GFP+ cells are separate populations, at P33, they would together still comprise only ∼2% (and ∼4% at P9) of all GFP+ cells. Consistent with our findings in the Ctx, GFP+ cells in the aPC of P33 Pdgfrα–GFP transgenic mice rarely expressed neuronal markers: a small number colabeled for PSA-NCAM (4 of 1820 GFP+ cells examined; 4 of 606 PSA-NCAM+ cells examined), and only 0.7 ± 0.2% of GFP+ cells coexpressed NeuN.
Figure 4.
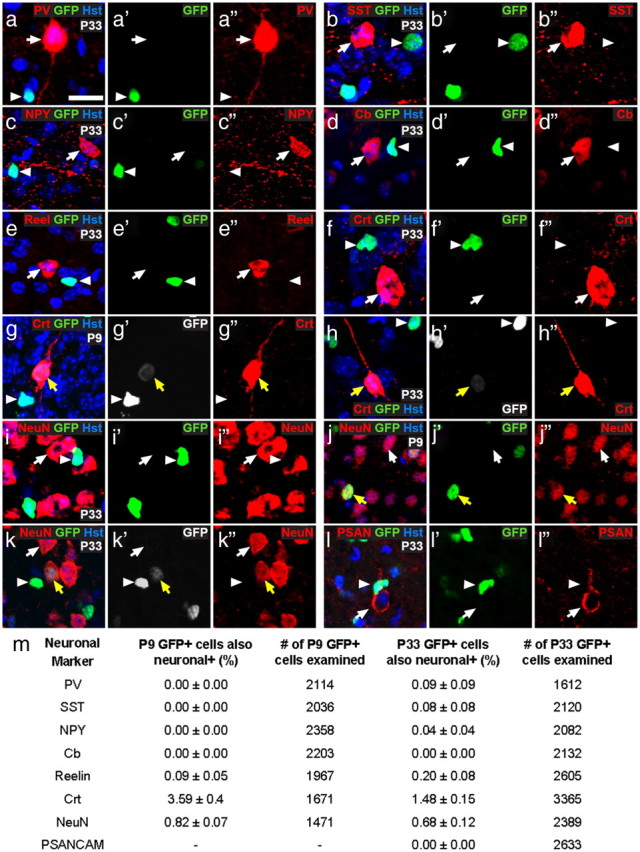
A small proportion of calretinin+ interneurons express GFP in the medial motor Ctx of Pdgfrα–GFP transgenic mice. a–l, Cryosections from P9 and P33 Pdgfrα–GFP transgenic mice were costained to detect GFP (green) and a variety of neuronal markers (red): parvalbumin (PV; a), SST (b), NPY (c), calbindin (cb; d), reelin (e), calretinin (crt; f–h), NeuN (i–k), and PSA-NCAM (l). m, The number of single-labeled GFP+ cells (white arrowheads), single-labeled neuronal cells (white arrows), and colabeled cells (yellow arrows) was counted. The table gives the proportion of GFP+ cells that coexpressed any of the neuronal markers. This was quantified in the Ctx for three mice and is expressed as the average percentage ± SEM. The total number of GFP+ cells examined is also given. The only neuronal markers expressed by subsets of cortical GFP+ cells were calretinin and NeuN. Scale bars: a, c–e, h, j–l, 15 μm; b, f, g, i, 10 μm.
Our finding that GFP (driven by an OL lineage cell promoter) is expressed by a small number of neuronal cells is reminiscent of a study from Belachew et al. (2003), who suggested that the brightly labeled GFP+ NG2+ OPCs that they observed in CNPase–GFP transgenic mice were able to generate faintly labeled GFP+ interneurons in the postnatal mouse hippocampus. Our data might suggest either that some interneurons are generated from OPCs and the GFP fluorescence is residual and indicative of their origin, or that a small subset of cortical neurons have a very low level of Pdgfrα promoter activity.
By counting the number of neurons labeled for calretinin and NeuN, we deduced that the GFP-labeled cells account for 11 ± 0.7% of calretinin+ and 0.14 ± 0.02% of NeuN+ neurons in the Ctx at P9 and 14 ± 2% of calretinin+ and 0.2 ± 0.02% of NeuN+ neurons in the Ctx at P33. The GFP+ neurons expressed extremely low levels of GFP that was approximately eight times lower in intensity (averaged over the nucleus) than was detected in NG2+ OPCs (neurons, 10.6 ± 1 arbitrary units; OPCs, 84.3 ± 0.1 arbitrary units) and approximately three times less than was detected in NG2neg OLs (35 ± 5 arbitrary units). The GFP+ neurons were difficult to detect even by antibody labeling, so it is highly unlikely that they would have been selected for patch clamping based on their intrinsic GFP fluorescence. Additionally, patch-clamp recordings were performed in brain hemisections; therefore, cells immediately bordering the cortical midline were not targeted, and the calretinin+ and NeuN+ cells were too few in number (<5%) to account for the significant number of GFP+ cells that we classify as excitable INa+ GFP+ cells (77 and 61% of the GFP+ cells that were studied expressed INa in the Ctx at P9 and P33, respectively).
All GFP+ cells that express NG2 strongly are proliferative and express INa
Based on GFP expression, we were able to identify OL lineage cells, but, as described above, we found that they were heterogeneous in terms of INa expression in all brain regions examined. The morphology of the Alexa Fluor-568-filled cells suggested that the expression of INa might be an OPC trait, whereas a lack of INa might be an OL trait. Morphology proved to be more useful for identifying putative OPCs in the white matter than in the gray matter. In white matter, GFP+ cells with OPC-like morphology (Fig. 2g) (INa+ GFP+ cell) could be easily distinguished from OLs with myelinating processes (Fig. 2h, INaneg GFP+ cell). OPCs were defined as having bipolar or stellate morphology and OLs as having multiple long processes aligned with axons. This distinction was less obvious in gray matter (Fig. 2i, INaneg GFP+, cell), because axons do not fasciculate in gray matter as they do in white matter tracts, and myelin internodes are not parallel with one another but oriented in all directions (Murtie et al., 2007; Vinet et al., 2010). Consequently, in the gray matter, some GFP+ differentiated OLs might easily be mistaken for OPCs. To identify OPCs unambiguously in both gray and white matter, it was necessary to perform post-recording immunohistochemistry for NG2.
