

Abstract
Elevated levels of Oncostatin M (OSM), an interleukin-6 family cytokine, have been observed in Multiple Sclerosis (MS), HIV-associated Neurocognitive Disorder (HAND) and glioblastoma (GBM); however, its effects within the CNS are not well understood. OSM regulates gene expression primarily by activating the JAK/STAT, NF-κB and/or MAPK pathways, in a cell-type specific manner. In our studies, OSM induces the production of the pro-inflammatory cytokine tumor necrosis factor-α (TNF-α) and inducible nitric oxide synthase (iNOS) from microglia in an NF-κB-dependent manner. This expression also partially requires the intermediate production of TNF-α and subsequent NF-κB activation via TNF-R1. We also demonstrate that OSM-induced TNF-α production from microglia is neurotoxic. The IL-12 family member, IL-27, suppresses OSM-mediated TNF-α and iNOS expression at the transcriptional level by inhibiting activation of the NF-κB pathway, and rescues the neurotoxicity induced by OSM-stimulated microglia. These studies are the first to demonstrate the pro-inflammatory effects of OSM in microglia, and also identify IL-27 as a novel inhibitor of inflammatory processes in these cells.
Keywords: Oncostatin M, Interleukin-27, NF-κB, neuroinflammation, neurotoxicity
INTRODUCTION
Microglia are myeloid lineage-derived innate immune cells in the brain that actively survey their local microenvironment and rapidly respond to pathogens and neuronal damage (Rivest 2009). In response to neuroinflammatory insults, these cells produce large quantities of cytokines and chemokines, activating nearby microglia and astrocytes, and recruiting peripheral immune cells into the brain parenchyma (D’Mello et al. 2009). In response to systemic and central nervous system (CNS) bacterial infection, neuronal injury, and neuroinflammation injury, microglia release tumor necrosis factor-α (TNF-α) (Rivest 2009), which stimulates the production of molecules including chemokine ligand 2 (CCL2), CCL5, interleukin-6 (IL-6), interleukin-1β (IL-1β), inducible nitric oxide synthase (iNOS) and TNF-α (Allan and Rothwell 2001; D’Mello et al. 2009; Nadeau and Rivest 2000). Expression of TNF-α is upregulated in the cerebrospinal fluid, brain and/or serum of patients with Multiple Sclerosis (MS), Alzheimer’s Disease (AD), HIV-associated Neurocognitive Disorder (HAND), Parkinson’s Disease (PD), and Amyotrophic Lateral Sclerosis (ALS) (Cereda et al. 2008; Sriram and O’Callaghan 2007). TNF-α expression is also elevated by traumatic brain injury, ischemia and bacterial infection (Sriram and O’Callaghan 2007). Blockade of TNF-α signaling abolishes the expression of proinflammatory cytokines and is protective in several animal models of neuroinflammation, including ischemia, traumatic brain injury, PD, AD and MS (Allan and Rothwell 2001; He et al. 2007; McCoy et al. 2006; McCoy et al. 2008; Nadeau and Rivest 2000; Selmaj et al. 1995). However, neuroprotective functions of TNF-α, including clearance of extracellular pathogens, aggregates and debris, have been reported in models of stroke, remyelination, AD and bacterial infection (Arnett et al. 2001; Lambertsen et al. 2009; Rivest 2009; Sriram and O’Callaghan 2007). Therefore, TNF-α production by microglia is a key regulator of inflammatory circuits within the brain.
TNF-α signals through the TNF-Type 1 receptor trimer, activating the TNF-R1-associated death domain (TRADD)/TNF-receptor-associated factor 2 (TRAF2)/receptor-interacting protein (RIP) complex (Aggarwal 2003). This results in activation of the nuclear factor-kappa B (NF-κB) pathway and initiation of inflammatory gene transcription, including that of TNF-α itself. Microglia also produce iNOS under neuroinflammatory conditions (Kawahara et al. 2009; Saha and Pahan 2006). As a member of the nitric oxide synthase family, iNOS catalyzes the production of nitric oxide (NO) from L-Arginine (Korhonen et al. 2005). Expression of iNOS and subsequent NO production is a critical component of the innate immune response. At lower concentrations, NO serves as a signaling molecule between neurons. At higher concentrations, NO functions as a cytotoxic molecule, aiding in clearance of pathogens and immune cell migration, proliferation and cytokine production (Korhonen et al. 2005), but can also inhibit neuronal respiration, causing neuronal depolarization and subsequent glutamate-mediated excitotoxicity (Bal-Price and Brown 2001).
Oncostatin M (OSM) is a member of the IL-6 family of cytokines that has been shown to both promote and inhibit cell proliferation, and exhibits both pro- and anti-inflammatory properties depending on the cell type or disease context (Chen and Benveniste 2004). In a cell-type dependent manner, OSM activates several signaling cascades including the JAK/STAT, MAPK and NF-κB pathways by binding to the gp130/LIFR or gp130/OSMRβ complex. OSM is expressed by microglia and macrophages in response to prostaglandin E2 (PGE2) through a cAMP/PKA-dependent pathway. Norepinephrine, PGE1, complement component C5a, IL-1β and TNF-α also induce OSM in cells of monocyte lineage (Kastl et al. 2008; Repovic and Benveniste 2002). While not expressed in normal brain tissue, OSM immunoreactivity has been detected in microglia, reactive astrocytes and infiltrating leukocytes in MS lesions (Ruprecht et al. 2001). OSM production is elevated in peripheral blood mononuclear cells from MS patients (Ensoli et al. 2002; Ruprecht et al. 2001). HIV-1 patients with HAND have higher levels of OSM in the peripheral blood than in patients without neurological symptoms (Vecchiet et al. 2003). Increased levels of OSM have also been detected in glioblastoma tissue (Repovic et al. 2003). In addition to increased levels in neurologic diseases, OSM itself induces the expression of IL-6, ICAM-1 and CCL2 in cerebral endothelial cells and causes disruption of the tight junctions between brain capillary endothelial cells. This effect may contribute to breakdown of the blood-brain-barrier (Ruprecht et al. 2001; Takata et al. 2008).
IL-27 is a member of the IL-12 family of cytokines and is composed of two subunits, Epstein-Barr virus-induced gene 3 (EBI-3) and p28, which signal through the WSX-1/gp130 complex, activating STATs 1, 3, 4 and 5 (Sonobe et al. 2005; Stumhofer and Hunter 2008; Yoshida and Miyazaki 2008). IL-27 is inducibly expressed by dendritic cells, macrophages and microglia, and is a powerful inhibitor of Th17 development and Experimental Autoimmune Encephalomyelitis (EAE), a mouse model of MS (Fitzgerald et al. 2007a). Specifically, mice deficient in the WSX-1 receptor demonstrate increased production of pro-inflammatory molecules such as TNF-α, IL-6, IL-17 and IL-1β from CNS immune cells after EAE challenge (Batten et al. 2006). Thus far, anti-inflammatory properties of IL-27 have only been described in T cells, and the effect of IL-27 on microglia is not known.
