

Abstract
The neurodevelopmental disorder Williams–Beuren syndrome is caused by spontaneous ∼1.5 Mb deletions comprising 25 genes on human chromosome 7q11.23. To functionally dissect the deletion and identify dosage-sensitive genes, we created two half-deletions of the conserved syntenic region on mouse chromosome 5G2. Proximal deletion (PD) mice lack Gtf2i to Limk1, distal deletion (DD) mice lack Limk1 to Fkbp6, and the double heterozygotes (D/P) model the complete human deletion. Gene transcript levels in brain are generally consistent with gene dosage. Increased sociability and acoustic startle response are associated with PD, and cognitive defects with DD. Both PD and D/P males are growth-retarded, while skulls are shortened and brains are smaller in DD and D/P. Lateral ventricle (LV) volumes are reduced, and neuronal cell density in the somatosensory cortex is increased, in PD and D/P. Motor skills are most impaired in D/P. Together, these partial deletion mice replicate crucial aspects of the human disorder and serve to identify genes and gene networks contributing to the neural substrates of complex behaviours and behavioural disorders.
Keywords: genomic disorder, haploinsufficiency phenotypes, mouse chromosome deletion models, Williams syndrome
INTRODUCTION
The relationship between genes and behaviour is a topic of great current interest. Genome-wide association studies report an increasing number of associations between behavioural traits and genomic regions identified by single nucleotide polymorphisms (SNP). But the genes responsible and the mechanism underlying most of these associations are still unknown. As an alternative approach, the study of a defined human disease caused by a recurrent structural genomic abnormality such as a microdeletion, can provide direct insights into the relationship between genes and behaviour. The microdeletion disorder Williams–Beuren syndrome (WBS; OMIM 194050) is widely studied because of its unique cognitive/neuropsychiatric profile and distinct dysmorphic phenotype (Kaplan et al, 2001; Pober & Dykens, 1996). Investigations in psychology, cognitive neuroscience, dysmorphology and brain imaging have discovered distinct functional and morphological features: uneven cognitive abilities, with relatively preserved expressive language and face-processing skills but significantly impaired visuo-spatial and numerical abilities, hypersensitivity to certain sounds, craniofacial dysmorphic features, vascular stenoses, mild to moderate mental retardation and increased anxiety. A characteristic outgoing personality leads to inappropriate social behaviour, such as overfriendliness towards strangers (Doyle et al, 2004; Jarvinen-Pasley et al, 2008).
WBS is caused by recurrent genomic deletions of ∼1.5 Mb in chromosome band 7q11.23. Deletion formation is mediated by large region-specific low-copy repeat elements (LCR) that flank the WBS critical region (WBSCR) and, when misaligned in meiosis, may lead to non-allelic homologous recombination (NAHR) (Bayes et al, 2003; Peoples et al, 2000). Such microdeletions on other chromosomes, mediated by LCRs as well, also result in distinct clinical syndromes (Lupski & Stankiewicz, 2005). The NAHR mechanism creates microduplications of the identical regions as well, although at lower frequency (Turner et al, 2008). The microduplication corresponding to the WBS deletion causes delay in speech and motor development with a relatively spared visuo-spatial cognition (Berg et al, 2007). Strikingly, the phenotypes of the WBS region duplication and deletion are opposite to each other with respect to visuo-spatial and language abilities, as well as behaviour. Therefore, we hypothesize that the WBS phenotype, including the gregarious talkative personality, is due to haploinsufficiency for a limited number of genes that are dosage-sensitive, and that extra copies of those genes lead to an opposite phenotype that falls within the autism spectrum.
A typical WBS deletion contains 25 genes encoding transcriptional regulators GTF2I, GTF2IRD1, BAZ1B, MLXIPL, signalling molecules FZD9, TBL2, LIMK1 and other molecules functioning in various cellular processes such as STX1A, CYLN2, FKBP6, EIF4H, CLDN3, CLDN4, VPS39D and RFC2 (Francke, 1999) (UCSC Genome browser March 2006 NCBI Build 36.1) (Fig 1A). Most of these genes are expressed in the brain. With the exception of the elastin (ELN) gene, whose deletion or disruption is responsible for supravalvar aortic stenosis (SVAS), the vascular phenotype of WBS (Ewart et al, 1993b), no specific gene–phenotype correlations have been established. Although the majority of individuals with WBS have molecularly identical deletions, they do show phenotypic differences. These may be attributed to variation in the non-deleted alleles on the normal chromosome 7 and/or to alleles at modifier loci located elsewhere in the genome. Therefore, attempts to resolve the gene/function relationship by studying single individuals with unusual smaller deletions are considered problematic, especially if a lacking phenotype is attributed to genes that are not deleted (Doyle et al, 2004; Tassabehji et al, 2005).
Figure 1. Generation of Wbscr deletion mice that model the human WBS.
