

Abstract
Cytochrome c oxidase (COX) deficiency is associated with a wide spectrum of clinical conditions, ranging from early onset devastating encephalomyopathy and cardiomyopathy, to neurological diseases in adulthood and in the elderly. No method of compensating successfully for COX deficiency has been reported so far. In vitro, COX-deficient human cells require additional glucose, pyruvate and uridine for normal growth and are specifically sensitive to oxidative stress. Here, we have tested whether the expression of a mitochondrially targeted, cyanide-resistant, alternative oxidase (AOX) from Ciona intestinalis could alleviate the metabolic abnormalities of COX-deficient human cells either from a patient harbouring a COX15 pathological mutation or rendered deficient by silencing the COX10 gene using shRNA. We demonstrate that the expression of the AOX, well-tolerated by the cells, compensates for both the growth defect and the pronounced oxidant-sensitivity of COX-deficient human cells.
Keywords: alternative oxidase, cytochrome c oxidase, human skin fibroblasts, superoxides
INTRODUCTION
The alternative oxidase (AOX), absent from mammals, bypasses the cytochrome (cyt) pathway to shuttle electrons directly from the quinone pool to molecular oxygen in numerous plants, microorganisms and some metazoans, including the sea squirt Ciona intestinalis (Fig 1A) (McDonald & Vanlerberghe, 2004). In a recent report, we demonstrated in cultured human cells that C. intestinalis AOX could be targeted to mitochondria and leads to metabolic changes as predicted by its known ability to bypass the cyt segment of the respiratory chain (Hakkaart et al, 2005). In short, AOX expression enabled human cells to respire in the presence of concentrations of cyanide which completely block control cell respiration. The cyanide-insensitive respiration was inhibited by n-propyl gallate (n-PG), which specifically inhibits AOX (Stegink & Siedow, 1986). Conversely, oxygen consumption in the absence of respiratory chain poisons was insensitive to n-PG, indicating that AOX does not significantly contribute to electron flow when the respiratory chain is functional. AOX expression also blocked the induction of superoxide dismutase in the presence of respiratory poisons such as antimycin, indicating that it alleviates enhanced superoxide production when the respiratory chain becomes inappropriately reduced. Furthermore, it greatly diminished the acidification of the medium caused by culturing cells overnight in the presence of cyanide, which results from a dependence on lactate production to reoxidize NADH if the respiratory chain is unavailable.
Figure 1. Alternative oxidase (AOX) bypasses the cyt segment of the respiratory chain.
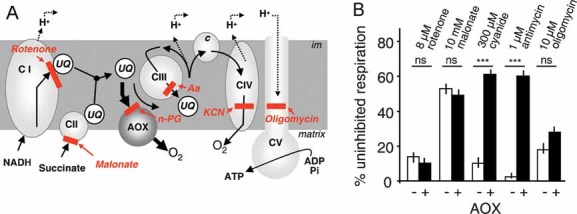
- The cyt pathway bypass provided by AOX and the targets of different respiratory chain inhibitors. Aa, antimycin; c, cyt c; CI–V, the various respiratory chain complexes; im, inner membrane; UQ, ubiquinone.
- The effect of AOX expression on respiration of HEK-293 cells treated with various OXPHOS inhibitors.
Altogether these results indicate that AOX expression can have a beneficial effect in combating the deleterious effects of a deficiency of the cyt chain, especially cytochrome c oxidase (COX). COX defect is associated with a wide spectrum of clinical presentations, ranging from early onset devastating encephalomyopathy and cardiomyopathy, to neurological diseases in adulthood and in the elderly (McFarland et al, 2007). Despite the major progresses made during the last decade in the determination of the molecular bases of COX defect, no method to compensate successfully for such deficiency has been reported so far (Turnbull & Lightowlers, 2002).
This prompted us to investigate the ability of the AOX to complement COX deficiency in human cells. To this end, we expressed the C. intestinalis AOX in COX15- and COX10-depleted human cells, both proteins being required for biosynthesis of the heme moiety of COX (Fontanesi et al, 2006). We demonstrate that AOX expression compensates for the growth defect and the pronounced oxidant-sensitivity of human cells presenting a COX defect.