Individual GFP+ cells in the CC, Ctx, and aPC were whole-cell patch clamped to determine whether or not they exhibited INa and then dye filled with Alexa Fluor-568. Dye filling not only revealed the cellular morphology but also permitted identification of the recorded cell after NG2 immunolabeling. Three categories of dye-filled cells were identified: (1) cells expressing NG2 strongly (NG2+ cells) that we define to be OPCs (Fig. 5a,c,e), (2) cells expressing low levels of NG2 limited to the soma or the base of some processes (NG2low cells) that were presumed to be early differentiating OLs (Fig. 5g), and (3) cells with no detectable NG2 (NG2neg cells) that were assumed to be OLs (Fig. 5i).
At both P9 and P33 and in all brain regions examined, we found that the NG2+ GFP+ cell population was the same as the population of INa+ GFP+ excitable cells (Fig. 5b,d,f). As expected, these NG2+ INa+ presumed OPCs were found to coexpress PDGFRα (five cells examined in the Ctx and seven cells in the aPC), confirming them as OPCs. The magnitude of the net inward current generated by OPCs on depolarization from −70 to 10 mV was ∼130–320 pA in all areas (Fig. 5k) and decreased with age, falling by 21, 61, or 31% from P9 to P33 in the CC, Ctx, or aPC, respectively (Fig. 5k). Similarly, in all brain areas, a reduction in membrane resistance (Fig. 5l) and peak outward current (Fig. 5n) was observed with age, whereas no consistent change in cell capacitance (a measure of cell size) could be detected with age (Fig. 5m). OPCs were consistently larger in gray matter than in white matter, regardless of age (Fig. 5m). The resting membrane potential of OPCs ranged from between approximately −85 and −94 mV: at P9, the resting potential of OPCs was −94 ± 3.8, −93 ± 2.6, and −92 ± 1.6 mV in the CC (n = 14), Ctx (n = 14), and aPC (n = 10), respectively, whereas at P33, the resting potential of OPCs was −89 ± 1.9, −86 ± 3.3, and −85 ± 2.3 mV in the CC (n = 22), Ctx (n = 18), and aPC (n = 17), respectively. These data suggest that the intrinsic membrane properties of OPCs are subject to change during development, but INa expression is a universal property of OPCs in all brain areas throughout postnatal life.
The second category of GFP+ cells, NG2low cells, had limited expression of NG2 (Fig. 5g). These cells had little to no INa (Fig. 5h; mean net inward current, 24 ± 5pA in nine cells) so we refer to these as NG2low INalow cells. We assume that they correspond to early differentiating OLs that are downregulating their INa and NG2 expression, as described previously (De Biase et al., 2010; Kukley et al., 2010), and are similar to the NG2+ INanegative cells reported in rat cerebellum by Káradóttir et al. (2008). The final category of GFP+ cells (NG2neg OLs) lacked both NG2 labeling (Fig. 5I) and INa expression (Fig. 5j) and therefore correspond to premyelinating or myelinating OLs (44 cells; NG2neg INaneg OLs), which we refer to collectively as OLs.
A number of studies have indicated the presence of a postmitotic population of OPCs (defined by NG2 immunolabeling) within the postnatal mouse brain (for review, see Richardson et al., 2011). In the piriform cortex, these postmitotic OPCs have been suggested to comprise a substantial fraction of the OPC population and to differentiate over time, providing a source of new projection neurons (Rivers et al., 2008; Guo et al., 2010). To assess the electrophysiological properties of dividing oligodendrogenic OPCs and the nondividing putative neurogenic OPCs, we administered the thymidine analog EdU to Pdgfrα–GFP mice for 5 d before making patch-clamp recordings at P9 and for 12–13 d before making patch-clamp recordings at P33. In addition to NG2 immunolabeling, slices were processed to detect EdU. These dosing regimens were selected to label the proliferating OPCs in the white and gray matter of the brain, which were predicted from Psachoulia et al. (2009) to be approximately half of all OPCs. Unexpectedly, given this prediction, the vast majority of GFP+, NG2+, INa+ OPCs that we analyzed were EdU+, regardless of brain region or age. At P9, ∼90% of NG2+ INa+ OPCs had divided in the previous 5 days (Ctx, 8 of 9 OPCs; CC, 12 of 13 OPCs). At P33, 70–100% of all GFP+, NG2+, INa+ OPCs were EdU labeled in all regions (Ctx, 17 of 24 OPCs; CC, 4 of 4 OPCs; aPC, 29 of 31 OPCs) (Fig. 5a–f). In contrast, none of the NG2low INalow cells analyzed in the Ctx of P33 mice (after ∼13 d of EdU administration) had incorporated EdU (nine cells analyzed), consistent with in vitro data from Sugiarto et al. (2011), who suggested that, after an asymmetric OPC division, one cell begins to downregulate mitogenic receptors, commits to differentiating, and will no longer incorporate BrdU, whereas the other retains the expression of mitogenic receptors, remains an OPC, and reenters the cell cycle. This supports the idea that the NG2low INalow cells are early differentiating OLs, formed from a cell division that occurred >13 d earlier. The large fraction of individual OPCs that were EdU+ after patch-clamp recordings accurately reflected the total fraction of EdU+ OPCs quantified in each brain region when confocal analysis was performed on large numbers of OPCs immunolabeled for NG2. In Ctx at P9, 90 ± 2% of NG2+ OPCs were EdU+, and, at P33, 83 ± 1% of NG2+ OPCs were EdU+, indicating that the high fraction of OPCs identified as proliferating was not the result of a cell-selection bias when patch clamping but instead indicated that a larger proportion of OPCs underwent cell division than was detected previously by Psachoulia et al. (2009) and Guo et al. (2010).
These data undermine previous reports, based on cumulative BrdU labeling of dividing OPCs in wild-type mice, which concluded that there is a postmitotic population of OPCs in the brain (Rivers et al., 2008; Psachoulia et al., 2009; Guo et al., 2010; Simon et al., 2011). This in turn raises questions about the origin of new projection neurons that accumulate in the piriform cortex of adult Pdgfrα–CreERT2 : R26R–YFP and Plp–CreERT2 : R26R–YFP transgenic mice but do so without incorporating BrdU even after long labeling periods and thus are unlikely to be derived from dividing progenitors (Rivers et al., 2008; Guo et al., 2010). However, our EdU labeling experiments on Ctx described above left open the possibility that there might be a local population of postmitotic OPCs specifically in the aPC. To test this, we administered EdU to P21 and P60 mice via their drinking water for various time periods up to 35 d. Coronal brain sections were immunolabeled for PDGFRα and NG2 to detect OPCs and were costained for EdU (Fig. 6a). When administering EdU from P21, we found that the proportion of OPCs in the aPC that became EdU+ increased linearly with time (r2 > 0.96) at a rate of 7.6% per day, until >98% of all OPCs were stably labeled after ∼13 d (Fig. 6b). From P60, we found that the fraction of EdU-labeled OPCs increased linearly with time (r2 > 0.97) at a rate of 3.15% per day until >98% of OPCs were labeled after ∼30 d. From these data, we were able to calculate the cell-cycle time for OPCs in the aPC as being ∼12.9 ± 0.8 d at P21 and 31.3 ± 1.7 d at P60 (using the methods described by Psachoulia et al., 2009). These data show that all OPCs (defined by strong NG2 and PDGFRα expression) in the aPC are mitotically active, as in other regions of the forebrain.