We wished to determine the influence of OSM on microglia with respect to promoting pro- or anti-inflammatory responses in these important CNS immune cells. In these studies, we identify OSM as a pro-inflammatory molecule in microglia, due to its induction of TNF-α and NO production. This expression is NF-κB-dependent and partially depends on intermediate TNF-α production and signaling through TNF-R1. IL-27 suppresses OSM-induced TNF-α and iNOS expression at the transcriptional level by reducing OSM-induced NF-κB activation and recruitment to the TNF-α and iNOS promoters. Conditioned media from OSM-stimulated microglia is toxic to neurons, further supporting the idea that OSM promotes inflammatory responses from microglia. Treatment of microglia with IL-27 partially rescued OSM-mediated neuronal toxicity. These studies are the first to provide evidence of the anti-inflammatory effects of IL-27 in microglia, and identify IL-27 as a possible therapeutic target in neuroinflammatory conditions.
MATERIALS AND METHODS
Recombinant Proteins and Reagents
Recombinant murine OSM, IL-27, IFN-γ and anti-TNF-α antibodies were purchased from R&D Systems® (Minneapolis, MN). The Bay-11 7085 compound was purchased from Calbiochem (Gibbstown, NJ). Abs against phosphorylated (p)-p65Ser276, p-p65Ser536, total p65, p-STAT1Tyr701, p-STAT3Tyr705, total STAT3, p-p38, p-p44/42Thr202/Tyr204 and p-Akt were obtained from Cell Signaling Technology, Inc. (Danvers, MA). Abs against p38, TNF-R1 and total mouse IgG were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Actin abs were obtained from Sigma-Aldrich Corp. (St. Louis, MO).
Cells
Primary microglia were prepared from WT and TNF-R1−/− 1-day old neonatal C57Bl/6J mice (The Jackson Laboratory, Bar Harbor, ME) as described previously (Nguyen and Benveniste 2002). After 14 days in culture, microglia were mechanically separated from astrocytes by shaking, pelleted by centrifugation at 1000g for 5 min, and plated at a density of 2 × 105 cells/ml. BV2 murine microglial cells were maintained as described previously (Petrova et al. 1999). Cells were seeded at a density of 105 cells/ml. Primary murine cortical neurons were prepared from embryonic day 17 C57BL/6 brains, and the neocortices were mechanically triturated as described previously (Vitvitsky et al. 2006). Dissociated cells were plated on Poly-L-Lysine-coated 24-well plates at a density of 2 × 105 cells/well. Plating media consisted of Neurobasal Medium (NBM, Gibco, Carlsbad, CA) supplemented with 2% B-27, 0.5 mM GlutaMax, and penicillin-streptomycin. At 2–3 days in vitro (DIV), the media was replaced with fresh media. After 8 DIV, the purity of the cultures was confirmed by immunostaining and western blot analysis with MAP-2 for neurons and GFAP for astrocytes. Cultures were routinely > 95% pure for neurons; 4–5% for astrocytes.
ELISA
Supernatants were collected and centrifuged at 12,000g for 15 min to remove cellular debris. Supernatants were then analyzed for TNF-α protein levels using the Mouse TNF-α Quantikine® Immunoassay (R&D Systems®, Minneapolis, MN) per the manufacturer’s instructions and as described previously (Wesemann et al. 2002).
NO Detection
Supernatants from unstimulated or 72 h OSM-treated microglia were collected and analyzed for nitrite, a stable end-product of NO metabolism, using the Griess reaction (Weissman and Gross 2001). The Griess reagent was prepared by combining 0.1% napthylenthylene diamine dichloride with 1% sulfanilamide in a 2.5% phosphoric acid-containing solution immediately prior to incubation with supernatants. 100 μl of the Griess reagent was added to 100 μl nitrite standards or supernatants and incubated for 15 min at room temperature. Samples were then read at 540 nm to measure nitrite levels.
RNA Isolation and RT-PCR (Quantitative and Semi-Quantitative)
Total RNA was isolated from primary microglia or BV2 cells using the TRIzol© reagent (Invitrogen, Carlsbad, CA) as described previously (Qin et al. 2006). One μg of purified RNA was reverse transcribed in a 20 μl reaction containing 4 μl RT 5X Buffer, 1 μl oligo(dt)15 primer, 1 μl 10mM dNTPs, 0.5 μl rRNasin and 1 μl Moloney Murine Leukemia Virus Reverse Transcriptase (Promega, Madison, WI), followed by semi-quantitative PCR using primers for the murine TNF-α, iNOS and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) genes, as described previously (Lee et al. 2007). Primer sequences were as follows: iNOS Fwd (TCCTACACCACACCAAAC); iNOS Rev (CTCCAATCTCTGCCTATCC); TNF-α Fwd (CCGATGGGTTGTACCTTGTC); TNF-α Rev (CTGGAAGACTCCTCCCAGGT); IL-6 Fwd (AGTTGCCTTCTTGGGACTGA); IL-6 Rev (CAGAATTGCCATTGCACAAC); GAPDH Fwd (TGAAGGTCGGTGTGAACGGATTTGG); GAPDH Rev (ACGACATACTCAGCACCGGCCTCAC). Quantitative PCR was performed using SYBR® Green I (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions (Applied Biosystems Inc, Foster City, CA) using an ABI7900HT Fast Real Time PCR system, as previously described (Atkinson et al. 2009). Mastermix reactions containing gene-specific primers, and sterile water were added to the micro-wells of a MicroAmp 96-well plate. Each gene was analyzed in triplicate. Fluorescence measurements corresponding to SYBR® Green I binding to synthesized double-stranded DNA were collected after each amplification cycle and used to determine Relative Quantity (RQ) values for each sample using Applied Biosystems StepOne™ Instrument software.
Immunoblotting
Thirty μg of total cellular protein was separated on 10% polyacrylamide gels by electrophoresis, transferred to nitrocellulose membranes, and probed with antibodies against p-p65Ser276, p-p65Ser536, total p65, p-STAT3Tyr705, total STAT3, TNF-R1 and actin, as described previously (Qin et al. 2006). Immunoreactivity was assessed using the Pierce® ECL or SuperSignal® West Dura substrate (Thermo Scientific, Rockford, IL).
MTT Assay
Primary microglia were seeded at a density of 2 × 105 cells/ml and incubated under various conditions for 24 h. Conditioned media from microglia were collected and, when indicated, antibodies against TNF-α or total mouse IgG were added for 2 h at 37°C. Conditioned media (400 μl) was then applied to primary murine neurons for 24 h. MTT Reagent (ATCC, Manassas, VA) (1 μl/100 μl media) was then added to neuronal cultures and incubated for 4 h at 37°C. Supernatants were then aspirated and 400 μl of DMSO was added to solubilize neurons. After 15 min, 100 μl of this solution was added to 96-well plate and the absorbance read at 570 nm.
Chromatin Immunoprecipitation (ChIP) Assays
ChIP analysis was performed as described previously (Baker et al. 2008; Qin et al. 2006). For each experimental condition, 12.5 × 106 BV2 microglia were plated in four 20-cm dishes for a total of 5 × 107 BV2 cells per condition. For treatment, BV2 cells were bathed in 1% FBS-containing media alone, treated with OSM (10 ng/ml) for 2 h, treated with IL-27 (5 ng/ml) for 6 h, or treated with IL-27 for 6 h and OSM for the last 2 h of IL-27 treatment. After stimulation, cells were fixed with 1% paraformaldehyde for 20 min at room temperature to crosslink DNA and proteins. Nuclei were isolated and chromatin was sheared by sonication. Samples were measured, aliquoted and precleared with salmon sperm DNA-saturated protein A sepharose beads for 1 h at 4°C. Samples were then incubated with 5 μg of Abs or isotype-matched control IgG overnight at 4°C. Immunoprecipitated chromatin was incubated with 200 mM NaCl at 65°C overnight to reverse cross-links. Protein was then digested with proteinase K, DNA purified over a Qiagen column, and then analyzed by PCR with GoTaq Flexi DNA polymerase. Primer sequences were designed to amplify NF-κB elements in the murine TNF-α and iNOS promoters using the Primer3 on-line software (Kuprash et al. 1999; Rozen and Skaletsky 2000; Xie et al. 1994). Primer sequences were as follows: iNOS Fwd (CCTCCCTCCCTAGTGAGTCC); iNOS Rev (GGGGCCAGAGTCTCAGTCTT); TNF-α Fwd (AAGGAGAAGGCTTGTGAGGTC) and TNF-α Rev (TTGTAGAAAGACCATGCCTGTG).