RESULTS
AOX acts as a bypass only for the cytochrome chain
Although the respiration of human embryonic kidney (HEK)-293-derived cells expressing AOX is significantly resistant to antimycin and cyanide, respective inhibitors of complex III and IV, it is still sensitive to rotenone, malonate and oligomycin, inhibitors of complex I, II and V, respectively (Fig 1B). Similar conclusions were reached when studying the sensitivity of freely accessible mitochondria in digitonin (0.004%)-permeabilized cells expressing the AOX. This is in accordance with the postulated function of AOX as a bypass for the cyt chain, but not for other OXPHOS complexes. In plants, working in concert with the non-proton motive rotenone-resistant NADH dehydrogenase, AOX can also bypass the control exerted on electron flow by complex V (Rustin & Moreau, 1979).
AOX expression in human COX10-depleted HEK-293 cells
HEK-293-derived cells harbouring tetracycline-inducible AOX were made COX-deficient using shRNA technology targeted against COX10, encoding the mitochondrial enzyme hemeA : farnesyltransferase involved in COX assembly (Barrientos et al, 2002). This resulted in a more than 90% decrease in the amount of COX10 gene product and a 60% decrease in COX activity (Fig 2A). COX10 depletion also resulted in a partial decrease of the respiratory rate (about 30%) which was partially compensated by AOX expression (Fig 2B). The proper mitochondrial location and functionality of the AOX gene product were previously established in these cells (Hakkaart et al, 2005). The present study confirmed that AOX expression did not affect the enzymatic activities of the respiratory chain complexes in the COX10-depleted (COX10−) cells (see Table SI of Supporting Information). The inhibitory effect of n-PG further confirmed AOX involvement in the recovery of respiration (Fig 2B). In a high glucose (4.5 g/l) medium, growth of these COX-deficient cells was indistinguishable from that of control cells (Fig 2C, D), whereas they showed a significant growth defect in OXPHOS-selective medium (low glucose, 1 g/l; Fig 2D). AOX induction fully eliminated the growth defect of COX10− cells (Fig 2D).
Figure 2. AOX compensates for the effects COX deficiency caused by COX10 knockdown in HEK-293 cells.
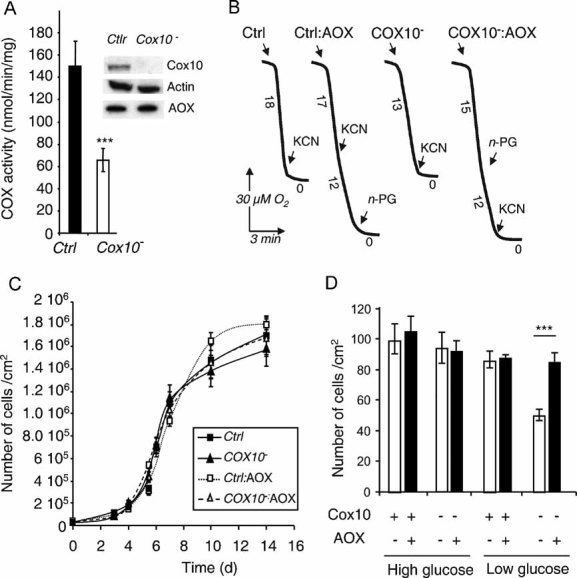
- COX activity in control (AOX-transgenic) and HEK-293 cells expressing COX10-targeted shRNA (clone selected after transfection with pSM2C-COX10 construct). Values are means of triplicates performed on three different clones. Inset shows immunoblot of 100 µg total cell lysates. Specificity of the anti-COX10 shRNA was established by using a scrambled shRNA which did not affect COX10 level or COX activity (data not shown).
- Cell growth of either control (AOX-transgenic) or COX10− HEK-293 cells, with or without doxycyclin induction of AOX expression, in non-selective medium. Cells were seeded at 10,000 cells/cm2.
- Number of cells after 7 days of growth (end of the exponential growth phase) of either control (AOX-transgenic) or COX10− cells, with or without doxycycline induction of AOX expression, in selective medium (i.e. 1 g/l glucose).
- Respiration of intact HEK control and COX10− cells ± AOX. KCN was 300 µM, n-propyl gallate (n-PG) 50 µM. Numbers along the traces represent nmol O2/min per mg protein (mean values from three experiments).