Figure 6.

All NG2+ OPCs in the aPC proliferate and incorporate EdU. a, Coronal brain sections containing the aPC immunolabeled to detect PDGFRα (red) and NG2 (green) to identify OPCs and then developed to visualize EdU incorporation (blue). b, The proportion of OPCs (PDGFRα+ NG2+) that had incorporated EdU as a function of EdU administration time (mean ± SEM, n = 3 mice for each time point). Scale bar, 35 μm.
OPCs generate exclusively OLs in the postnatal mouse brain
To determine what cell types are produced by OPCs in the CC, Ctx, and aPC, we labeled and traced the fates of OPCs using two cell tracing methods simultaneously: (1) extended EdU exposure to label all proliferating cells (including all OPCs and their differentiating progeny) and (2) Cre–lox fate mapping using Pdgfrα–CreERT2 mice. EdU was administered via the drinking water to Pdgfrα–CreERT2 : R26R–YFP mice continuously after P25 to prelabel all proliferating OPCs. Tamoxifen was administered to these same mice on 4 consecutive days (P45–P48 inclusive) to activate Cre recombination and induce YFP expression in PDGFRα-positive OPCs (see experimental time line in Fig. 7a).
Figure 7.
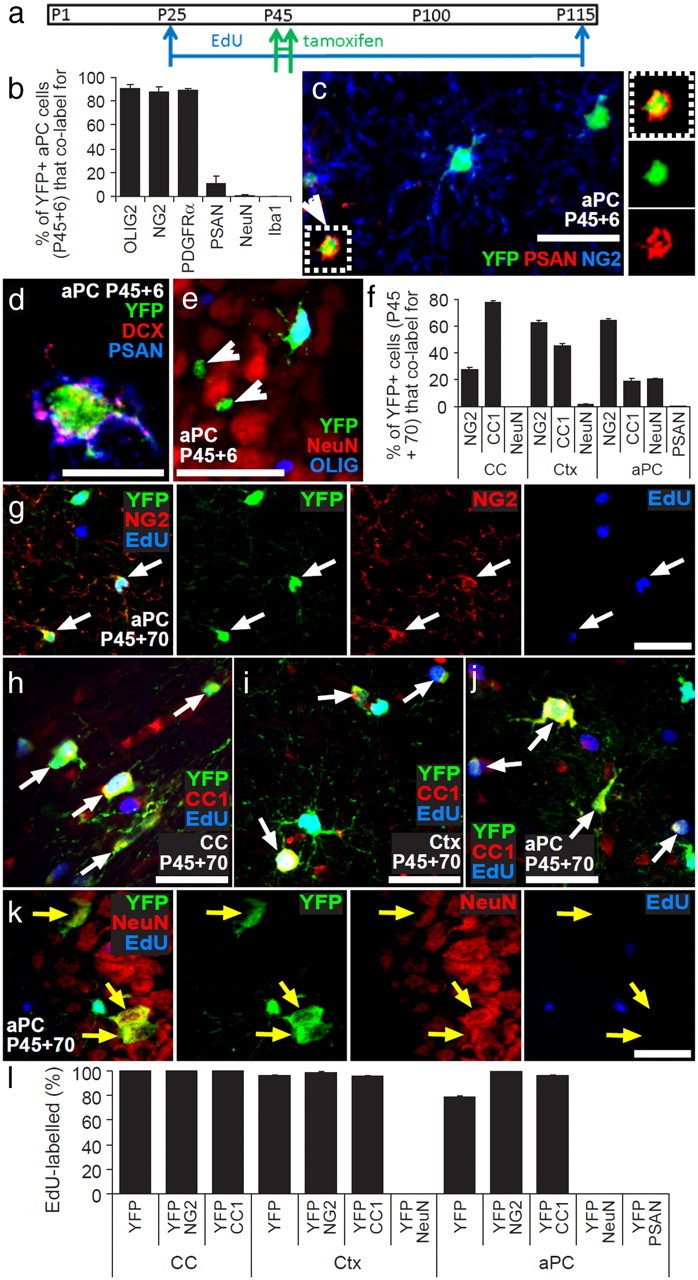
OPCs generate new OLs but not neurons in the postnatal mouse brain. a, Pdgfrα–CreERT2 : R26R–YFP transgenic mice received EdU from P25, tamoxifen from P45 to P48, and were perfusion fixed at P115 (P45 + 70). b, Proportions of YFP+ cells in aPC of P45 + 6 Pdgfrα–CreERT2 : R26R–YFP mouse brains that were OPCs (NG2+), OLs (CC1+), neuroblasts [PSA-NCAM+ (PSAN)], neurons (NeuN+), or microglia (Iba1+) (mean ± SEM, n = 3 mice). c, Immunolabeling of the aPC of P45 + 6 Pdgfrα–CreERT2 : R26R–YFP transgenic mice for NG2 (blue), GFP (green), and PSA-NCAM (red). d, A single-scan (1 μm) confocal image through a cell in the aPC of P45 + 6 Pdgfrα–CreERT2 : R26R–YFP transgenic mice that was immunolabeled for PSA-NCAM (blue), YFP (green), and DCX (red). e, Immunolabeling of the aPC of P45 + 6 Pdgfrα–CreERT2 : R26R–YFP transgenic mice for OLIG2 (blue), YFP (green), and NeuN (red). White arrowheads indicate unidentified YFP+ cells in the aPC. f, Proportions of YFP+ cells in the CC, Ctx, and aPC of P45 + 70 Pdgfrα–CreERT2 : R26R–YFP mice that were OPCs (NG2+), OLs (CC1+), or neurons (NeuN+) (mean ± SEM, n = 3 mice) and proportion in aPC that were PSA-NCAM+. g, Immunolabeling of the aPC for NG2 (red), GFP (green), and detection of EdU (blue). h–j, Immunolabeling of the CC (h), Ctx (i), and aPC (j) for CC1 antigen (red), GFP (green), and detection of EdU (blue). k, Immunolabeling of the aPC for NeuN (red), GFP (green), and detection of EdU (blue). l, Percentage of YFP+ OPCs (YFP+ NG2+), YFP+ OLs (YFP+ CC1+), and YFP+ neurons (YFP+ NeuN+) that incorporated EdU (mean ± SEM, n = 3 mice). White arrows indicate triple-labeled cells. Yellow arrows indicate double-positive but EdU-negative cells. Scale bars: c, e–k, 30 μm; d, 10 μm.