Statistical Analysis
All assays were performed a minimum of two times. Levels of significance between samples were determined by the student’s t test distribution. A value of p ≤ 0.05 was considered to be statistically significant.
RESULTS
OSM induces TNF-α and iNOS expression in microglia
OSM can upregulate expression of pro- and anti-inflammatory genes depending on the cell type and microenvironment (Chen and Benveniste 2004). The effect of OSM on gene expression in microglia has not been characterized. After screening numerous pro- and anti-inflammatory genes, we found that OSM induces expression of TNF-α and iNOS mRNA in primary microglia and in the BV2 microglial cell line (Figures 1A and B). TNF-α protein expression was detected in supernatants from OSM-treated microglia as early as 4 h after OSM treatment, and could still be detected at 48 h (Figure 1C, Figure 3C and data not shown). In BV2 microglia, TNF-α protein levels in supernatants increased between 4 and 24 h after OSM treatment, from an average of 15.1 to 74.0 pg/ml (Figure 1D). When iNOS is expressed, it quickly generates NO from L-Arginine (Korhonen et al. 2005). Therefore, we examined levels of nitrite, a stable end product of NO production, in primary microglia supernatants. Nitrite production was detected after 72 h incubation with OSM (Figure 1E). Furthermore, OSM-induction of NO occurs in a dose-dependent manner in BV2 microglia (Figure 1F). Together, these results demonstrate that OSM mediates pro-inflammatory responses in microglia by promoting the production of TNF-α and NO.
Figure 1.

OSM induces TNF-α and iNOS expression in microglia. A. B. Primary microglia or BV2 microglial cells were treated with OSM (10 ng/ml) for 0, 2, 4 or 8 h, and analyzed for TNF-α and iNOS mRNA expression by RT-PCR. GAPDH levels were assessed as a control for total mRNA. Representative of 3 independent experiments. C. Primary microglia were treated with OSM (10 ng/ml) for 14 h and supernatants assessed for TNF-α protein expression by ELISA. Mean +/− S.D. of 3 independent experiments. D. BV2 cells were treated with OSM (10 ng/ml) for 0, 4, 8, 16 or 24 h and supernatants analyzed for TNF-α protein expression by ELISA. Mean +/− S.D. of 2 independent experiments. E. Primary microglia were treated with OSM (10 ng/ml) for 72 h and supernatants were analyzed for production of nitrite, a stable end product of NO production, using the Griess reagent. Mean +/− S.D. of 3 independent experiments. F. BV2 cells were stimulated with increasing concentrations of OSM (0–200 ng/ml) for 72 h and supernatants were analyzed for nitrite production. Mean +/− S.D. of 3 independent experiments. * p-values < 0.05 were considered statistically significant.
Figure 3.

OSM-induced TNF-α and NO production requires activation of the NF-κB pathway. A. Primary microglia were pre-treated with 0.1% DMSO or 10 μM BAY 11-7085 for 1 h, followed by treatment with OSM (10 ng/ml) for 30 min. Protein lysates were collected and analyzed for P-p65Ser276, total p65 and actin by immunoblotting. Representative of 2 independent experiments. B. Primary microglia were pre-treated with 0.1% DMSO, 0.1 μM BAY 11-7085 or 1 μM BAY 11-7085 for 1 h, then treated with OSM (10 ng/ml) for 4 h. Total mRNA was collected and analyzed for TNF-α and iNOS gene expression by RT-PCR. Representative of 3 independent experiments. C, D. Primary microglia were pre-treated with 0.1% DMSO, 0.1 μM BAY 11-7085, or 1 μM BAY 11-7085 for 1 h, then treated with OSM (10 ng/ml) for 4 h (C) or 72 h (D). Supernatants were collected and analyzed for TNF-α protein production by ELISA (C) or for nitrite production using the Griess reagent (D). Mean +/− S.D. of 2 independent experiments. * p-values < 0.05 were considered statistically significant.
OSM activates NF-κB p65 in microglia
In mouse cells, OSM signals through a receptor complex composed of the gp130 and OSMRβ subunits (Chen and Benveniste 2004). OSM signaling through this receptor leads to activation of the JAK/STAT, MAPK and NF-κB pathways in many cell types (Baker et al. 2008; Boing et al. 2006; Halfter et al. 1999). The signaling pathways activated by OSM in microglia have not been studied. In our studies, OSM induces robust activation of NF-κB p65 in both primary microglia and BV2 cells (Figures 2A and B). In these experiments, p65 phosphorylation could be detected as early as 15 min after OSM treatment and remained activated at 4 h. Surprisingly, OSM did not activate the JAK/STAT pathway in microglia as determined by analyzing the phosphorylation status of STAT3 (Figures 2A and 2B) and STAT1 (data not shown). To demonstrate that STAT3 is capable of being phosphorylated in primary microglia, cells were stimulated with IFN-γ as a positive control, and with OSM. As shown in Supplemental Figure 1, IFN-γ induced STAT3 phosphorylation, while OSM was without effect.
Figure 2.

OSM activates the NF-κB p65 pathway in microglia. A. B. Primary microglia and BV2 cells were treated with OSM (10 ng/ml) for the indicated times. Protein lysates were analyzed for P-p65ser276 (primary microglia), P-p65ser536 (BV2 cells), total p65, P-STAT3Tyr705, total STAT3 and actin (as a loading control) by immunoblotting. Representative of 3 independent experiments.
OSM-induced TNF-α and NO production requires NF-κB pathway activation
OSM strongly activates the NF-κB pathway in microglia (Figure 2), and previous studies have demonstrated a role for the NF-κB pathway in TNF-α and iNOS gene expression (Collart et al. 1990; Gavrilyuk et al. 2001; Xie et al. 1994). Therefore, we assessed the involvement of this pathway in OSM-induced TNF-α and iNOS expression in primary microglia using a pharmacological inhibitor of the NF-κB pathway, BAY 11-7085. Pre-treatment of primary microglia with BAY 11 for 1 h inhibited OSM-mediated phosphorylation of NF-κB p65 (Figure 3A). Blockade of the NF-κB pathway with 0.1 μM BAY 11 strongly inhibited OSM-induced TNF-α and iNOS mRNA expression, and this expression was completely inhibited by 1 μM BAY 11 (Figure 3B). Production of the TNF-α protein in response to OSM treatment for 4 h was significantly inhibited by pretreatment with 0.1 μM BAY 11 (from 65.7 to 23.8 pg/ml), and completely abolished with 1 μM BAY 11 (Figure 3C). Pre-treatment with 1 μM BAY 11 also completely blocked OSM-mediated induction of NO, as determined by assessing nitrite production (Figure 3D). These data demonstrate the requirement of NF-κB signaling in OSM-mediated expression of TNF-α and iNOS in microglia.