AOX expression in human COX15-mutant skin fibroblasts
We next expressed the C. intestinalis AOX transgene in immortalized COX-defective fibroblasts derived from a patient presenting an early onset fatal hypertrophic cardiomyopathy due to a deleterious COX15 gene mutation (Antonicka et al, 2003). With 35–40% residual COX activity (Fig 3A), these cells exhibited a partial defect of respiration (about 30% decrease as compared to control fibroblasts). The cells were transduced by a packaged lentiviral construct containing the C. intestinalis AOX coding sequence under the control of the EF1α promoter, and in cis to the internal ribosome entry site-green fluorescent protein (IRES-GFP). Owing to the green fluorescence, about 80% of the COX15− cells were successfully transduced (not shown). Using selection on antimycin, which rapidly triggers COX15− cell death, we isolated three clones of COX15−, AOX-expressing (COX15− : AOX) cells. The presence of AOX was confirmed by Western blotting (Fig 3B) and by immunocytofluorescence using, respectively, AOX antibodies raised against the Sauromatum guttatum enzyme (Elthon et al, 1989) and against a mixture of two peptides of the C. intestinalis enzyme which also revealed a clear mitochondrial localization (Fig 3C). The respiration of COX15−: AOX cells was increased by 25–30% compared with parental COX15− cells, and was largely cyanide-resistant (55% residual respiration in the presence of cyanide; Fig 3D). When cultured under restrictive conditions (low glucose), the growth of COX15− cells was significantly impaired, but restored to levels observed in high glucose by the expression of AOX (Fig 3E). Similar results were obtained with three COX15− clones expressing the AOX (data not shown).
Figure 3. AOX compensates for COX deficiency in immortalized fibroblasts derived from a patient harbouring a pathological mutation in the COX15 gene.
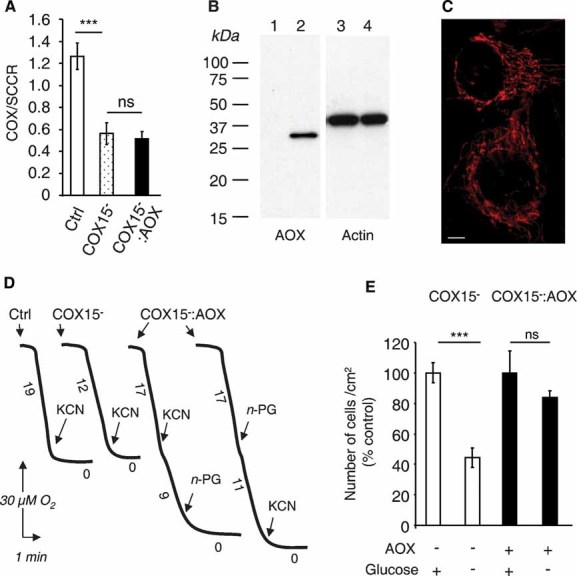
- COX and SCCR activity ratios in immortalized control, COX15− and COX15−: AOX fibroblasts.
- Respiration of intact control, COX15− and COX15−: AOX cells measured using a Clark electrode. KCN was 300 µM, n-PG 50µM. Numbers along the traces represent nmol O2/min per mg protein (mean values from three experiments).
- Immunoblot of 25 µg whole cell lysates from COX15-mutant fibroblasts transduced (or not) with AOX, probed with antibodies against lily AOX (lanes 1 and 2), and actin (lanes 3 and 4).
- AOX mitochondrial localization assessed by AOX antibody. Scale bar, 20 µm.
- Numbers of cells after 7 day growth in selective or non-selective media, expressed as percentage of cell number obtained under non-selective condition. Cells seeded at 15,000 cells/cm2.
AOX protects human cells from oxidative insult
OXPHOS-deficient cells, e.g. those harbouring the MELAS mutation in mtDNA, exhibit a pronounced sensitivity to oxidative insult (Wong & Cortopassi, 1997). This was attributed to an increased oxidant-susceptibility of the calcium-dependent mitochondrial permeability transition pore (Wong & Cortopassi, 1997). We thus measured the oxidant-sensitivity of cultured human fibroblasts rendered COX deficient by mutations in SURF1 (Von Kleist-Retzow et al, 2001), COX10 (Valnot et al, 2000) and COX15 (Antonicka et al, 2003),—and of HEK cells made COX-deficient by shRNA-silencing of COX10. With this aim, we treated cells grown in non-selective medium plus antimycin, which causes endogenous oxidative insult, or with H2O2 resulting in an exogenous oxidative insult. Assessing cell viability revealed that COX-deficient cells were significantly more sensitive to both types of oxidative insult. As compared to control cells, the viability of COX-deficient antimycin-treated cells was reduced to 70–80% for SURF1, COX10 or COX15 mutated fibroblasts and 85% for COX-deficient HEK cells, while antimycin did not affect the viability of control cells (Table I). Similarly, the viability of control fibroblasts was not affected by a 2 day treatment with 200 µM H2O2, while a similar treatment significantly affected the viability of all the COX-deficient cells, from 21 to 13%. Treating HEK cells with 800 µM H2O2 for 6 h only mildly affected viability (94%), but decreased the viability of COX-deficient HEK cells to 63% of untreated cells. Comparable results were observed in a low glucose medium (see discussion below).