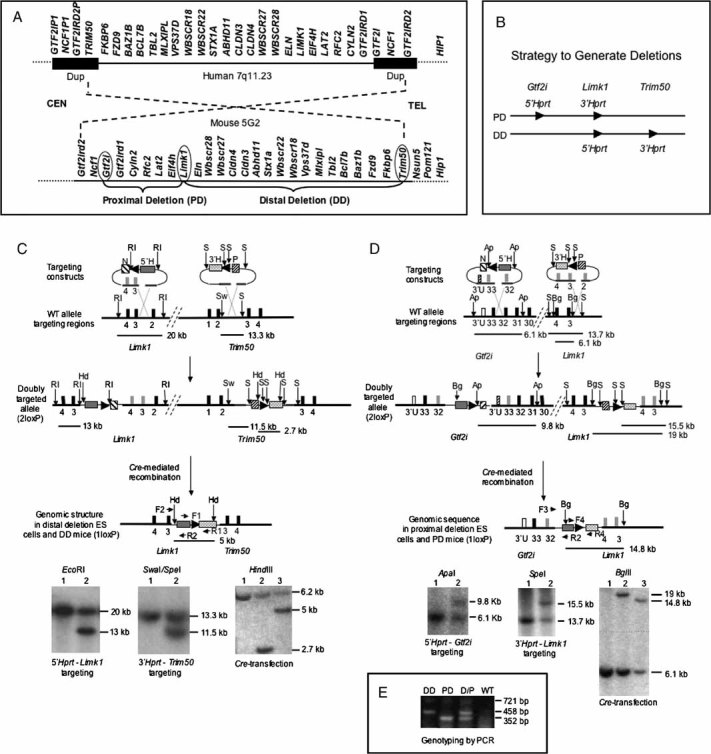
- Comparison of WBSCR genomic regions in human and mouse shows conservation of gene order and the area inverted with respect to centromere (CEN) and telomere (TEL).
- To generate partial deletion mice that model the WBS deletion, constructs containing loxP sites (arrowheads) and partial Hprt sequences were inserted by homologous recombination at three locations. Cre-mediated recombination between the loxP sites generates the desired deletions.
-
To generate DD mice, a Limk1/neomycin resistance (N)/ 5′-Hprt (5′H) construct was first introduced into HPRT-deficient ES cells. Correctly targeted neomycin-resistant clones were then transfected with the Fkbp6/puromycin resistance (P)/ 3′-Hprt (3′H) construct. Doubly targeted (2loxP) cells were transiently transfected with a Cre-recombinase expression vector. Recombination between the two loxP sites in cis generates a restored Hprt minigene whose expression enabled selection for DD containing ES cells in HAT-medium. Arrowheads indicate the location and orientation of loxP sites; black blocks represent numbered genomic exons; grey blocks symbolize exons in the vector. Restriction sites: EcoRI (RI), SpeI (S), SwaI (Sw), HindIII (Hd). Primer pair F1/R1 amplifies the restored Hprt sequence. Primer pair F2/R2 is used for PCR genotyping of DD. Horizontal lines indicate location and size of restriction fragments.Lower left: Southern blot confirms correct targeting of 5′-Hprt construct. Lane 1, WT ES cells; lane 2, targeted ES cells. Lower middle: Southern blot confirms correct insertion of 3′-Hprt construct. Lane 1, WT ES cells; lane 2, targeted ES cells. Lower right: Southern blot confirms the Cre-mediated deletion in 1loxP cells. Following HindIII digestion, a PCR amplified fragment within 3′-Hprt was used as probe. Lane 1, WT ES cells; lane 2, 2loxP ES cells; lane 3, 1loxP ES cells. The ∼6.2 kb band in all lanes is due to non-specific cross-hybridization.
- To generate the PD mice, the Gtf2i locus was targeted with a 5′-Hprt construct, and subsequent transfection with a Limk1 3′-Hprt construct yielded doubly targeted 2loxP ES cells. All symbols are as in (C) and open blocks represent Gtf2i 3′ UTR sequences (3′U). Restriction sites: ApaI (Ap), BglII (Bg). Primer pair F3/R2 is used to genotype PD mice by PCR. Primer pair F4/R4 amplifies the loxP/Hprt sequence. Lower left: Southern blot confirms correct targeting of 5′-Hprt construct. Lane 1, WT ES cells; Lane 2, targeted ES cells. Lower middle: Southern blot confirms correct insertion of 3′-Hprt construct. Lane 1, WT ES cells; lane 2, targeted ES cells. Lower right: Southern blot confirms Cre-mediated deletion in 1loxP cells. Lane 1, WT ES cells; lane 2, 2loxP ES cells; lane 3, 1lox ES cells.
- Multiplex PCR for genotyping of DD, PD and double heterozygous (D/P) mice with primers F2 (for DD), F3 (for PD), and R2 (for both). F4/R4 generate a721 bp fragment from the inserted Hprt sequences, and its absence confirms WT.