OSM-induced TNF-α and iNOS partially requires the TNF-R1
TNF-α signaling can induce iNOS expression as well as its own expression (Benveniste and Benos 1995; Combs et al. 2001; Rivest 2009). We detected TNF-α protein expression as early as 4 h following OSM treatment in primary microglia (Figure 3C), which was still detectable at 48 h (data not shown). Also, iNOS gene expression and NO production occurred with delayed kinetics compared to TNF-α mRNA and protein expression (Figure 1). Therefore, we tested if OSM-induced TNF-α production was contributing to OSM-mediated expression of iNOS and TNF-α. For this purpose, we utilized microglia deficient in the TNF-R1 (Peschon et al. 1998). Microglia from WT and TNF-R1−/− mice (Figure 4A) were treated with OSM for 4 h and mRNA analyzed for TNF-α and iNOS by qRT-PCR (Figures 4B and 4C). In WT microglia, OSM-induced expression of TNF-α mRNA was ~ 35.2-fold over untreated values, while TNF-α expression in TNF-R1−/− microglia was only increased by 26.1-fold, a statistical difference. For iNOS, the average fold changes were 31.4 and 19.1 in WT versus TNF-R1−/− microglia, respectively. TNF-α protein expression in TNF-R1−/− microglia after 4 h of OSM treatment was 69% of that in WT microglia (Figure 4D). After 8 h of OSM treatment, this dropped to 13.7% of WT microglia. Examination of another OSM-induced gene, IL-6, revealed that IL-6 mRNA expression was actually enhanced and prolonged in TNF-R1−/− microglia (Supplemental Figure 2), indicating the specificity of the inhibitory effect for OSM-induced TNF-α and iNOS expression. To determine the mechanism of this inhibition, we assessed the activation status of NF-κB p65. Indeed, OSM-induced phosphorylation of p65 was reduced in TNF-R1−/− microglia compared to WT cells, especially at the 4 h time point (Figure 4E). These results suggest that TNF-α produced in response to OSM signals through TNF-R1, contributing to NF-κB p65 activation and subsequent TNF-α and iNOS gene expression.
Figure 4.

OSM-induced TNF-α and iNOS partially requires the TNF-R1. A. Primary microglia from WT or TNF-R1−/− mice were analyzed for expression of TNF-R1 by immunoblotting. B. WT and TNF-R1−/− microglia were treated with OSM (10 ng/ml) for 4 h and mRNA was analyzed for expression of TNF-α (B) and iNOS (C) by quantitative RT-PCR. Threshold cycle values were used to calculate relative quantities (RQ), or fold induction, of each gene. Mean +/− S.D. of 2 independent experiments. D. WT and TNF-R1/− microglia were treated with OSM (10 ng/ml) for 4 h or 8 h and supernatants were analyzed for TNF-α protein expression by ELISA. TNF-α levels are expressed as a percentage of TNF-α levels in OSM-treated WT supernatants. Mean +/− S.D. of 3 independent experiments. E. WT and TNF-R1−/− microglia were treated with OSM (10 ng/ml) for the indicated times and analyzed for P-p65Ser276 and total p65 by immunoblotting. Representative of 2 independent experiments. * p-values < 0.05 were considered statistically significant.
OSM-induced release of TNF-α from microglia is neurotoxic
Because neurotoxic effects of both TNF-α and NO have been reported (He et al. 2002; Pacher et al. 2007), we tested if supernatants from OSM-treated microglia were toxic to neurons. As measured by the MTT assay, 24 h OSM-treated supernatants from primary microglia reduced primary neuron cell viability by 40% (Figure 5). Neurotoxicity was blocked by incubating supernatants from OSM-stimulated microglia with an anti-TNF-α antibody for 2 h at 37°C before adding them to primary cortical neuron cultures. Supernatants incubated with anti-IgG antibodies induced a similar degree of neurotoxicity as supernatants from OSM-stimulated microglia.
Figure 5.
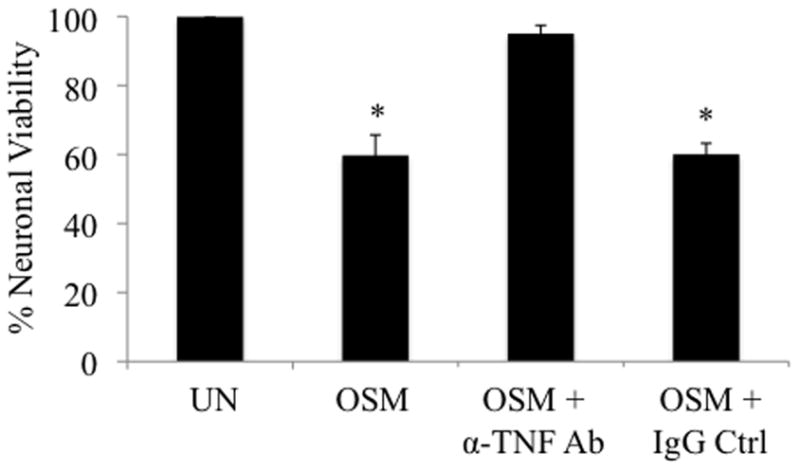
OSM-induced release of TNF-α from microglia is neurotoxic. Primary microglia were incubated with media alone (UN) or with OSM (10 ng/ml) for 24 h. Four hundred μl of supernatant from each condition was added to primary cortical neuron cultures and incubated for 24 h at 37°C. Where indicated, supernatants were incubated with antibodies against TNF-α or total mouse IgG for 2 h at 37°C before being added to neurons. After 24 h, neuronal viability was assessed using the MTT Reagent. Absorbance values of each sample were measured in duplicate at 570 nm. Cell viability readings of neurons incubated with supernatants from media alone-treated microglia were set to 100%. Results show the average % cell viability for each condition +/− S.D. from 2 independent experiments. * p-values < 0.05 were considered statistically significant.
IL-27 inhibits OSM-induced TNF-α and iNOS expression and reduces neurotoxicity
Microglial production of TNF-α and NO has been shown to be toxic to neurons (Bal-Price and Brown 2001; He et al. 2002; Rivest 2009; Wen et al. 2006). IL-27 is protective in EAE, the mouse model of MS, and dampens expression of the pro-inflammatory cytokines IL-17, IL-6 and TNF-α (Batten et al. 2006). However, the anti-inflammatory effects of IL-27 in microglia have not been studied. Pre-treatment of primary microglia with IL-27 (5 ng/ml) for 4 h, followed by 4 h of OSM (10 ng/ml), inhibited mRNA expression of TNF-α and iNOS from 14- to 3.2-fold and from 5.4- to 1.8-fold, respectively (Figure 6A). IL-27 alone had no effect on TNF-α or iNOS mRNA expression (Figure 6A). Pre-treatment of BV2 cells with IL-27 for either 4 or 2 h inhibited OSM-induced TNF-α and iNOS mRNA expression, however, co-treatment of IL-27 with OSM was without effect (Figure 6B). Using a 4 h IL-27 pre-treatment time, we examined the effect of increasing doses of IL-27 on OSM-induced TNF-α and iNOS gene expression in BV2 cells (Figure 6C). While a 1 ng/ml dose of IL-27 was capable of inhibiting OSM-induced TNF-α mRNA expression, inhibition of iNOS expression required at least 5 ng/ml of IL-27 (Figure 6C). Interestingly, this dose of IL-27 completely abolished OSM-induced iNOS expression, while complete inhibition of TNF-α expression required 20 ng/ml of IL-27. Pre-treatment of primary microglia with IL-27 for 4 h inhibited OSM-induced TNF-α protein and nitrite production to 22.0% and 27.8% of OSM-treated microglia, respectively (Figures 6D and 6E).