Table I.
Hypersensitivity of COX deficient cells to oxidative insults. Viability of cells exposed to 5 µM antimycin and 200 or 800 µM H2O2 for various periods of time
| Cell viability | ||||||
|---|---|---|---|---|---|---|
| Antimycin (7 days) | H2O2 (6 h) | H2O2 (2 days) | ||||
| –a | 5 µM | –b | 800 µM | –b | 200 µM | |
| SURF1− (n = 6) | 93.2 ± 7.1c | 65.3 ± 7.5***(70)d | nde | nd | 91.8 ± 5.8 | 72.6 ± 12.9**(79) |
| COX10− (n = 3) | 93.4 ± 2.6 | 64.8 ± 10.1***(69) | nd | nd | nd | nd |
| COX15− (n = 3) | 96.3 ± 0.2 | 79.2 ± 2.4**(82) | nd | nd | 95.9 ± 1.2 | 83.6 ± 2.0***(87) |
| Mean viability of COX-defective fibroblasts | 91.2 ± 6.9 | 68.6 ± 7.9***(75) | nd | nd | 93.1 ± 5.6 | 79.2 ± 12.3**(85) |
| HEK COX10− | 97.0 ± 0.3 | 82.7 ± 1.4***(85) | 92.1 ± 2.1 | 58.1 ± 5.6***(63) | nd | nd |
| Control fibroblasts (n = 9) | 96.0 ± 0.4 | 91.2 ± 6.8ns(95) | nd | nd | 96.8 ± 0.6 | 96.1 ± 1.2ns(99) |
| Control HEK cells (n = 3) | 97.3 ± 0.4 | 92.9 ± 1.9**(95) | 97.7 ± 6.0 | 91.5 ± 2.0ns(94) | nd | nd |
Measured in the absence of antimycin.
Measured in the absence of H2O2.
Viability of cells expressed as a percentage of cells excluding the Trypan blue. Values are means ± SD of three independent experiments; ns, not significant.
Percentage of untreated cells.
nd, not determined.
This provided a rationale to challenge the COX15− cells by mitochondrial superoxide overproduction triggered by antimycin (Robinson et al, 2006). Antimycin caused massive cell death under restrictive conditions (100% at 3 d) which was counteracted by the AOX expression (60% of growth in the absence of antimycin; 3 d) (Fig 4A). The rescue of cell growth by AOX expression was abolished if antimycin-treated cells were further treated with 1 mM n-PG (Fig 4A). AOX expression also strongly reduces the excess superoxide production (700%) seen in COX15− cells in the presence of antimycin as measured by MitoSOX™ labelling (Fig 4B–C). Under basal conditions, COX15− cells, endowed with or without with AOX, did not produce a detectable amount of superoxides (Fig 4B, 1). Accordingly, superoxide dismutase activity, induced in the case of significant superoxide production, was not increased in these cells (38 ± 7 U/mg prot, control fibroblasts versus 40 ± 10 U/mg prot, COX15− cells versus 38 ± 8 U/mg prot, COX15− : AOX cells). Cell death induced by antimycin was also partially prevented (about 35%) upon a 24 h treatment with a spin trap, 1 mM TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy) (Soule et al, 2007) (Fig 4A). This indicated that cell death triggered by antimycin was in part due to superoxide overproduction which was overcome by AOX expression.
Figure 4. AOX compensates for the hypersensitivity of COX15-mutant fibroblasts to oxidative insult.
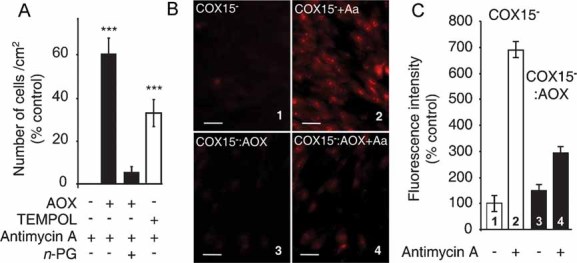
- Growth of AOX-expressing or non-expressing cells (3 d) in low glucose medium in the presence or absence of 5 µM antimycin ±1 mM TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy) (percentage of cells in the absence of antimycin). Cells seeded at 15,000 cells/cm2.
- Fluorescence microscopy images using MitoSOX™ probe, with or without 5 µM antimycin (30 min). Panels 1 and 2, COX15− cells (1 untreated; 2 antimycin treated), Panels 3 and 4, COX15− : AOX cells (3 untreated; 4 antimycin treated) (scale bars represent 40 µm).