The identity of the starting population of YFP-labeled cells was confirmed by immunolabeling sections of Pdgfrα–CreERT2 : R26R–YFP mouse brains at P45 + 6, 2 days after the final dose of tamoxifen. Sections were immunolabeled for YFP and different combinations of OLIG2, NG2, PDGFRα, PSA-NCAM, NeuN, or the microglial marker Iba1 (Fig. 7b). In the aPC, ∼90% of all YFP+ cells colabeled for OLIG2 (>500 YFP+ cells counted), most of which were OPCs, because 88 ± 4% of YFP+ cells also colabeled for NG2 (>600 YFP+ cells counted) and 89 ± 1% colabeled for PDGFRα (>400 YFP+ cells counted). At P45 + 6, <1% of YFP+ cells in the aPC colabeled for NeuN (>500 YFP+ cells counted). These data confirmed that tamoxifen induced the YFP labeling of OPCs and an insignificant number of NeuN+ neurons. In addition to OPCs, we also detected a population of YFP+ PSA-NCAM+ cells in the aPC (Fig. 7c), which constituted 11 ± 6% of all YFP+ cells (>400 YFP+ cells counted). The YFP+ PSA-NCAM+ cells were smaller than OPCs, having cell bodies 5.2 ± 0.3 μm in diameter compared with 10.1 ± 0.5 μm for YFP+ NG2+ OPCs (20 cells measured in each case). Both the OPCs in the aPC and the very small YFP+ PSA-NCAM+ cells were found to express the microtubule-associated protein DCX (80 NG2+ OPCs and 15 YFP+ PSA-NCAM+ cells examined) (Fig. 7d); however, the small YFP+ PSA-NCAM+ cells were distinguishable from OL lineage cells in the same region because they did not express OLIG2 (Fig. 7e). Despite their small size, the YFP+ PSA-NCAM+ cells were not microglial cells because they did not express Iba1; indeed, no YFP+ Iba1+ cells were found in the aPC (>400 YFP+ cells counted; Fig. 7b). Importantly, the YFP+ PSA-NCAM+ cells did not become EdU labeled even when EdU was administered for 90 d (from P25 to P115; Fig. 7l). Therefore, OPCs are the only EdU+ YFP+ double-labeled cells within the aPC, allowing us to follow their fate specifically in vivo.
Pdgfrα–CreERT2 : R26R–YFP mice were labeled with EdU from P25, tamoxifen was administered from P45 to P48 (Fig. 7a), and the mice were analyzed on P115 (P45 + 70). Coronal brain sections were double immunolabeled for YFP and NG2 (OPCs), CC1 (OLs), or NeuN (neurons) (Fig. 7f). The sections were also processed to detect EdU. The vast majority of YFP+ cells in the CC and Ctx were OL lineage cells that expressed NG2 or CC1. However, as reported previously (Rivers et al., 2008), YFP+ NeuN+ neurons accumulated in the aPC in which they were about as numerous as YFP+, CC1+ OLs (Fig. 7f). As expected, essentially all of the YFP+ OPCs were EdU+ in all brain regions examined (Fig. 7g,l) (<2% of YFP+ OPCs escaped EdU labeling). Therefore, all the differentiated progeny of YFP+ EdU+ OPCs would be expected to inherit both markers. This was true for YFP+ CC1+ OLs in the CC, Ctx, and aPC, which were clearly also EdU+ (<4% of YFP+ CC1+ OLs escaped EdU labeling) (Fig. 7h–j,l), identifying them as cells that were generated from OPCs at some point between P45 and P115. However, this was not true for the YFP+ NeuN+ neurons that accumulated in the aPC over the same time period. None of the YFP+ NeuN+ neurons had incorporated EdU (Fig. 7k,l) (>1000 YFP+, NeuN+ cells counted in five mice), indicating that they are not the progeny of YFP+, EdU+ OPCs in the postnatal mouse brain. This suggests that the YFP+ aPC neurons were generated after P45 (when tamoxifen was given) by direct differentiation of precursor cells, as yet unidentified, that had undergone their final division before P25 (i.e., before we administered EdU).
YFP+ PSA-NCAM+ cells in the aPC are postmitotic cells that are born before P25
The YFP+ EdUneg PSA-NCAM+ cells that we identified in the aPC of P45 + 6 Pdgfrα–CreERT2 : R26R–YFP mice could be the source of the YFP+ EdUneg aPC neurons detected at P45 + 70. None of the rare YFP+ PSA-NCAM+ cells remaining in the aPC at P45 + 70 (Fig. 7f) were EdU+ (Fig. 7l), indicating that, like the YFP+ NeuN+ cells, they were not the progeny of YFP+ EdU+ OPCs. In fact, no PSA-NCAM+ cells detected in the aPC at any age had incorporated EdU (>150 PSA-NCAM+ cells examined in three mice), indicating that they were born before EdU exposure began on P25. These cells were suggested previously to be an intermediate between OPCs and projection neurons, being generated by the direct differentiation of DCX+ OPCs and retaining DCX expression (Guo et al., 2010). However, although our data indicate that both OPCs and the small YFP+ PSA-NCAM+ cells express DCX (Fig. 7d), the small YFP+ PSA-NCAM+ cells are actually a postmitotic population and so are not generated after P25 from proliferating OPCs or proliferating neural stem cells. At P45 + 70, PSA-NCAM+ cells represented only 0.5 ± 0.1% of all YFP+ cells within the aPC (>1200 YFP+ cells examined in n = 3 mice; Fig. 7f). This fraction is 20-fold less than at P45 + 6 (Fig. 7b), reflecting their dilution by the accumulating number of YFP+ OLs and neurons. From Rivers et al. (2008), we know that YFP+ NeuN+ cells are added to the aPC of Pdgfrα–CreERT2 : R26R–YFP transgenic mice at a rate of ∼0.18 cells per 30 μm coronal brain section per day, starting from 30 d after tamoxifen administration. This equates to approximately seven YFP+ neurons per section by P45 + 70 [(70 d − 30 d) × 0.18 per day = 7.2]. In our study, the total number of YFP+ PSA-NCAM+ cells per 30 μm coronal brain section fell from approximately seven cells per section at P45 + 6 to an average of approximately one cell per section at P45 + 70 (average number in 12 sections across three mice from each time point), which suggests that YFP+ PSA-NCAM+ cells either die or differentiate over time. However, the fall of approximately six YFP+ PSA-NCAM+ cells per section can be seen to almost match the number of YFP+ NeuN+ neurons added to the aPC in the same timeframe.