Figure 6.

IL-27 inhibits OSM-induced TNF-α and iNOS expression and partially rescues neuronal cell death. A. Primary microglia were incubated in media alone, OSM (10 ng/ml) for 4 h, IL-27 (5 ng/ml) for 8 h, or pre-treated with IL-27 for 4 h, then exposed to OSM for an additional 4 h. Total mRNA was harvested and analyzed for TNF-α and iNOS gene expression by RT-PCR. GAPDH was included as a control. Values for the first lane (untreated condition) were set to 1 and fold induction for treatment conditions were determined by dividing each treatment value by the untreated value. Representative of 3 independent experiments. B. BV2 microglia were incubated in media alone, treated with OSM (10 ng/ml) for 4 h, or pre-treated with IL-27 (5 ng/ml) for 4, 2, or 0 h (co-treatment), then treated with OSM for an additional 4 h. Total mRNA was harvested and analyzed for TNF-α and iNOS gene expression by RT-PCR. Fold induction was calculated as in A. Representative of 3 independent experiments. C. BV2 cells were incubated in media alone, treated with OSM (10 ng/ml) for 4 h, or pre-treated with increasing concentrations (1, 5, 10 or 20 ng/ml) of IL-27 for 4 h, then treated with OSM for an additional 4 h. Total mRNA was harvested and analyzed for TNF-α and iNOS gene expression by RT-PCR. Fold induction was calculated as in A. Representative of 3 independent experiments. D, E. Primary microglia were pre-treated with IL-27 (5 ng/ml) for 4 h, followed by treatment with OSM (10 ng/ml) for 24 h (D) or 72 h (E). Supernatants were collected and analyzed for TNF-α protein (D) or nitrite production (E). Graphs in D and E show the average % inhibition in TNF-α and nitrite production +/−S.D., respectively, from 3 independent experiments. F. Primary microglia were incubated in 1% DMEM alone (UN), treated with OSM (10 ng/ml) for 24 h (OSM), treated with IL-27 (5 ng/ml) for 24 h (IL-27), or pre-treated with IL-27 for 4 h, and then treated with OSM for an additional 24 h (IL-27 + OSM). Four hundred μl of supernatant from each condition was added to primary cortical neuron cultures and incubated for 24 h. Neuronal viability was then assessed using the MTT Reagent. Absorbance values of each sample were measured in duplicate at 570 nm. Cell viability readings of neurons incubated with supernatants from media alone-treated microglia were set to 100%. Results show the average % cell viability for each condition +/− S.D., from 2 independent experiments. * p-values < 0.05 were considered statistically significant.
We have shown that TNF-α contributes to its own expression as well as to expression of iNOS, and blockade of TNF-α rescues neuron cell death induced by supernatants from OSM-stimulated microglia. Because IL-27 inhibits OSM-induced TNF-α and iNOS production, we tested if IL-27 could reverse the decrease in cell viability. In these experiments, supernatants from OSM-stimulated microglia reduced cell viability by 21% (Figure 6F). Supernatants from IL-27-treated microglia did not affect cell viability. However, supernatants from microglia pre-treated with IL-27 for 4 h, then exposed to OSM for 24 h, partially rescued neuron viability (from 79% to 90%). As a positive control for neurotoxicity, neurons were incubated in conditioned media from LPS-stimulated microglia, which is known to induce toxicity (He et al. 2002). Pre-treatment of microglia with IL-27 did not rescue the reduction in cell viability induced by LPS (data not shown).
IL-27 inhibits OSM-mediated p65 activation and recruitment to the TNF-α and iNOS promoters
We next sought to determine the mechanism by which IL-27 inhibits OSM induction of TNF-α and iNOS. Our studies have demonstrated a role for the NF-κB pathway in the expression of OSM-induced TNF-α and iNOS expression. As such, we examined whether IL-27 inhibited OSM-induced p65 activation. In BV2 cells, a 15 min treatment with OSM (10 ng/ml) activated p65 (Figure 7A, lane 2). Treatment with IL-27 for 4 h (5 ng/ml) did not induce p65 phosphorylation (lane 3). However, pre-treatment with IL-27 for 4 h blocked OSM-induced phosphorylation of p65 (lane 4). To further examine the molecular mechanism of IL-27 action, we assessed the recruitment of phosphorylated p65 (P-p65) to the TNF-α and iNOS promoters by ChIP analysis under different experimental conditions. At the TNF-α promoter, treatment with OSM for 2 h induced recruitment of P-p65, enhanced recruitment of total p65 and p300, and enhanced acetylation of histone H3 (AcH3) (Figure 6B, lane 2). Pre-treatment with IL-27 for 4 h inhibited OSM-induced recruitment of P-p65, total p65, p300 and H3 acetylation (compare lanes 2 and 4). IL-27 alone was capable of recruiting a modest amount of P-p65, but did not affect total p65, p300 or AcH3 at the TNF-α promoter (lane 3). IL-27 also recruited HDAC1, a transcriptional repressor, to the TNF-α promoter, both in the absence and presence of OSM (lanes 3 and 4). The data for the iNOS promoter was similar to that for the TNF-α promoter, with the exception of slight increases in p300 recruitment and H3 acetylation in the presence of IL-27 alone (lane 7). As with the TNF-α promoter, pre-treatment with IL-27 blocked OSM-mediated recruitment of P-p65, total p65 and p300, inhibited OSM-mediated H3 acetylation, and recruited HDAC1 to the iNOS promoter (compare lanes 6 and 8). Therefore, IL-27 blocks the permissive conformation of the TNF-α and iNOS promoters by inhibiting OSM-induced p65 recruitment, p300 recruitment and H3 acetylation, and by inducing recruitment of HDAC1, a transcriptional repressor.
Figure 7.

IL-27 inhibits OSM-mediated p65 activation and recruitment to the TNF-α and iNOS promoters. A. BV2 microglial cells were treated with media alone, OSM (10 ng/ml) for 0.25 h, IL-27 (5 ng/ml) for 4.25 h, or pre-treated with IL-27 for 4 h and then treated with OSM for 0.25 h. Protein lysates were collected and analyzed for P-p65Ser536 and total p65 by immunoblotting. Representative of 2 independent experiments. B. BV2 cells were treated with media alone, OSM (10 ng/ml) for 2 h, IL-27 (5 ng/ml) for 6 h, or pre-treated with IL-27 for 4 h and then treated with OSM for 2 h. The ChIP assay was performed to assess binding of P-p65 Ser536, total p65, AcH3, p300 and HDAC1 to NF-κB sites within the TNF-α and iNOS promoters. Total IgG shows no non-specific binding. Input serves as a control for total DNA levels.
DISCUSSION
Our studies demonstrate for the first time that OSM induces the expression of TNF-α and NO in microglia, indicating that OSM has pro-inflammatory effects in this CNS cell type. This expression is NF-κB-mediated, as inhibition of this pathway completely blocked TNF-α and iNOS expression. In microglia, OSM does not activate the JAK/STAT or MAPK pathways, as has been reported for other cell types (Baker et al. 2008; Hintzen et al. 2008; Korzus et al. 1997), but robustly activates the NF-κB pathway. The mechanism for this unique signaling pathway activation in microglia is not known. Future studies to understand how OSM signals in different cell types of the CNS will be critical for understanding the role of OSM in neuroinflammatory diseases.