- Quantification of MitoSOX™ staining (Fig 3B panels 1–4) expressed as a percentage of the labelling measured in control COX15− cells (panel 1).
DISCUSSION
So far, mutations in up to eight different genes, including nuclear genes SURF1, SCO1, SCO2, COX10 and COX15, plus the mitochondrial genes MTCOX I-III, have been shown specifically to cause COX defect in humans (MITOMAP, 2007). However, the physiological consequences of COX deficiency, including the tissue-specificity of the resulting phenotypes and the underlying pathological processes, remain poorly understood. This is certainly limiting the development of therapy (Smeitink et al, 2006).
We show here that expressing AOX rescues major impairments associated with COX deficiency especially the growth defect observed under selective conditions. We also established that AOX not only facilitates bypassing of the electron flow blockade in the cyt chain, but also provides a potent antioxidant defence to human cells. Although AOX has been lost during vertebrate evolution (McDonald & Vanlerberghe, 2004), we hardly detected any major drawback of its re-introduction in mammalian mitochondria, at least in cultured cells. Importantly, the expressed enzyme is essentially inert as long as the cyt chain is functional (Hakkaart et al, 2005). Obviously, AOX function will decrease, but not nullify, the yield of the phosphorylation associated with cell respiration. This decreased ATP yield might represent a disadvantage under some specific circumstances of high-energy requirement in fast-moving organisms such as mammals as compared to plants. Conversely, in plants, AOX plus the non-proton motive rotenone-resistant NADH dehydrogenase, should allow escape from control of mitochondrial function imposed by photosynthetic activity, allowing the organism to face the challenge of highly variable energy requirements encountered during the day–night cycle (Rustin & Lance, 1986). Ongoing research (Jacobs et al, 2008) suggests that AOX expression is benign even in fast moving animals such as flies, while the antioxidant capacity it represents could offer a definite advantage. AOX expression thus provides a useful tool to delineate the functional consequences of defects in the cyt chain. In particular, as we show here, it highlights the pathophysiological importance of the hypersensitivity of COX-deficient cells to oxidative insult.
We previously demonstrated that AOX expression in human cells protects respiration from the inhibitory effect of cyanide and antimycin (Hakkaart et al, 2005). We now show that it also alleviates many of the consequences resulting from inherited COX defect in cultured fibroblasts derived from patients. So far, all our experiments indicate that AOX is well-tolerated in human cells. Noticeably, due to its distinct kinetic properties, it does not readily compete with a functional cyt system; in other words, it remains mostly inactive so long as COX is working, and will be active only when needed, in COX-deficient cells. This suggests that AOX could be of benefit in human disorders resulting from impaired activity of the cyt segment of the respiratory chain, but not those involving complex I or II. Such benefit could either be targeted to specific organs or possibly throughout the organism. However, before concluding that AOX could have therapeautic utility, we obviously need to confirm that it is benign in development and in all organs of mammalian models.
MATERIALS AND METHODS
Construction of the AOX expressing vector
The C. intestinalis AOX cDNA (Hakkaart et al, 2005) was recloned as a SmaI fragment in the PmeI site of lentivector pWPI (Addgene, Cambridge, MA, USA) (Kvell et al, 2005), via blunt-end cloning, creating the construct pWPI-AOX. Virus production used standard procedures (Bovia et al, 2003) and the second-generation packaging system, which incorporates inbuilt safety features (Pellinen et al, 2004) (http://www.lentiweb.com/protocols_lentivectors.php).
Cell culture, transfection and infection
HEK-293-derived AOX-transgenic cells (Hakkaart et al, 2005) were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco Invitrogen, Cergy Pontoise, France) containing 4.5 g/l glucose supplemented by 2 mM glutamine (as glutamax™), 5% tetracycline-free foetal calf serum, 200 µM uridine, 1 mM pyruvate (King & Attardi, 1989), 100 µg/ml hygromycin and 10 µg/ml blasticidin. AOX transgene expression was induced by 1 µg/ml doxycyclin (Sigma–Aldrich; Saint Quentin, Flavier, France). Fibroblasts were grown in DMEM containing 4.5 g/l glucose supplemented by 2 mM glutamine (as glutamax™), 10% foetal calf serum, 200 µM uridine, 2 mM pyruvate and 10 mg/ml penicillin/streptomycin. HEK-293-derived AOX-transgenic cells were transfected with pSM2C plasmid from Open Biosystems (Huntsville, USA) expressing a shRNA directed against COX10 using lipofectamine (Invitrogen, Saint-Quentin en Yvelines, France) according to the manufacturer's instructions. The hairpin sequence was: 5′-TGCTGTTGACAGTGAGCGACCAGCCTATCTTTGTCCAGAATAGTGAAGCCACAGATGTATTCTGGACAAAGATAGGCTGGGTGCCTACTGCCTCGGA-3′, where sense and antisense sequences have been underlined. Lentivector transduction used standard methods (LENTIWEB-PROTOCOLS, 2008; Salmon et al, 2000). For determination of growth characteristics, HEK-293-derived cells and fibroblasts were seeded at 10,000 and 15,000 cells/cm2, respectively; plating efficiency was measured and the medium was changed every 2 days.