In mouse, the entire Wbscr is conserved on chromosome band 5G2 in reverse orientation with respect to the centromere (DeSilva et al, 2002) (Fig 1A). Single-gene knock-out mice for several WBS deletion loci have been reported; however, the phenotypes in heterozygotes were either normal, as for Limk1 (Meng et al, 2002), or not studied, with two exceptions: heterozygotes for Cyln2 and Gtf2ird1 null mutations have attenuated phenotypes when compared to homozygotes. Cyln2+/− mice exhibit mild growth retardation, mild brain abnormality, attenuated contextual fear, and reduced synaptic plasticity and motor function (Hoogenraad et al, 2002). Mice with a heterozygous deletion of exons 2–4 of Gtf2ird1 are less aggressive, more sociable and possibly less anxious (Young et al, 2008).
Mouse models that resemble the WBS genetically and phenotypically are crucial to understand the roles haploinsufficient genes play in development, cognition and behaviour. We generated two strains of mice with complementary half-deletions of the conserved Wbscr on mouse chromosome 5G2 by using chromosome-engineering techniques in mouse embryonic stem cells (ESCs) (Fig 1B). Double heterozygotes (D/P) model the complete WBS deletion, and mice with half-deletions (distal deletion, DD, and proximal deletion, PD) allow us to identify contributions of different subsets of genes. Morphologic and behavioural characterisation revealed that the mice replicate several features of WBS, including abnormal social interaction phenotypes.
RESULTS
Generation of Wbscr partial deletion mice
To create mouse models that reproduce the WBS deletion in humans, we started by making two half-deletions of the Wbscr on chromosome 5G2. To generate mice with DD, we first selected single ESC clones that harbour the 5′-Hprt-loxP-Limk1 insert. One clone was transfected with a 3′-Hprt-loxP vector containing part of Trim50 intron 2. Nine ESC clones that contained both vectors were transfected with a plasmid expressing Cre-recombinase and placed into HAT-selection medium. Two clones had undergone successful recombination restoring a functional Hprt minigene and survived HAT selection. To produce mice that carry the DD deletion, we injected ES cells containing the Cre-induced deletion (1loxP) into C57BL/6J blastocysts. We derived seven chimeras, but none of them transmitted the 1loxP deletion allele through the germ-line. Therefore, to obtain DD mice, we first generated mice from 2loxP ESC clones carrying the two Hprt-loxP insertions in cis, by injecting the clones into C57BL/6J blastocysts. One of six male chimeras transmitted the 2loxP allele to offspring. To induce the Cre-mediated deletion in the zygote, we bred male 2loxP mice with transgenic females that express Cre-recombinase in oocytes as reported previously (Ding et al, 2008). By using PCR primers specific for the deleted allele (F1/R1, Fig 1C), we screened 226 offspring and identified two that carried the DD deletion.
To generate mice with the PD half-deletion, we first produced 2loxP ES cells by consecutive transfections of 5′-Hprt-loxP-Gtf2i and 3′-Hprt-loxP-Limk1 vectors. When two of the four correctly targeted 2loxP clones were injected into mouse blastocysts, one of eight chimeras transmitted the 2loxP allele through the germ-line. To create 1loxP mice, we injected ESC with the Cre-induced deletion into blastocysts. One of seven chimeras transmitted the PD (1loxP) allele to offspring.
By inter-breeding DD/+ and PD/+ heterozygotes, we obtained mice with all four genotypes +/+ (WT) : DD/+ (DD) : PD/+ (PD) : DD/PD (D/P) within the same sibships. Genotyping was performed using a multiplex protocol (see Supporting Information). Littermates were used for comparative studies of gene expression, cranial and brain morphology as well as cognition and behaviour. This breeding scheme reduces the variability introduced by different genetic backgrounds and environmental (maternal) factors.
Reduced expression of genes within the deletions
To determine whether the heterozygous DD and PD deletions result in a corresponding reduction of transcripts, we isolated RNA from mouse brains and performed real-time quantitative RT-PCR (qRT-PCR) for 14 genes that are expressed in brain (see Methods in Supporting Information). In Fig. 2, we plotted the average transcript levels of nine genes within the DD region and six genes in the PD region relative to WT. Limk1 is partially deleted in both DD and PD and, therefore, is included in both graphs. Consistent with a gene-dosage effect, all nine genes in the DD region had reduced transcript levels (∼30–75% of WT levels) in both DD and D/P (Fig 2A). In PD brains, expression of the nine DD region genes were comparable to WT controls, except for Baz1b encoding a chromatin remodelling factor. The reduced level of Baz1b transcripts was nearly identical in DD, PD and D/P, suggesting a regulatory mechanism for Baz1b expression. The Baz1b results were reproduced in two independent sets of pooled samples, and by testing the individual samples separately. In Fig 2B, all PD genes tested were reduced to ∼50% of WT levels in both PD and D/P. As expected, Limk1 expression was reduced to nearly 50% of WT level in both half-deletions and was much lower in D/P. Hip1, encoding huntingtin-interacting protein-1 and flanking the DD distally, showed normal expression levels in deletion mice (data not shown).
Figure 2. Expression of genes measured by real-time qRT-PCR.