Discussion
OPCs were originally assumed to be a homogeneous class of immature oligodendroglial cell but have since been subdivided according to their electrical properties (Chittajallu et al., 2004; Káradóttir et al., 2008), gene expression profiles (Gensert and Goldman, 2001; Mallon et al., 2002; Lin et al., 2009), proliferation rates (Psachoulia et al., 2009), response to injury (Keirstead et al., 1998; Lytle et al., 2009), and their abilities to differentiate into myelinating OLs (Mallon et al., 2002; Dimou et al., 2008) or other types of cells, including neurons (Rivers et al., 2008; Zhu et al., 2008; Guo et al., 2010). There continues to be much debate over how many subtypes of OPC there might be and the most appropriate way to classify them. To advance our understanding of this issue, we performed experiments examining both the histological and electrophysiological properties of OPCs and determined that all postnatal OPCs defined by strong expression of the NG2 proteoglycan (1) coexpress the mitogenic receptor PDGFRα, (2) express INa and are electrically excitable, (3) are mitotically active and incorporate EdU, and (4) generate differentiated OLs but not neurons.
OPCs in the postnatal mouse brain are excitable but do not fire action potentials
Injecting current into OPCs led to action potential-like depolarization waves, consistent with previous studies in mice (Chittajallu et al., 2004; Ge et al., 2009; De Biase et al., 2010). These action potential-like waves differed from neuronal action potentials and the action potentials described previously for OPCs in the rat cerebellar white matter (Káradóttir et al., 2008), because they required a larger depolarization for activation, their amplitudes increased with larger current injections, and the timescale of the waves was longer. These regenerative events could be induced in OPCs in all brain areas and at all developmental stages investigated, suggesting they are a universal property of OPCs.
The likelihood that a cell will generate an action potential is determined by the magnitude of the voltage-gated sodium current and the cell membrane resistance (which can be adversely reduced by the conductance of the seal between the patch electrode and the cell membrane: Bakiri et al., 2009). Thus, the difference in spiking behavior we observed in mouse OPCs compared with rat OPCs (Káradóttir et al., 2008) might reflect the fact that mouse OPCs have a sixfold lower ratio of net inward current to membrane resistance than was found previously for rat INa OPCs (Káradóttir et al., 2008). It is conceivable that, even in mouse OPCs, more robust spiking behavior would be observed in vivo in the absence of shunting by the seal between the patch electrode and the cell.
We observed significant variation among OPCs in the amount of current required to initiate Na+ spikes (for the most excitable cells, the minimum current needed was 60 pA), in part reflecting differences in input resistances of the cells (Bakiri et al., 2009). White matter OPCs have been reported to receive ∼140 synapses (Kukley et al., 2007), for each of which the miniature EPSC at the resting potential is ∼6 pA (Kukley et al., 2007, 2010), implying that 10 simultaneous EPSCs would be needed to trigger this response in vivo (60 pA/6 pA). This is likely to happen more frequently during development when synchronized neuronal firing occurs in the cortex (Mao et al., 2001), hippocampus (Mohns and Blumberg, 2008), cerebellum (Watt et al., 2009), and retina (Meister et al., 1991; Demas et al., 2003). However, whether or not full action potentials are produced by mouse OPCs in vivo, activation of their voltage-gated sodium channels is likely to amplify excitatory synaptic inputs, raising intracellular calcium levels to regulate OPC development, so the expression of INa by OPCs might play a key role in regulating the developmental myelination of active circuits.
Is excitability a regulator of OPC proliferation?
All OPCs have INa, proliferate, and generate OLs in vivo (Figs. 5–7). However, the rate of OPC proliferation and OL production varies significantly between brain regions, with OPCs in the white matter dividing more rapidly than those in the gray matter (Power et al., 2002; Dimou et al., 2008; Rivers et al., 2008; Psachoulia et al., 2009; Simon et al., 2011). Many factors regulate OPC proliferation, such as PDGF-AA, basic fibroblast growth factor FGF2, and sonic hedgehog (Pringle et al., 1989; Collarini et al., 1992; Zhu et al., 1999). Furthermore, the expression of NG2 by OPCs has been reported to enhance ligand binding to PDGFRα and the fibroblast growth factor receptor (Goretzki et al., 1999). In the presence of growth factors, OPCs can proliferate in the absence of neurons (Collarini et al., 1992). However, there is evidence that neuronal activity also influences OPC proliferation in vivo. For example, injecting TTX into the eye, to block action potentials in retinal ganglion neurons, causes a large decrease in OPC proliferation in the optic nerve (Barres and Raff, 1993). This inhibitory effect of TTX could be alleviated by supplying exogenous PDGF to the optic nerve, suggesting that electrical activity stimulates release of PDGF within the nerve or enhances the responsiveness of OPCs to PDGF (Barres and Raff, 1993). There is other evidence that links growth factor signaling and electrical activity. PDGFRα signaling maintains the expression of delayed outward-rectifying K+ channels by OPCs (Chittajallu et al., 2005), and block of K+ channels as a consequence of glutamatergic signaling to OPCs inhibits their proliferation and lineage progression (Knutson et al., 1997).
We considered whether OPC proliferation rate might correlate with the density of voltage-gated potassium channels (Knutson et al., 1997). The cell cycle time for OPCs in the CC of the P21 mouse brain is ∼3 d, which is significantly shorter than the cell-cycle time for OPCs in the Ctx (∼18 d; Young, Psachoulia, and Richardson, unpublished observations) or aPC (∼13 d; Fig. 6). Perhaps coincidentally, in P33 mice, the peak outward current was twofold larger in the CC compared with the other regions (Fig. 5n), consistent with a potential role for IK in contributing to the differential proliferation rate in white versus gray matter areas at late developmental times.