The paper explained
PROBLEM:
Respiration is crucial for cellular energy production. Mutations in genes required for the proper functioning of the mitochondrial respiratory chain can therefore have severe deleterious effects. In fact, more than 10 of the 30 genes required for the functioning of the terminal portion of the mitochondrial respiratory chain have been found to be mutated in several devastating human diseases such as Leigh syndrome, an early onset encephalomyopathy, myo-, neuro-, gastrointestinal encephalopathy (MNGIE syndrome) or Kearns–Sayre syndrome (KSS). Unfortunately, these have remained untreatable thus far. Decreased activity of the chain has also been implicated in human ageing.
RESULTS:
Here, it is shown that the expression of an alternative oxidase (AOX) from the marine invertebrate C. intestinalis in cultured human cells overcomes the deleterious consequences associated with pathological mutations affecting the terminal portion of the respiratory chain (COX10, COX15 depletion). This single-chain, monomeric oxidase restores active electron flow when the function of the chain is impaired thus restoring respiration and protecting the cell from oxidative insult. Importantly, due to its very unusual kinetic properties, expression of AOX does not impede the normal function of the endogenous respiratory chain.
IMPACT:
The data suggest that this approach has promising clinical applications in a wide spectrum of human metabolic and degenerative diseases, arising from respiratory defects and/or major oxidative stress. We look forward to seeing whether AOX can be expressed in specific organs or in the whole organism and most importantly whether it can safely compensate for respiratory chain impairment in mammalian disease models.
Viability assays
Cells were grown in DMEM containing 4.5 g/l glucose in six-well plates to near confluence and exposed either to 200 or 800 µM hydrogen peroxide, made fresh daily, for 6 or 48 h or else seeded at 15,000 cells/cm2 and exposed to 5 µM antimycin for 7 days with the medium changed every 2 days. Cells were harvested by trypsin-EDTA treatment and resuspended in Hank's balanced salt solution (HBSS). At least three replicates of each treatment were used to calculate viability using the Trypan blue exclusion assay.
Biochemical methods
Cell lysates were prepared by the RIPA method. Western blots were carried out to confirm the effect of shRNA on COX10 and to check for AOX expression. Primary antibodies were rabbit polyclonal anti-COX10 (ProteinTech, Manchester, UK), mouse monoclonal anti-actin (Chemicon-Millipore, Saint Quentin en Yvelines, France) and mouse monoclonal anti-c-Myc, clone 9E10 (Roche, Meylan, France), mouse monoclonal anti-lily (Arum maculatum L.). AOX (Elthon et al, 1989) was kindly provided by A. Sainsard-Chanet. Peroxidase conjugated anti-mouse and anti-rabbit secondary antibodies (Amersham, Buckinghamshire, UK) were used at 10,000-fold dilution. COX and succinate cyt c reductase (SCCR) activities were measured according to the procedure described by Benit et al (2006). Cell respiration was measured using a Clark oxygen electrode (Hansatech, Norfolk, UK) fitted to a magnetically stirred 250 µl chamber maintained at 37°C in a medium consisting of 0.3 M mannitol, 5 mM KCl, 5 mM MgCl2, 10 mM phosphate buffer (pH 7.2) and 1 mg/ml bovine serum albumin, plus inhibitors as indicated. Total superoxide dismutase (SOD) activity (EC 1.15.1.1; Mn- and CuZn-dependent enzymes) was determined by the pyrogallol auto-oxidation assay, with 50% decrease of the auto-oxidation rate by SOD being defined as 1 U. Results were expressed as U/mg protein.