In addition to activation of the NF-κB pathway, we have shown that intermediary TNF-α expression induced by OSM contributes to the expression of both TNF-α and iNOS, as loss of the TNF-R1 partially attenuates their expression. The reduction in TNF-α protein expression in TNF-R1−/− microglia at 4 h, while statistically significant, was less striking than at 8 h. These results indicate that while other mechanisms initially contribute to TNF-α expression, TNF-α signaling through the TNF-R1 complex is important for sustaining its expression.
OSM is upregulated in neuroinflammatory diseases including MS and HAND (Repovic et al. 2003). Our results demonstrate that OSM, acting through microglia, promotes neurotoxicity. The neurotoxic phenotype of microglia is mediated by OSM-induced expression of TNF-α, as blockade of TNF-α signaling abolishes neurotoxicity. This does not exclude the possibility that OSM-mediated NO production also contributes to neurotoxicity. However, because TNF-α is an intermediate in iNOS expression, blockade of TNF-α signaling likely causes a reduction in NO production.
IL-27 is emerging as an important regulator of immune responses and is anti-inflammatory in T cells (Stumhofer and Hunter 2008; Stumhofer et al. 2007; Yoshida and Miyazaki 2008). We have identified IL-27 as a promising therapeutic agent in neuroinflammatory disease due to its ability to inhibit OSM-induced TNF-α and iNOS expression in microglia, and to limit neuron cell death. The protection conferred by IL-27 is likely due to its ability to block TNF-α expression. IL-27 inhibits both TNF-α and iNOS expression by reducing OSM-induced p65 phosphorylation and subsequent recruitment to their respective promoters. The mechanism of IL-27-mediated inhibition of p65 activation is not clear. One possibility is that IL-27, signaling though a gp130/WSX-1 dimer, acts as a molecular sink, binding up the gp130 receptor, preventing OSM binding. Future studies are planned to test this hypothesis.
While IL-27 induces modest p65 activation and P-p65 recruitment to the TNF-α and iNOS promoters, IL-27 also reduces OSM-mediated recruitment of the coactivator p300 and acetylation of histone H3, which are transcriptionally permissive modifications, and promotes the recruitment of the transcriptional corepressor HDAC1. Furthermore, studies have shown that the transcriptional activity of p65 is enhanced by its association with p300 and is repressed by association with HDAC1 (Ashburner et al. 2001; Zhong et al. 1998). Importantly, phosphorylated p65 binds p300, while unphosphorylated p65 recruits HDAC1 (Zhong et al. 2002). These results demonstrate that IL-27 blocks OSM-mediated p65 phosphorylation and recruitment, which is associated with decreased recruitment of p300 and increased recruitment of HDAC1 to the TNF-α and iNOS promoters (Figure 8).
Figure 8.

Model of OSM-mediated expression of TNF-α and iNOS in microglia: Regulation by IL-27. OSM induces activation of NF-κB p65, which activates TNF-α and iNOS transcription. TNF-α protein is then translated, released, and signals through the TNF-R1 to sustain NF-κB p65 activation, contributing to subsequent TNF-α and iNOS gene expression. OSM-induced secretion of soluble factors, including TNF-α and possibly iNOS, induce neuron cell death. In the presence of IL-27, OSM-induced NF-κB p65 activation and recruitment to the TNF-α and iNOS promoters is reduced. Also, the presence of p300 at these promoters is replaced by HDAC1. Together, this leads to diminished expression of TNF-α and iNOS and protection against neurotoxicity.
Some studies have shown that the anti-inflammatory effects of IL-27 are mediated through its induction of SOCS-3 or IL-10 in T-cells (Awasthi et al. 2007; Fitzgerald et al. 2007b; Owaki et al. 2006). However, we did not detect SOCS-3 or IL-10 expression in microglia in response to IL-27 (data not shown). Interestingly, the expression of these genes in T-cells occurred via the JAK/STAT pathway and IL-27 does not activate STAT1 or STAT3 in microglia. However, preliminary studies show that IL-27 activates the p38 pathway in microglia (data not shown). A particularly interesting function of the p38 pathway is to induce the expression and phosphorylation of an RNA-binding protein called tristetrapolin (TTP) (Mahtani et al. 2001). TTP has been shown to interact directly with p65, interfering with its nuclear transport and inhibiting NF-κB-dependent gene expression (Liang et al. 2009; Schichl et al. 2009). TTP was also found to interact with HDAC1 and its inhibitory effects on gene expression depended on the presence and deacetylase activity of HDAC1. This observation correlates with the increased recruitment of HDAC1 that we see in the presence of IL-27. Future studies will test whether IL-27 induces recruitment of TTP to the TNF-α and iNOS promoters. Our studies are the first to demonstrate an anti-inflammatory and neuro-protective role for IL-27 in microglia.
Our data demonstrate that in microglia, OSM activates the NF-κB p65 pathway and promotes recruitment of P-p65 to the TNF-α and iNOS promoters (Figure 8). OSM stimulation also enhances recruitment of the co-activator p300, which likely associates with P-p65, and helps drive TNF-α and iNOS gene expression. OSM-induced TNF-α further contributes to p65 activation and expression of TNF-α and iNOS by signaling through TNF-R1. OSM-mediated production of TNF-α (and possibly iNOS) from microglia is neurotoxic. In the presence of IL-27, however, activation of p65 by OSM is reduced, leading to decreased recruitment of P-p65 to the TNF-α and iNOS promoters and a subsequent decrease in gene expression. IL-27 also induces recruitment of HDAC1, which may associate with p65, inducing formation of a repressor complex that prevents active gene transcription (Figure 8). This decrease in inflammatory gene expression from microglia in the presence of IL-27 also protects neurons against OSM-mediated cell death. Thus, we have identified OSM as a pro-inflammatory molecule in microglia that, through the release of soluble factors, promotes neurotoxicity. IL-27 is anti-inflammatory in microglia by blocking inflammatory gene expression and subsequent neuron cell death. Future studies will further assess the functions of OSM and IL-27 within microglia in the hopes of identifying better molecular targets to treat neuroinflammatory diseases.
Supplementary Material
IFN-γ, but not OSM, activates STAT3 in primary microglia. Primary microglia were incubated in media alone (−), or treated with OSM (10 ng/ml) or IFN-γ (10 U/ml) for 15 min. Protein lysates were collected and analyzed for P-STAT3Tyr705 and total STAT3 by immunoblotting. Representative of 2 independent experiments.
OSM induction of TNF-α, but not IL-6, is impaired in TNF-R1−/− microglia. WT and TNF-R1−/− microglia were incubated in media alone (0) or treated with OSM (10 ng/ml) for 2, 4 or 8 h. Total mRNA was harvested and analyzed for IL-6 and TNF-α gene expression by RT-PCR. GAPDH was included as a control. Representative of 2 independent experiments.
Acknowledgments
This work was funded in part by grants from the National Institutes of Health (NS-57563, NS-50665, E.N.B.), a Collaborative MS Research Center Award from the National Multiple Sclerosis Society (CA1059-A-13, E.N.B.), and NMSS grant (RG 3892-A-12, E.N.B.). B.J.B. is supported by NIH T32-NS-48039 and X. Ma is supported by the China Scholarship Council (2008622086).