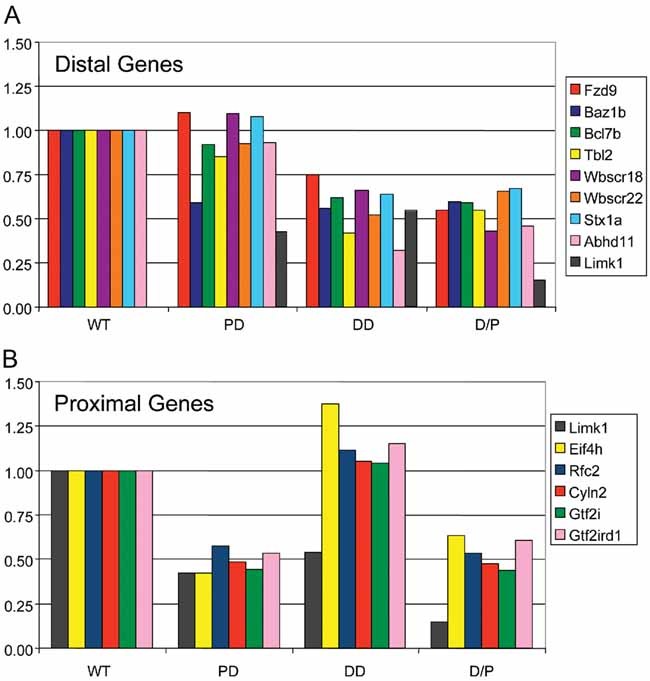
- Within the DD and
- Within the PD. Pooled brain RNA samples from four mice per genotype were analysed. Bars represent the average of triplicate measurements. WT levels are set as 1.00.
Viability, somatic growth and connective tissue phenotypes
Lifespan is normal for all three types of deletion mice. Fertility is normal for DD and PD and somewhat reduced for D/P. Matings between DD/+ and PD/+ of either gender yielded a total of 645 offspring with a genotype distribution of WT 31.9%, DD 24.2%, PD 24.7% and D/P 19.2%. The deviation from the expected Mendelian ratio for WT and D/P was significant by Chi-square analysis (p < 0.05). No gender × genotype distribution difference was detected.
Reduced growth from birth and short stature as adults are characteristics of individuals with WBS. Growth parameters for males and females with WBS are not significantly different, they both follow the 3rd percentile of the general growth curves. To evaluate the growth of the Wbscr deletion mice, we recorded weekly body weights up to 32 weeks of age. From 1 to 5 postnatal weeks onward, all types of deletion mice weighed less than the WT controls (see Fig S1 and statistics in Supporting Information). Males were more severely growth-retarded than females, DD males were less affected than PD, and D/P were most affected. These data indicate an additive effect of the deleted genes, as well as a sex-specific effect on growth.
People with WBS develop connective tissue abnormalities such as hernias, diverticulosis and rectal prolapse. We observed hernias in DD and D/P mice starting at 2 months of age and worsening over time. In severe cases, females developed rectal or vaginal prolapse and had to be killed. The percentage of mice affected was clearly related to strain background. In early generations (N1 or N2) of backcrossing to C57BL/6J, up to 40% of males and 25% of females with D/P had hernias. For DD mice, 10% of males and 5% of females were affected by age 6 months. In later generations when the C57BL/6J background had increased to >80%, only ∼18% of D/P females, and rarely DD mice, still developed hernias. The DD region includes the ELN gene. Haploinsufficiency for ELN is responsible for the connective tissue and vascular abnormalities seen in WBS, in particular, the SVAS and peripheral pulmonary stenosis (Ewart et al, 1993b). Although no cardiovascular abnormalities were detected during autopsies in any deletion mice, real-time M-mode ultrasound studies of abdominal aortic wall motion revealed significantly reduced anterior wall motion in DD and D/P compared to WT and PD (in collaboration with Craig J. Goergen and Charles A. Taylor, Department of Bioengineering, Stanford University).
DD and D/P have foreshortened skulls
WBS individuals have an abnormal craniofacial appearance and a short cranial base. The facial features include a flat nasal bridge, periorbital fullness, malar flattening, a short up-turned nose with anteverted nostrils, a long flat filtrum, full cheeks, prominent lips, a wide mouth and a small chin. These features are documented by three-dimensional facial imaging (Hammond et al, 2005). Upon inspection, Wbscr deletion mice appeared to have a shortened face. To obtain quantitative data, we produced CT scans of the head and reconstructed 3D skull images. We measured distances between landmarks and compared the data for each deletion type to WT (Fig. 3). The most striking abnormalities are observed for D/P. The distances between landmarks along the anterior/posterior axis are shorter in mice of both genders, with females more severely affected. The posterior width of the skull is reduced for both genders. In males, skull widths in the anterior and mid-portions are also reduced. Interestingly, while DD exhibit patterns of changes similar to D/P in skull structure, although with lower significance, PD males had longer skulls than WT males, and PD females were almost normal. These results suggest that haploinsufficient genes in the DD deletion contribute primarily to the craniofacial abnormalities.
Figure 3. Cranial dysmorphology of deletion mice.