Fluorescence microscopy
Cells were seeded at 15,000 cells/cm2 and grown for 3 days in standard conditions on Lab-Tek® chambered coverglass (Nunc, Rochester, NY, USA). For MitoSOX™ staining, cells were washed three times with pre-warmed HBSS (Invitrogen, Saint Quentin en Yvelines, France) buffer and incubated for 30 min in HBSS supplemented with 3 µM DMSO-dissolved MitoSOX™ with or without 5 µM antimycin. Finally, cells were washed three times in HBSS plus MitoSOX™. Fibroblasts washed with phosphate buffered saline (PBS) were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 (w/v) for 5 min and washed three times with PBS. After blocking with 5% goat serum in PBS/0.05% Tween (w/v) for 30 min, cells were treated for 2 h with anti-AOX serum (5,000-fold dilution, rabbit polyclonal antibody raised against C. intestinalis AOX peptides CVNHDLGSRKPDEQNPYPPGQ and FKIETNDSTDEPNIEVENFPC (21st Century Biochemicals, Marlboro, MA, USA). Cells were washed three times with PBS/Tween, treated for 1 h with Alexa 562-conjugated anti-rabbit secondary antibody (1,000-fold dilution; Invitrogen, Cergy-Pontoise, France), washed again three times and mounted with Fluorescent mounting medium (Dako, Trappes, France; all procedures at room temperature). Fluorescence was visualized using an eclipse TE300 microscope (Nikon, Champigny sur Marne, France).
Statistical treatment
All data were analysed by one-way ANOVA using SigmaStat software (Sigma, St. Louis, USA). Data shown are means ± 1 SD. **p < 0.01 and ***p < 0.001 and 'ns' indicates non-significant differences.
Acknowledgments
We thank Tea Tuomela, Marika Vähä-Jaakkola, Päivi Turunen and Merja Jokela for technical assistance; Eric Shoubridge for the gift of COX15 mutated immortalized patient fibroblasts, stimulating comments and for carefully reading the manuscript; AFM (Association Française contre les Myopathies), AMMI (Association contre les Maladies Mitochondriales) and the Leducq Foundation for financial support to E.P.D. and P.R. Academy of Finland, Tampere University Hospital Medical Research Fund and Juselius Foundation for financial support to H.T.J.
Supporting Information is available at EMBO Molecular Medicine online (http://www.embomolmed.org).
The authors declare that they have no conflict of interest.
For more information
The Children's Mitochondrial Disease Network:
http://www.emdn-mitonet.co.uk/
United Mitochondrial Disease Foundation:
Association contre les maladies mitochondriales:
http://www.association-ammi.org/
Mitochondrial Medicine Society:
Mitochondrial Research Society:
MitoMap, A human mitochondrial genome database:
Author's website:
Pierre Rustin:
http://www.u676.org/html/mitochondrie.html (in french)
Howy Jacobs:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Antonicka H, Mattman A, Carlson CG, Glerum DM, Hoffbuhr KC, Leary SC, Kennaway NG, Shoubridge EA. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am J Hum Genet. 2003;72:101–114. doi: 10.1086/345489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos A, Barros MH, Valnot I, Rotig A, Rustin P, Tzagoloff A. Cytochrome oxidase in health and disease. Gene. 2002;286:53–63. doi: 10.1016/s0378-1119(01)00803-4. [DOI] [PubMed] [Google Scholar]
- Benit P, Goncalves S, Philippe Dassa E, Briere JJ, Martin G, Rustin P. Three spectrophotometric assays for the measurement of the five respiratory chain complexes in minuscule biological samples. Clin Chim Acta. 2006;374:81–88. doi: 10.1016/j.cca.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Bovia F, Salmon P, Matthes T, Kvell K, Nguyen TH, Werner-Favre C, Barnet M, Nagy M, Leuba F, Arrighi JF, Piguet V, Trono D, Zubler RH. Efficient transduction of primary human B lymphocytes and nondividing myeloma B cells with HIV-1-derived lentiviral vectors. Blood. 2003;101:1727–1733. doi: 10.1182/blood-2001-12-0249. [DOI] [PubMed] [Google Scholar]
- Elthon TE, Nickels RL, McIntosh L. Monoclonal antibodies to the alternative oxidase of higher plant mitochondria. Plant Physiol. 1989;89:1311–1317. doi: 10.1104/pp.89.4.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanesi F, Soto IC, Horn D, Barrientos A. Assembly of mitochondrial cytochrome c-oxidase, a complicated and highly regulated cellular process. Am J Physiol Cell Physiol. 2006;291:C1129–1147. doi: 10.1152/ajpcell.00233.2006. [DOI] [PubMed] [Google Scholar]