References
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3(9):745–56. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2(10):734–44. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4(11):1116–22. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21(20):7065–77. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson GP, Nozell SE, Harrison DK, Stonecypher MS, Chen D, Benveniste EN. The prolyl isomerase Pin1 regulates the NF-kappaB signaling pathway and interleukin-8 expression in glioblastoma. Oncogene. 2009;28(42):3735–45. doi: 10.1038/onc.2009.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, Weiner HL. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8(12):1380–9. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- Baker BJ, Qin H, Benveniste EN. Molecular basis of oncostatin M-induced SOCS-3 expression in astrocytes. Glia. 2008;56(11):1250–62. doi: 10.1002/glia.20694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21(17):6480–91. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7(9):929–36. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- Benveniste EN, Benos DJ. TNF-alpha- and IFN-gamma-mediated signal transduction pathways: effects on glial cell gene expression and function. FASEB J. 1995;9(15):1577–84. doi: 10.1096/fasebj.9.15.8529837. [DOI] [PubMed] [Google Scholar]
- Boing I, Stross C, Radtke S, Lippok BE, Heinrich PC, Hermanns HM. Oncostatin M-induced activation of stress-activated MAP kinases depends on tyrosine 861 in the OSM receptor and requires Jak1 but not Src kinases. Cell Signal. 2006;18(1):50–61. doi: 10.1016/j.cellsig.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Cereda C, Baiocchi C, Bongioanni P, Cova E, Guareschi S, Metelli MR, Rossi B, Sbalsi I, Cuccia MC, Ceroni M. TNF and sTNFR1/2 plasma levels in ALS patients. J Neuroimmunol. 2008;194(1–2):123–31. doi: 10.1016/j.jneuroim.2007.10.028. [DOI] [PubMed] [Google Scholar]
- Chen SH, Benveniste EN. Oncostatin M: a pleiotropic cytokine in the central nervous system. Cytokine GrowthFactor Rev. 2004;15(5):379–91. doi: 10.1016/j.cytogfr.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. 1990;10(4):1498–506. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Karlo JC, Kao SC, Landreth GE. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21(4):1179–88. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Mello C, Le T, Swain MG. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29(7):2089–102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensoli F, Fiorelli V, Lugaresi A, Farina D, De Cristofaro M, Collacchi B, Muratori DS, Scala E, Di Gioacchino M, Paganelli R, et al. Lymphomononuclear cells from multiple sclerosis patients spontaneously produce high levels of oncostatin M, tumor necrosis factors alpha and beta, and interferon gamma. Mult Scler. 2002;8(4):284–8. doi: 10.1191/1352458502ms817oa. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DC, Ciric B, Touil T, Harle H, Grammatikopolou J, Das Sarma J, Gran B, Zhang GX, Rostami A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007a;179(5):3268–75. doi: 10.4049/jimmunol.179.5.3268. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DC, Zhang GX, El-Behi M, Fonseca-Kelly Z, Li H, Yu S, Saris CJ, Gran B, Ciric B, Rostami A. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol. 2007b;8(12):1372–9. doi: 10.1038/ni1540. [DOI] [PubMed] [Google Scholar]
- Gavrilyuk V, Horvath P, Weinberg G, Feinstein DL. A 27-bp region of the inducible nitric oxide synthase promoter regulates expression in glial cells. J Neurochem. 2001;78(1):129–40. doi: 10.1046/j.1471-4159.2001.00375.x. [DOI] [PubMed] [Google Scholar]
- Halfter H, Friedrich M, Postert C, Ringelstein EB, Stogbauer F. Activation of Jak-Stat and MAPK2 pathways by oncostatin M leads to growth inhibition of human glioma cells. Mol Cell Biol Res Commun. 1999;1(2):109–16. doi: 10.1006/mcbr.1999.0117. [DOI] [PubMed] [Google Scholar]
- He BP, Wen W, Strong MJ. Activated microglia (BV-2) facilitation of TNF-alpha-mediated motor neuron death in vitro. J Neuroimmunol. 2002;128(1–2):31–8. doi: 10.1016/s0165-5728(02)00141-8. [DOI] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learningand memory deficits in Alzheimer’s mice. J Cell Biol. 2007;178(5):829–41. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintzen C, Evers C, Lippok BE, Volkmer R, Heinrich PC, Radtke S, Hermanns HM. Box 2 region of the oncostatin M receptor determines specificity for recruitment of Janus kinases and STAT5 activation. J Biol Chem. 2008;283(28):19465–77. doi: 10.1074/jbc.M710157200. [DOI] [PubMed] [Google Scholar]
- Kastl SP, Speidl WS, Kaun C, Katsaros KM, Rega G, Afonyushkin T, Bochkov VN, Valent P, Assadian A, Hagmueller GW, et al. In human macrophages the complement component C5a induces the expression of oncostatin M via AP-1 activation. Arterioscler Thromb Vasc Biol. 2008;28(3):498–503. doi: 10.1161/ATVBAHA.107.160580. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Yoshida A, Koga K, Yokoo S, Kuniyasu A, Gotoh T, Sawada M, Nakayama H. Marked induction of inducible nitric oxide synthase and tumor necrosis factor-alpha in rat CD40+ microglia by comparison to CD40-microglia. J Neuroimmunol. 2009;208(1–2):70–9. doi: 10.1016/j.jneuroim.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Korhonen R, Lahti A, Kankaanranta H, Moilanen E. Nitric oxide production and signaling in inflammation. Curr Drug Targets Inflamm Allergy. 2005;4(4):471–9. doi: 10.2174/1568010054526359. [DOI] [PubMed] [Google Scholar]
- Korzus E, Nagase H, Rydell R, Travis J. The mitogen-activated protein kinase and JAK-STAT signaling pathways are required for an oncostatin M-responsive element-mediated activation of matrix metalloproteinase 1 gene expression. J Biol Chem. 1997;272(2):1188–96. doi: 10.1074/jbc.272.2.1188. [DOI] [PubMed] [Google Scholar]
- Kuprash DV, Udalova IA, Turetskaya RL, Kwiatkowski D, Rice NR, Nedospasov SA. Similarities and differences between human and murine TNF promoters in their response to lipopolysaccharide. J Immunol. 1999;162(7):4045–52. [PubMed] [Google Scholar]
- Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, Haugaard LS, Wirenfeldt M, Nielsen M, Dagnaes-Hansen F, et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci. 2009;29(5):1319–30. doi: 10.1523/JNEUROSCI.5505-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Qin H, Benveniste EN. Simvastatin inhibits IFN-gamma-induced CD40 gene expression by suppressing STAT-1alpha. J Leukoc Biol. 2007;82(2):436–47. doi: 10.1189/jlb.1206739. [DOI] [PubMed] [Google Scholar]