OPCs generate OLs but not neurons in the postnatal mouse forebrain
We and others have reported previously that OPCs generate new neurons in the postnatal mouse brain (Rivers et al., 2008; Guo et al., 2010). We observed very small numbers of YFP+, NeuN+ neurons throughout the forebrain of Pdgfrα–CreERT2 : R26R–YFP mice at short times after tamoxifen, but, in general, these neurons did not accumulate with time after tamoxifen, as one would predict if they were produced continually from YFP+ precursors and survived long term. Similar observations were made using an independently generated Pdgfrα–CreERT2 line (Kang et al., 2010) and an Ng2–CreER line (Zhu et al., 2011), prompting the suggestion that the rare labeled neurons were the result of sporadic activation of the CreER transgenes in a subset of neurons (Kang et al., 2010; Zhu et al., 2011). However, we also observed YFP-labeled neurons in the aPC (but not the posterior piriform cortex; Young and Richardson, unpublished observations) that apparently survived and accumulated for months after tamoxifen treatment (Rivers et al., 2008). We observed a similar accumulation of mGFP+ neurons with time in the aPC of Pdgfrα–CreERT2 : Tau–mGFP transgenic mice (Fig. 8). It is difficult to understand how this could come about unless these particular neurons are produced continuously in the postnatal aPC from a pool of precursors that become YFP labeled at the time of tamoxifen treatment. In the previous studies, the YFP-labeled neurons were consistently BrdU negative, even after extended BrdU exposure, as were ∼50% of OPCs, leading us to speculate that a subpopulation of mitotically quiescent OPCs might differentiate directly into new aPC neurons without an intervening cell division. Now, using an alternative thymidine analog (EdU), we find that all OPCs in the forebrain are mitotically active (Fig. 6) and so cannot be the source of new neurons in the aPC, which are consistently EdU negative (Fig. 7). Therefore, we now agree with Kang et al. (2010) who found that all OPCs, but no neurons, incorporated BrdU after a long labeling period. Nevertheless, the fact that YFP+ neurons accumulate in the aPC after tamoxifen treatment of our Pdgfrα–CreERT2 : R26R–YFP mice still requires explanation, as does the apparent discrepancy between our line of Pdgfrα–CreER mice and the line made by Kang et al. (2010) in which YFP+ neurons did not accumulate in the aPC. It is conceivable that our line, but not theirs, marks an unidentified population of Pdgfrα+ neurogenic precursor/stem cells that either resides in the CNS or arrives there via the blood. Additional experiments, e.g., with Pdgfrα–CreER knock-in mice, will be required to resolve this.
Figure 8.

GFP+ neurons accumulate in the aPC of postnatal Pdgfrα–CreERT2 : Tau–mGFP transgenic mice. a–c, Pdgfrα–CreERT2 transgenic mice were crossed with a different reporter mouse that, after recombination, expresses mGFP under the control of the Tau promoter (Tau–mGFP reporter mice; Hippenmeyer et al., 2005). The double-heterozygous Pdgfrα–CreERT2 : Tau–mGFP transgenic mice received tamoxifen from P45 to P48 to induce recombination in PDGFRα+ cells and were analyzed at P45 + 14, 27, 70, and 150 d, by immunolabeling coronal brain sections to detect mGFP (green) and NeuN (red). Cell nuclei were stained with Hoechst 33258 (Hst; blue). a, A maximum intensity projection of a confocal image stack through a 30 μm brain section, demonstrating the location of mGFP+ NeuN+ neurons (white arrows) in layer II of the aPC. b, A single-scan (1 μm) confocal image through an mGFP+ NeuN+ neuron. c, Numbers of mGFP+ NeuN+ cells in the aPC of each 30 μm coronal brain section. We observed a significant accumulation of mGFP+ neurons with time. However, it was smaller in magnitude than our previous report examining Pdgfrα–CreERT2 : R26R–YFP transgenic mice (Rivers et al., 2008), which probably reflects a difference in the recombination efficiencies of the two reporter lines.
In summary, our data show that OPCs throughout the postnatal brain are electrically excitable, proliferative, and are the precursors of new OLs but do not make neurons at any time that we examined (after P25).
Footnotes
L.E.C. was supported by a 4-year PhD studentship from the Wellcome Trust. K.M.Y. was supported by a fellowship and grant from the Alzheimer's Society, UK and the Bupa Foundation. This work was funded by research grants from the Wellcome Trust, the Medical Research Council, National Health and Medical Research Council Grant 1030939, the National Institutes of Health, and the European Union (Leukotreat). We thank Ulla Dennehy for technical help.
References
- Arisi GM, Foresti ML, Mukherjee S, Shapiro LA. The role of olfactory stimulus in adult mammalian neurogenesis. Behav Brain Res. 2012;227:356–362. doi: 10.1016/j.bbr.2011.03.050. [DOI] [PubMed] [Google Scholar]
- Bakiri Y, Attwell D, Káradóttir R. Electrical signalling properties of oligodendrocyte precursor cells. Neuron Glia Biol. 2009;5:3–11. doi: 10.1017/S1740925X09990202. [DOI] [PubMed] [Google Scholar]
- Barres BA, Raff MC. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature. 1993;361:258–260. doi: 10.1038/361258a0. [DOI] [PubMed] [Google Scholar]
- Belachew S, Chittajallu R, Aguirre AA, Yuan X, Kirby M, Anderson S, Gallo V. Postnatal NG2 proteoglycan-expressing progenitor cells are intrinsically multipotent and generate functional neurons. J Cell Biol. 2003;161:169–186. doi: 10.1083/jcb.200210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187–191. doi: 10.1038/35012083. [DOI] [PubMed] [Google Scholar]
- Bernier PJ, Bedard A, Vinet J, Levesque M, Parent A. Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc Natl Acad Sci U S A. 2002;99:11464–11469. doi: 10.1073/pnas.172403999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullmann T, Härtig W, Holzer M, Arendt T. Expression of the embryonal isoform (0N/3R) of the microtubule-associated protein tau in the adult rat central nervous system. J Comp Neurol. 2010;518:2538–2553. doi: 10.1002/cne.22351. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Aguirre A, Gallo V. NG2-positive cells in the mouse white and grey matter display distinct physiological properties. J Physiol. 2004;561:109–122. doi: 10.1113/jphysiol.2004.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, Aguirre AA, Gallo V. Downregulation of platelet-derived growth factor-α receptor-mediated tyrosine kinase activity as a cellular mechanism for K+-channel regulation during oligodendrocyte development in situ. J Neurosci. 2005;25:8601–8610. doi: 10.1523/JNEUROSCI.2122-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collarini EJ, Kuhn R, Marshall CJ, Monuki ES, Lemke G, Richardson WD. Down-regulation of the POU transcription factor SCIP is an early event in oligodendrocyte differentiation in vitro. Development. 1992;116:193–200. doi: 10.1242/dev.116.1.193. [DOI] [PubMed] [Google Scholar]