- Hakkaart GA, Dassa EP, Jacobs HT, Rustin P. Allotopic expression of a mitochondrial alternative oxidase confers cyanide resistance to human cell respiration. EMBO Rep. 2005;7:341–345. doi: 10.1038/sj.embor.7400601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs H, Vartiainem S, Dassa E, Moreno D, Minkkinen K, Sanz A, Babusiak M, Dufour E, Hakkart G, Tuomela T, O'Dell KM, Rustin P. AOX therapy for mitochondrial diseases. Euromit 7 – Mitochondrial Pathology, From Basic Mechanisms to Disease and Aging. 2008:6. Abstract book. [Google Scholar]
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Kvell K, Nguyen TH, Salmon P, Glauser F, Werner-Favre C, Barnet M, Schneider P, Trono D, Zubler RH. Transduction of CpG DNA-stimulated primary human B cells with bicistronic lentivectors. Mol Ther. 2005;12:892–899. doi: 10.1016/j.ymthe.2005.05.010. [DOI] [PubMed] [Google Scholar]
- McDonald A, Vanlerberghe G. Branched mitochondrial electron transport in the Animalia: presence of alternative oxidase in several animal phyla. IUBMB Life. 2004;56:333–341. doi: 10.1080/1521-6540400000876. [DOI] [PubMed] [Google Scholar]
- McFarland R, Taylor RW, Turnbull DM. Mitochondrial disease – its impact, etiology, and pathology. Curr Top Dev Biol. 2007;77:113–155. doi: 10.1016/S0070-2153(06)77005-3. [DOI] [PubMed] [Google Scholar]
- MITOMAP. A Human Mitochondrial Genome Database. 2007. http://www.mitomap.org. [DOI] [PMC free article] [PubMed]
- Pellinen R, Hakkarainen T, Wahlfors T, Tulimaki K, Ketola A, Tenhunen A, Salonen T, Wahlfors J. Cancer cells as targets for lentivirus-mediated gene transfer and gene therapy. Int J Oncol. 2004;25:1753–1762. [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivousing ethidium-based probes. Proc Natl Acad Sci USA. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustin P, Lance C. Malate metabolism in leaf mitochondria from the crassulacean acid metabolism plant Kalanchoe blossfeldiana Poelln. Plant Physiol. 1986;81:1039–1043. doi: 10.1104/pp.81.4.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustin P, Moreau F. Malic enzyme activity and cyanide-insensitive electron transport in plant mitochondria. Biochem Biophys Res Commun. 1979;88:1125–1131. doi: 10.1016/0006-291x(79)91525-0. [DOI] [PubMed] [Google Scholar]
- Salmon P, Kindler V, Ducrey O, Chapuis B, Zubler RH, Trono D. High-level transgene expression in human hematopoietic progenitors and differentiated blood lineages after transduction with improved lentiviral vectors. Blood. 2000;96:3392–3398. [PubMed] [Google Scholar]
- Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT. Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 2006;3:9–13. doi: 10.1016/j.cmet.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Soule BP, Hyodo F, Matsumoto K, Simone NL, Cook JA, Krishna MC, Mitchell JB. The chemistry and biology of nitroxide compounds. Free Radic Biol Med. 2007;42:1632–1650. doi: 10.1016/j.freeradbiomed.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegink SJ, Siedow JN. Binding of butyl gallate to plant mitochondria: II. Relationship to the presence or absence of the alternative pathway. Plant Physiol. 1986;80:196–201. doi: 10.1104/pp.80.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull DM, Lightowlers RN. A roundabout route to gene therapy. Nat Genet. 2002;30:345–346. doi: 10.1038/ng0402-345. [DOI] [PubMed] [Google Scholar]
- Valnot I, von Kleist-Retzow JC, Barrientos A, Gorbatyuk M, Taanman JW, Mehaye B, Rustin P, Tzagoloff A, Munnich A, Rotig A. A mutation in the human heme A: farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum Mol Genet. 2000;9:1245–1249. doi: 10.1093/hmg/9.8.1245. [DOI] [PubMed] [Google Scholar]
- Von Kleist-Retzow JC, Yao J, Taanman JW, Chantrel K, Chretien D, Cormier-Daire V, Rotig A, Munnich A, Rustin P, Shoubridge EA. Mutations in SURF1 are not specifically associated with Leigh syndrome. J Med Genet. 2001;38:109–113. doi: 10.1136/jmg.38.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A, Cortopassi G. mtDNA mutations confer cellular sensitivity to oxidant stress that is partially rescued by calcium depletion and cyclosporin A. Biochem Biophys Res Commun. 1997;239:139–145. doi: 10.1006/bbrc.1997.7443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.