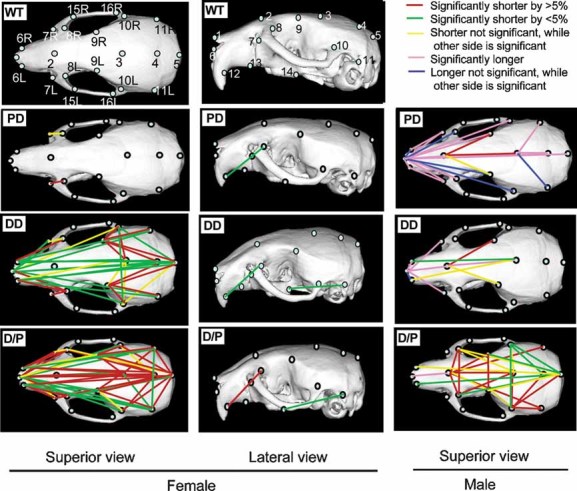
Superior and lateral views of skulls after 3D reconstruction of CT images. Top two panels represent a WT female skull defining the landmarks used for measurements. Colour-coded lines on CT images of deletion mice indicate the differences in length between deletion mice and WT littermates.
Reduced brain size and abnormal brain morphology
People with WBS have smaller brains, with whole brain volume reduced by ∼13% (Thompson et al, 2005). In addition, increased neuronal packing density is observed in primary visual cortex (Galaburda et al, 2002). We compared adult mouse brains with Wbscr deletions to WT controls (Fig 4A, left). Brain weights are reduced by 9.7% in DD, and by ∼14% in D/P. In PD, only female brains are smaller than WT. Images of male brains (Fig 4A, right) illustrate that the entire brain is smaller in DD and D/P, while PD males have normal brain size. Thus, brain sizes appear to be correlated with skull dimensions.
Figure 4. Reduced brain size and left-ventricular (LV) volume, and increased neuronal density.
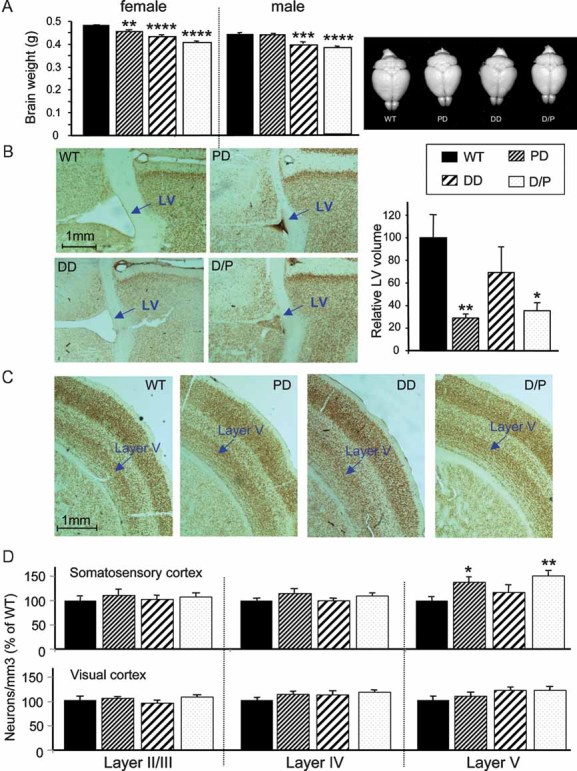
- Comparison of adult brain weights. N = 9–11/genotype for females, N = 6/genotype for males. Bars represent SEM. Right: Brains from male littermates are structurally normal but smaller in DD and P/D.
- Representative images of sections including the left LV at 0.14 mm from bregma after NeuN staining (left); bar graph presents the relative LV volumes for N = 4–5 mice/group (right).
- Representative sections of somatosensory cortex reveal increased neuronal packing in PD and D/P mice.
- Neuronal density in different layers of somatosensory and primary visual cortex shows significant increase in PD and D/P in layer V of somatosensory cortex. N = 4–5/group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To study brain histology, we stained coronal sections with an antibody against NeuN, a marker for mature neurons. The most striking difference between the deletion and control brains involves the size of the lateral ventricle (LV). Representative sections show that PD and D/P brains exhibit strikingly reduced LV size, while DD brains do not appear to differ from WT (Fig 4B, left). For quantification, we reconstructed the LV volumes for each mouse and normalized the data to WT (Fig 4B, right).
When we counted the number of neurons and calculated the neuronal density, we discovered clear differences at layer V of the somatosensory cortex (Fig 4C and Fig 4D, top). The number of neurons in a given area are significantly increased in PD and D/P compared to WT controls. No significant difference was seen in layers II/III and IV of the somatosensory cortex or in the visual cortex (Fig 4D, bottom). Together, these results suggest that genes within the DD affect overall brain growth, while genes in the PD are associated with reduced LV volume and increased neuronal packaging in the somatosensory cortex.