- Dayer AG, Cleaver KM, Abouantoun T, Cameron HA. New GABAergic interneurons in the adult neocortex and striatum are generated from different precursors. J Cell Biol. 2005;168:415–427. doi: 10.1083/jcb.200407053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biase LM, Nishiyama A, Bergles DE. Excitability and synaptic communication within the oligodendrocyte lineage. J Neurosci. 2010;30:3600–3611. doi: 10.1523/JNEUROSCI.6000-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demas J, Eglen SJ, Wong RO. Developmental loss of synchronous spontaneous activity in the mouse retina is independent of visual experience. J Neurosci. 2003;23:2851–2860. doi: 10.1523/JNEUROSCI.23-07-02851.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demerens C, Stankoff B, Logak M, Anglade P, Allinquant B, Couraud F, Zalc B, Lubetzki C. Induction of myelination in the central nervous system by electrical activity. Proc Natl Acad Sci U S A. 1996;93:9887–9892. doi: 10.1073/pnas.93.18.9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimou L, Simon C, Kirchhoff F, Takebayashi H, Götz M. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci. 2008;28:10434–10442. doi: 10.1523/JNEUROSCI.2831-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Wang LP, Klempin F, Römer B, Kettenmann H, Kempermann G. Enriched environment and physical activity reduce microglia and influence the fate of NG2 cells in the amygdala of adult mice. Cell Tissue Res. 2011;345:69–86. doi: 10.1007/s00441-011-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge WP, Zhou W, Luo Q, Jan LY, Jan YN. Dividing glial cells maintain differentiated properties including complex morphology and functional synapses. Proc Natl Acad Sci U S A. 2009;106:328–333. doi: 10.1073/pnas.0811353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensert JM, Goldman JE. Heterogeneity of cycling glial progenitors in the adult mammalian cortex and white matter. J Neurobiol. 2001;48:75–86. [PubMed] [Google Scholar]
- Goretzki L, Burg MA, Grako KA, Stallcup WB. High-affinity binding of basic fibroblast growth factor and platelet-derived growth factor-AA to the core protein of the NG2 proteoglycan. J Biol Chem. 1999;274:16831–16837. doi: 10.1074/jbc.274.24.16831. [DOI] [PubMed] [Google Scholar]
- Gudz TI, Komuro H, Macklin WB. Glutamate stimulates oligodendrocyte progenitor migration mediated via an αv integrin/myelin proteolipid protein complex. J Neurosci. 2006;26:2458–2466. doi: 10.1523/JNEUROSCI.4054-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Maeda Y, Ma J, Xu J, Horiuchi M, Miers L, Vaccarino F, Pleasure D. Pyramidal neurons are generated from oligodendroglial progenitor cells in adult piriform cortex. J Neurosci. 2010;30:12036–12049. doi: 10.1523/JNEUROSCI.1360-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol. 2003;23:4013–4025. doi: 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Seki T, Sato K, Iwai M, Zhang WR, Manabe Y, Abe K. Expression of polysialylated neural cell adhesion molecule in rat brain after transient middle cerebral artery occlusion. Brain Res. 2001;907:130–133. doi: 10.1016/s0006-8993(01)02543-4. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biol. 2005;35:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káradóttir R, Hamilton NB, Bakiri Y, Attwell D. Spiking and nonspiking classes of oligodendrocyte precursor glia in CNS white matter. Nat Neurosci. 2008;11:450–456. doi: 10.1038/nn2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keirstead HS, Levine JM, Blakemore WF. Response of the oligodendrocyte progenitor cell population (defined by NG2 labelling) to demyelination of the adult spinal cord. Glia. 1998;22:161–170. [PubMed] [Google Scholar]
- Knutson P, Ghiani CA, Zhou JM, Gallo V, McBain CJ. K+ channel expression and cell proliferation are regulated by intracellular sodium and membrane depolarization in oligodendrocyte progenitor cells. J Neurosci. 1997;17:2669–2682. doi: 10.1523/JNEUROSCI.17-08-02669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukley M, Capetillo-Zarate E, Dietrich D. Vesicular glutamate release from axons in white matter. Nat Neurosci. 2007;10:311–320. doi: 10.1038/nn1850. [DOI] [PubMed] [Google Scholar]
- Kukley M, Kiladze M, Tognatta R, Hans M, Swandulla D, Schramm J, Dietrich D. Glial cells are born with synapses. FASEB J. 2008;22:2957–2969. doi: 10.1096/fj.07-090985. [DOI] [PubMed] [Google Scholar]
- Kukley M, Nishiyama A, Dietrich D. The fate of synaptic input to NG2 glial cells: neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J Neurosci. 2010;30:8320–8331. doi: 10.1523/JNEUROSCI.0854-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brus-Ramer M, Martin JH, McDonald JW. Electrical stimulation of the medullary pyramid promotes proliferation and differentiation of oligodendrocyte progenitor cells in the corticospinal tract of the adult rat. Neurosci Lett. 2010;479:128–133. doi: 10.1016/j.neulet.2010.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Mela A, Guilfoyle EM, Goldman JE. Neonatal and adult O4(+) oligodendrocyte lineage cells display different growth factor responses and different gene expression patterns. J Neurosci Res. 2009;87:3390–3402. doi: 10.1002/jnr.22065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Bergles DE. Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nat Neurosci. 2004;7:24–32. doi: 10.1038/nn1162. [DOI] [PubMed] [Google Scholar]
- Lytle JM, Chittajallu R, Wrathall JR, Gallo V. NG2 cell response in the CNP-EGFP mouse after contusive spinal cord injury. Glia. 2009;57:270–285. doi: 10.1002/glia.20755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallon BS, Shick HE, Kidd GJ, Macklin WB. Proteolipid promoter activity distinguishes two populations of NG2-positive cells throughout neonatal cortical development. J Neurosci. 2002;22:876–885. doi: 10.1523/JNEUROSCI.22-03-00876.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao BQ, Hamzei-Sichani F, Aronov D, Froemke RC, Yuste R. Dynamics of spontaneous activity in neocortical slices. Neuron. 2001;32:883–898. doi: 10.1016/s0896-6273(01)00518-9. [DOI] [PubMed] [Google Scholar]