- Liang J, Lei T, Song Y, Yanes N, Qi Y, Fu M. RNA destabilizing factor tristetraprolin negatively regulates NF-kappaB signaling. J Biol Chem. 2009;284(43):29383–90. doi: 10.1074/jbc.M109.024745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR. Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. MolCell Biol. 2001;21(19):6461–9. doi: 10.1128/MCB.21.9.6461-6469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Martinez TN, Ruhn KA, Szymkowski DE, Smith CG, Botterman BR, Tansey KE, Tansey MG. Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J Neurosci. 2006;26(37):9365–75. doi: 10.1523/JNEUROSCI.1504-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Ruhn KA, Martinez TN, McAlpine FE, Blesch A, Tansey MG. Intranigral lentiviral delivery of dominant-negative TNF attenuates neurodegeneration and behavioral deficits in hemiparkinsonian rats. Mol Ther. 2008;16(9):1572–9. doi: 10.1038/mt.2008.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Role of microglial-derived tumor necrosis factor in mediating CD14 transcription and nuclear factor kappa B activity in the brain during endotoxemia. J Neurosci. 2000;20(9):3456–68. doi: 10.1523/JNEUROSCI.20-09-03456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VT, Benveniste EN. Critical role of tumor necrosis factor-alpha and NF-kappa B in interferon-gamma -induced CD40 expression in microglia/macrophages. J Biol Chem. 2002;277(16):13796–803. doi: 10.1074/jbc.M111906200. [DOI] [PubMed] [Google Scholar]
- Owaki T, Asakawa M, Kamiya S, Takeda K, Fukai F, Mizuguchi J, Yoshimoto T. IL-27 suppresses CD28-mediated [correction of medicated] IL-2 production through suppressor of cytokine signaling 3. J Immunol. 2006;176(5):2773–80. doi: 10.4049/jimmunol.176.5.2773. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160(2):943–52. [PubMed] [Google Scholar]
- Petrova TV, Akama KT, Van Eldik LJ. Selective modulation of BV-2 microglial activation by prostaglandin E(2). Differential effects on endotoxin-stimulated cytokine induction. J Biol Chem. 1999;274(40):28823–7. doi: 10.1074/jbc.274.40.28823. [DOI] [PubMed] [Google Scholar]
- Qin H, Wilson CA, Roberts KL, Baker BJ, Zhao X, Benveniste EN. IL-10 inhibits lipopolysaccharide-induced CD40 gene expression through induction of suppressor of cytokine signaling-3. J Immunol. 2006;177(11):7761–71. doi: 10.4049/jimmunol.177.11.7761. [DOI] [PubMed] [Google Scholar]
- Repovic P, Benveniste EN. Prostaglandin E2 is a novel inducer of oncostatin-M expression in macrophages and microglia. J Neurosci. 2002;22(13):5334–43. doi: 10.1523/JNEUROSCI.22-13-05334.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repovic P, Fears CY, Gladson CL, Benveniste EN. Oncostatin-M induction of vascular endothelial growth factor expression in astroglioma cells. Oncogene. 2003;22(50):8117–24. doi: 10.1038/sj.onc.1206922. [DOI] [PubMed] [Google Scholar]
- Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9(6):429–39. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Ruprecht K, Kuhlmann T, Seif F, Hummel V, Kruse N, Bruck W, Rieckmann P. Effects of oncostatin M on human cerebral endothelial cells and expression in inflammatory brain lesions. J Neuropathol Exp Neurol. 2001;60(11):1087–98. doi: 10.1093/jnen/60.11.1087. [DOI] [PubMed] [Google Scholar]
- Saha RN, Pahan K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid Redox Signal. 2006;8(5–6):929–47. doi: 10.1089/ars.2006.8.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schichl YM, Resch U, Hofer-Warbinek R, de Martin R. Tristetraprolin impairs NF-kappaB/p65 nuclear translocation. J Biol Chem. 2009;284(43):29571–81. doi: 10.1074/jbc.M109.031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmaj K, Papierz W, Glabinski A, Kohno T. Prevention of chronic relapsing experimental autoimmune encephalomyelitis by soluble tumor necrosis factor receptor I. J Neuroimmunol. 1995;56(2):135–41. doi: 10.1016/0165-5728(94)00139-f. [DOI] [PubMed] [Google Scholar]
- Sonobe Y, Yawata I, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A. Production of IL-27 and other IL-12 family cytokines by microglia and their subpopulations. Brain Res. 2005;1040(1–2):202–7. doi: 10.1016/j.brainres.2005.01.100. [DOI] [PubMed] [Google Scholar]
- Sriram K, O’Callaghan JP. Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol. 2007;2(2):140–53. doi: 10.1007/s11481-007-9070-6. [DOI] [PubMed] [Google Scholar]
- Stumhofer JS, Hunter CA. Advances in understanding the anti-inflammatory properties of IL-27. Immunol Lett. 2008;117(2):123–30. doi: 10.1016/j.imlet.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O’Shea JJ, Hunter CA. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol. 2007;8(12):1363–71. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- Takata F, Sumi N, Nishioku T, Harada E, Wakigawa T, Shuto H, Yamauchi A, Kataoka Y. Oncostatin M induces functional and structural impairment of blood-brain barriers comprised of rat brain capillary endothelial cells. Neurosci Lett. 2008;441(2):163–6. doi: 10.1016/j.neulet.2008.06.030. [DOI] [PubMed] [Google Scholar]
- Vecchiet J, Dalessandro M, Falasca K, Di Iorio A, Travasi F, Zingariello P, Schiavone C, Ensoli F, Pizzigallo E, Paganelli R. Increased production of oncostatin-M by lymphomononuclear cells from HIV-1-infected patients with neuroAIDS. J Acquir Immune Defic Syndr. 2003;32(4):464–5. doi: 10.1097/00126334-200304010-00019. [DOI] [PubMed] [Google Scholar]
- Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee R. A functional transsulfuration pathway in the brain links to glutathione homeostasis. J Biol Chem. 2006;281(47):35785–93. doi: 10.1074/jbc.M602799200. [DOI] [PubMed] [Google Scholar]
- Weissman BA, Gross SS. Measurement of NO and NO synthase. Curr Protoc Neurosci. 2001;Chapter 7(Unit 7):13. doi: 10.1002/0471142301.ns0713s05. [DOI] [PubMed] [Google Scholar]
- Wen W, Sanelli T, Ge W, Strong W, Strong MJ. Activated microglial supernatant induced motor neuron cytotoxicity is associated with upregulation of the TNFR1 receptor. Neurosci Res. 2006;55(1):87–95. doi: 10.1016/j.neures.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Wesemann DR, Dong Y, O’Keefe GM, Nguyen VT, Benveniste EN. Suppressor of cytokine signaling 1 inhibits cytokine induction of CD40 expression in macrophages. J Immunol. 2002;169(5):2354–60. doi: 10.4049/jimmunol.169.5.2354. [DOI] [PubMed] [Google Scholar]
- Xie QW, Kashiwabara Y, Nathan C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem. 1994;269(7):4705–8. [PubMed] [Google Scholar]
- Yoshida H, Miyazaki Y. Regulation of immune responses by interleukin-27. Immunol Rev. 2008;226:234–47. doi: 10.1111/j.1600-065X.2008.00710.x. [DOI] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9(3):625–36. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1(5):661–71. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
IFN-γ, but not OSM, activates STAT3 in primary microglia. Primary microglia were incubated in media alone (−), or treated with OSM (10 ng/ml) or IFN-γ (10 U/ml) for 15 min. Protein lysates were collected and analyzed for P-STAT3Tyr705 and total STAT3 by immunoblotting. Representative of 2 independent experiments.
OSM induction of TNF-α, but not IL-6, is impaired in TNF-R1−/− microglia. WT and TNF-R1−/− microglia were incubated in media alone (0) or treated with OSM (10 ng/ml) for 2, 4 or 8 h. Total mRNA was harvested and analyzed for IL-6 and TNF-α gene expression by RT-PCR. GAPDH was included as a control. Representative of 2 independent experiments.