Cognitive and behavioural phenotypes
Behavioural testing was performed independently at Baylor College of Medicine (BCM) and Stanford University Medical Center (SUMC) using methodologies and cohorts of mice that were different and included both genders at BCM and only males at SUMC. Despite these differences, the results are generally consistent. Of note, we observed no differences in test results of offspring derived from DD/+ or PD/+ mothers.
Social interaction tests
Individuals with WBS have inappropriate social approach behaviour. In particular, children with WBS do not develop stranger-anxiety and continue to display overly social and outgoing behaviour towards strangers as adults. To explore sociability in the Wbscr deletion mice, we performed a variety of social interaction tests, including the partition test, direct social interaction test and tube test (at BCM) and the social choice paradigm (at SUMC).
Partition test To assess basic social recognition as well as social interest, we scored the behaviour and level of interest in partner mice at a clear plastic partition between cages that contained small holes. Following a 5 min baseline test with their overnight familiar partners, mice were given a 5 min test with an unfamiliar partner and then another 5 min test with their original familiar partner. Degree of social interest can be reflected by either the total duration of time spent at the partition or by the average (or mean) time spent per visit to the partition (Kudryavtseva, 2003). Deletion mice did not differ from WT in their reaction to the unfamiliar partner relative to the familiar partner, as measured by mean time at the partition (Fig 5A, top). Repeated measures analysis of variance (ANOVA) for genotypes across all three tests, however, reveal that DD, PD and D/P show greater overall social interest relative to WT (p < 0.003) by the mean time measure (Fig 5A, bottom). Genotypes differ for the time spent at the partition (p = 0.050), but not for the number of approaches (p = 0.786). Repeated measures ANOVA for gender across all three tests indicate that male mice made more approaches to the partition (p = 0.003) and spent more time at the partition (p = 0.007) than female mice. There are no significant genotype × gender interactions.
Figure 5. Deletion mice have abnormal social behaviour.
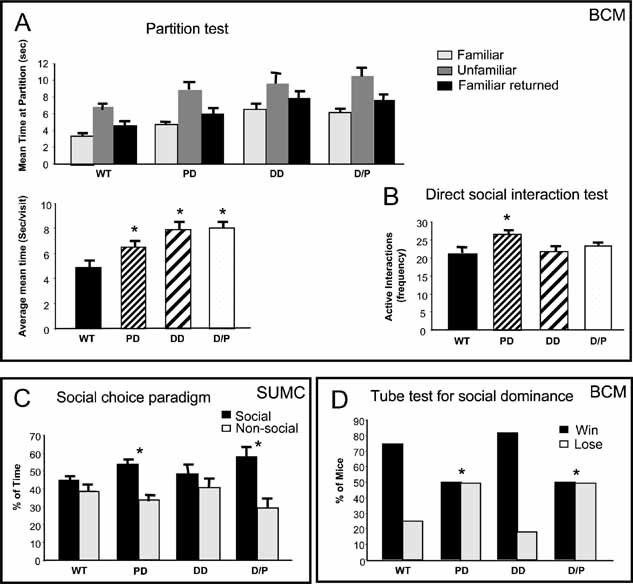
- Partition test measures the time a test mouse spends at the partition that separates it from a partner mouse during three sessions: with a familiar partner, an unfamiliar partner and finally the familiar partner returned. The mean time at the partition per approach to the partition is shown for each test (top panel). The mean time per approach averaged across all three tests (bottom panel) shows that PD, DD and D/P mice exhibited greater social interest at the partition than WT. N = 12–17 per genotype, both sexes.
- In a direct social interaction test, only PD mice showed an increased frequency of interactions during the 10 min test period. N = 12–17 per genotype.
- In the social choice test, mice are placed in a three-chambered apparatus with a stimulus mouse in one chamber, and the percentage of time spent in the ‘social’ versus the ‘non-social’ chambers is recorded. Male PD and D/P demonstrated increased sociability. N = 8–10 per genotype.
- Abnormal social dominance behaviour of PD and D/P in a tube test. Tested mice are released into a tube against a control mouse. The one who backs out of the tube first is considered the loser. N = 12–17 per genotype.
Direct social interaction test After two additional days of housing in the partitioned cages with C57BL/6J partners, mice were scored for active social, passive social and non-social behaviour in a 10 min direct social interaction test (Fig 5B). We found a main effect of genotype for the number of active social approaches by the subject (p = 0.036). Post hoc comparisons revealed that PD made more active social approaches than WT and DD littermates (p = 0.01). The difference between PD and D/P was not significant (p = 0.070). Overall, males spent more time in active social interactions than females (p = 0.043). No genotype × gender interactions were observed for any variable in this test.
Social choice paradigm At SUMC, we used a three-chambered apparatus in which the subject mouse was given the choice to spend time in the ‘social’ chamber and approach a stimulus mouse confined to a restricted enclosure or spend time in the ‘non-social’ chamber where a similar enclosure was placed, but with no animal inside. Male PD and D/P spent significantly greater percentages of total time in the ‘social’ chamber (p < 0.016) (Fig 5C). In contrast, WT and DD spent similar times in both the social and non-social chambers (p > 0.05). Consistent with the percentage time measure, comparisons of the distances travelled in the social and non-social chambers also indicate that PD and D/P, but not WT or DD, travel a greater distance in the social compared to the non-social chamber (p < 0.05). The overall distances travelled in the test chamber were not different among the four groups, indicating similar levels of exploratory and motor activity (p > 0.05).