- Meister M, Wong RO, Baylor DA, Shatz CJ. Synchronous bursts of action potentials in ganglion cells of the developing mammalian retina. Science. 1991;252:939–943. doi: 10.1126/science.2035024. [DOI] [PubMed] [Google Scholar]
- Mohns EJ, Blumberg MS. Synchronous bursts of neuronal activity in the developing hippocampus: modulation by active sleep and association with emerging gamma and theta rhythms. J Neurosci. 2008;28:10134–10144. doi: 10.1523/JNEUROSCI.1967-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtie JC, Macklin WB, Corfas G. Morphometric analysis of oligodendrocytes in the adult mouse frontal cortex. J Neurosci Res. 2007;85:2080–2086. doi: 10.1002/jnr.21339. [DOI] [PubMed] [Google Scholar]
- Nacher J, Alonso-Llosa G, Rosell D, McEwen B. PSA-NCAM expression in the piriform cortex of the adult rat. Modulation by NMDA receptor antagonist administration. Brain Res. 2002;927:111–121. doi: 10.1016/s0006-8993(01)03241-3. [DOI] [PubMed] [Google Scholar]
- Pekcec A, Löscher W, Potschka H. Neurogenesis in the adult rat piriform cortex. Neuroreport. 2006;17:571–574. doi: 10.1097/00001756-200604240-00003. [DOI] [PubMed] [Google Scholar]
- Power J, Mayer-Pröschel M, Smith J, Noble M. Oligodendrocyte precursor cells from different brain regions express divergent properties consistent with the differing time courses of myelination in these regions. Dev Biol. 2002;245:362–375. doi: 10.1006/dbio.2002.0610. [DOI] [PubMed] [Google Scholar]
- Pringle N, Collarini EJ, Mosley MJ, Heldin CH, Westermark B, Richardson WD. PDGF A chain homodimers drive proliferation of bipotential (O-2A) glial progenitor cells in the developing rat optic nerve. EMBO J. 1989;8:1049–1056. doi: 10.1002/j.1460-2075.1989.tb03472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psachoulia K, Jamen F, Young KM, Richardson WD. Cell cycle dynamics of NG2 cells in the postnatal and ageing brain. Neuron Glia Biol. 2009;5:57–67. doi: 10.1017/S1740925X09990354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson WD, Young KM, Tripathi RB, McKenzie I. NG2-glia as multipotent neural stem cells: fact or fantasy? Neuron. 2011;70:661–673. doi: 10.1016/j.neuron.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, Arai Y. Temporal and spacial relationships between PSA-NCAM-expressing, newly generated granule cells, and radial glia-like cells in the adult dentate gyrus. J Comp Neurol. 1999;410:503–513. doi: 10.1002/(sici)1096-9861(19990802)410:3<503::aid-cne11>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Shapiro LA, Ng KL, Kinyamu R, Whitaker-Azmitia P, Geisert EE, Blurton-Jones M, Zhou QY, Ribak CE. Origin, migration and fate of newly generated neurons in the adult rodent piriform cortex. Brain Struct Funct. 2007;212:133–148. doi: 10.1007/s00429-007-0151-3. [DOI] [PubMed] [Google Scholar]
- Simon C, Götz M, Dimou L. Progenitors in the adult cerebral cortex: cell cycle properties and regulation by physiological stimuli and injury. Glia. 2011;59:869–881. doi: 10.1002/glia.21156. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiarto S, Persson AI, Munoz EG, Waldhuber M, Lamagna C, Andor N, Hanecker P, Ayers-Ringler J, Phillips J, Siu J, Lim DA, Vandenberg S, Stallcup W, Berger MS, Bergers G, Weiss WA, Petritsch C. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20:328–340. doi: 10.1016/j.ccr.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Kataoka Y, Cui Y, Takamori Y, Watanabe Y, Yamada H. Multi-directional differentiation of doublecortin- and NG2-immunopositive progenitor cells in the adult rat neocortex in vivo. Eur J Neurosci. 2007;25:3489–3498. doi: 10.1111/j.1460-9568.2007.05617.x. [DOI] [PubMed] [Google Scholar]
- Tong XP, Li XY, Zhou B, Shen W, Zhang ZJ, Xu TL, Duan S. Ca2+ signaling evoked by activation of Na+ channels and Na+/Ca2+ exchangers is required for GABA-induced NG2 cell migration. J Cell Biol. 2009;186:113–128. doi: 10.1083/jcb.200811071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi RB, Clarke LE, Burzomato V, Kessaris N, Anderson PN, Attwell D, Richardson WD. Dorsally and ventrally derived oligodendrocytes have similar electrical properties but myelinate preferred tracts. J Neurosci. 2011;31:6809–6819. doi: 10.1523/JNEUROSCI.6474-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinet J, Lemieux P, Tamburri A, Tiesinga P, Scafidi J, Gallo V, Sík A. Subclasses of oligodendrocytes populate the mouse hippocampus. Eur J Neurosci. 2010;31:425–438. doi: 10.1111/j.1460-9568.2010.07082.x. [DOI] [PubMed] [Google Scholar]
- Wake H, Lee PR, Fields RD. Control of local protein synthesis and initial events in myelination by action potentials. Science. 2011;333:1647–1651. doi: 10.1126/science.1206998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, Cuntz H, Mori M, Nusser Z, Sjöström PJ, Häusser M. Traveling waves in developing cerebellar cortex mediated by asymmetrical Purkinje cell connectivity. Nat Neurosci. 2009;12:463–473. doi: 10.1038/nn.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KM, Merson TD, Sotthibundhu A, Coulson EJ, Bartlett PF. p75 neurotrophin receptor expression defines a population of BDNF-responsive neurogenic precursor cells. J Neurosci. 2007;27:5146–5155. doi: 10.1523/JNEUROSCI.0654-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Eisen AM, McBain CJ, Gallo V. A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development. 1998;125:2901–2914. doi: 10.1242/dev.125.15.2901. [DOI] [PubMed] [Google Scholar]
- Zhu G, Mehler MF, Zhao J, Yu Yung S, Kessler JA. Sonic hedgehog and BMP2 exert opposing actions on proliferation and differentiation of embryonic neural progenitor cells. Dev Biol. 1999;215:118–129. doi: 10.1006/dbio.1999.9431. [DOI] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 2008;135:145–157. doi: 10.1242/dev.004895. [DOI] [PubMed] [Google Scholar]
- Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, Nishiyama A. Age-dependent fate and lineage restriction of single NG2 cells. Development. 2011;138:745–753. doi: 10.1242/dev.047951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]