Tube test In this test at BCM, an experimental mouse is placed at one end of a tube and a C57BL/6J partner mouse at the other end. When the two mice are released, they enter the tube. The mouse that backs out of the tube first is considered the ‘loser’. WT and DD both ‘won’ approximately 70% of their matches (Fig 5D). In contrast, PD and D/P only ‘won’ 50% of their matches. Chi-square analyses confirm that PD and D/P display an abnormal ‘win/loss’ distribution compared to WT. Since differences in body weight may affect social interaction, we weighed the mice daily during the social testing. Repeated measures two-way ANOVA indicate main effects of genotype (p = 0.038) and gender (p < 0.0005), but no genotype × gender interaction (p = 0.448). Post hoc comparisons reveal that D/P mice are significantly smaller than all other genotypes (p < 0.005) and DD and PD are significantly smaller than WT (p < 0.02), consistent with the weight curves established at SUMC (see Fig S1 of Supporting Information). Since the weights of DD and PD are very similar, however, weight differences cannot explain the strikingly different tube test results for DD and PD versus the same partner mouse. Results from all social interaction tests, taken together, provide evidence for PD region gene(s) being the main contributor(s) to abnormal sociability phenotypes.
Exploratory activity and anxiety-related responses
Since anxiety is one of the clinical manifestations of WBS, we assessed exploratory activity and anxiety-related responses using the open-field test, as well as a light–dark box and marble-burying assays (Supporting Information).
Open-field test At BCM, there was an overall difference among genotypes in the total distance travelled (p < 0.005) (Fig 6A, top left). Post hoc comparisons revealed that DD and D/P moved significantly less than WT and PD (p < 0.02). D/P showed increased anxiety-related behaviour, as measured by the centre ratio (Fig 5A, top right), and D/P spent less of the total distance travelled in the centre of the arena, compared to WT, PD and DD mice (p < 0.02). There was a significant genotype × gender interaction in the centre ratio data (p < 0.03). Simple effects post hoc analysis of the interaction revealed that D/P and PD males had decreased centre ratio compared to WT mice (p < 0.01), and that D/P males had a lower centre ratio compared to DD males (p < 0.02). In females, only D/P had a lower centre ratio compared to PD (p < 0.01).
Figure 6. D/P mice have decreased motor function.
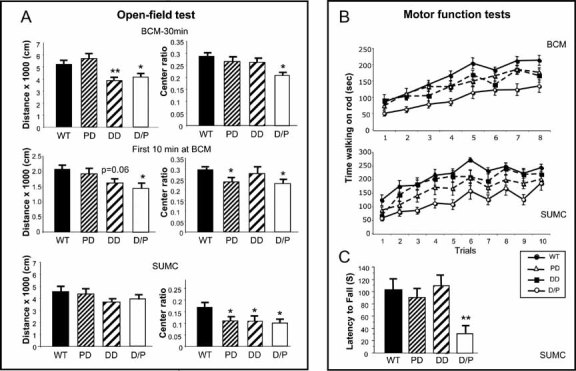
- Exploratory activity and anxiety-related responses were measured with open-field tests performed at BCM (top and middle panels) and SUMC (bottom panel). The total distance travelled (left) and percentage of time spent in the centre of the arena versus the entire open field (right, Centre ratio) showed similar trends for each genotype.
- To assess motor function and motor learning, rotarod tests were performed at BCM and SUMC. Time of walking on the rod is recorded. At BCM, mice were tested for four trials/day on two consecutive days. At SUMC, mice were tested for five consecutive days with two trials/day. The results reveal very similar patterns, with D/P mice performing poorly, and DD and PD mice falling in between D/P and WT, although the SUMC data suggest that DD are closer to WT than PD mice.
- In a wire hanging test, mice are placed upside down on a hanging metal grid, and the latency to fall is recorded. The time for D/P mice was significantly shortened.
The overall pattern of the data obtained at SUMC is similar, although not identical, to those at BCM (Fig 6A, bottom). There are no significant differences in activity, as measured by the distance travelled between the four genotypes tested (p > 0.05). But the WT mice spent significantly more of their exploratory activity, measured in both distance and time, in the centre of the arena compared to DD, PD and D/P (p < 0.05). Among the many factors that could contribute to minor differences between the BCM and SUMC data, such as exact genetic background, gender difference, testing protocol and housing environment, we focused on the testing time. At BCM, mice were tested for 30 min, and at SUMC for 10 min. We, therefore, evaluated only the first 10 min of the BCM data (Fig 6A, middle), and found a pattern more consistent with SUMC data, suggesting that test time was a significant contributor to the differences between sites. Loss of genes in the DD region appears to contribute to the decreased activity phenotype, and BCM data suggest that genes in the PD region are responsible for the anxiety-related response. The SUMC data indicate, however, that the DD region may also contribute to the anxiety response, at least in males.
Sensory and motor function
Children with WBS are hypotonic and have delayed developmental motor milestones. We assessed motor coordination and motor skill learning in the Wbscr deletion mice by using the rotarod and wire hanging tests.
The rotarod test was performed at both BCM and SUMC with very similar results (Fig 6B). At BCM, we found an overall significant difference in the amount of time mice spent walking on top of the rotating rod (p < 0.001). Post hoc comparisons revealed that D/P were impaired relative to WT, DD and PD (p < 0.01). Although not significant (p = 0.08), there was also a trend for DD to perform more poorly than WT. At SUMC, the overall difference in time on top of the rotarod was highly significant as well (p = 0.001). In follow-up comparisons, D/P were impaired relative to WT (p < 0.001) and DD (p = 0.003). Significantly, PD also did not walk on the rotarod as long as WT mice (p = 0.018). Although not significant (p = 0.058), D/P performed more poorly than PD. In a wire hanging test at SUMC (Fig 6C), the latency to fall was significantly shorter for D/P mice than for the other three groups (p < 0.014). Thus, motor coordination, as assessed by the rotarod test, is impaired in D/P mice, and both PD and DD regions contribute to this phenotype. The SUMC rotarod data suggest that the PD genes contribute more than DD region genes, which agrees with the non-significant trend seen in the wire hanging test.
Prepulse inhibition and GAP detection tests The majority (85–95%) of individuals with WBS are hypersensitive to certain types of noises (Van Borsel et al, 1997), most likely due to abnormal auditory processing. Both the prepulse inhibition (PPI) and Gap detection tests can be used to assess sensorimotor processing. The only difference is the presence of an auditory stimulus in the PPI test, and the absence (or gap in sound) in the Gap test, before a loud startling stimulus is presented. In both tests at BCM, we first assessed the baseline startle response to the loud startling stimulus. The overall startle responses during the PPI and Gap tests were different (p = 0.01 and p = 0.02, respectively). Post hoc comparisons revealed that PD displays a significantly greater startle response compared to WT, DD and D/P (p < 0.05) (Fig 7A, left). The percentage inhibition of the startle response by either a prepulse or a gap is also significantly different between genotypes (p < 0.05), but the nature of the differences is stimulus-specific (Fig 7A, right). In the PPI test, the percentage inhibition of startle is significantly lower in PD compared to WT, DD and D/P mice (p < 0.05). The apparent increase in PPI at 78 dB seen in DD is not significant (p > 0.05). In contrast, the percentage inhibition of startle caused by the presence of a gap in sound was significantly increased in DD relative to WT, PD and D/P. Remarkably, the performance of D/P was indistinguishable from WT in both tests. Genes in the DD and PD region appear to regulate sensorimotor processing in different ways, depending on the nature of the stimulus. The PPI abnormality in PD and the GAP detection abnormality in DD are both normalized in D/P, suggesting a complementation between these defects. Of note, PPI abnormalities are not known to be part of WBS in humans.
Figure 7. Abnormal responses to noise and pain, PPI and conditioned fear stimuli.
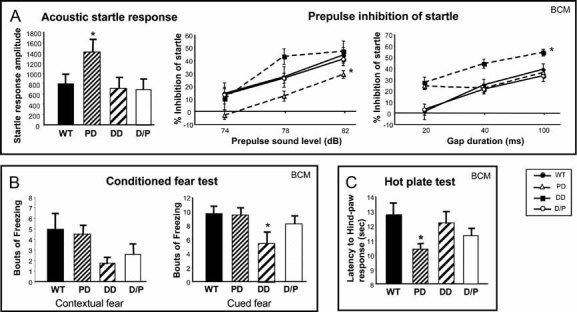
- Deletion mice have abnormal startle response to PPI. First, responses to 120 dB white noise sound were recorded (acoustic startle response, left). Then, percentage inhibition of startle by three prepulses (middle) or by three gaps in sound (right) were recorded.
- DD mice have reduced fear response in an associated learning test. In both contextual and cued fear tests, DD mice exhibited reduced freezing compared to WT littermates.
- PD mice exhibit an accelerated response to thermal pain.
All comparisons were between deletion mice and WT. *p < 0.05, **p < 0.01. Data are presented as mean ± SEM. Mice used at BCM are N = 12–17 per genotype (male and female). Mice used at SUMC are N = 9–11, all male.
Hotplate test The hotplate test provides insight into analgesia-related responses. PD mice have a shorter latency to respond relative to WT and DD (p < 0.05), suggesting that they may have increased sensitivity to painful stimuli (Fig 7C).
Learning and memory
People with WBS exhibit developmental delay and cognitive impairment, usually in the mild to moderate range, with full-scale IQs ranging from severe mental retardation to low-normal.
Conditioned fear test Learning and memory processes in the Wbscr deletion mice were assessed using the conditioned fear test at BCM. The levels of freezing measured during the context and cued fear (conditioned stimulus, CS) tests show an overall difference among the genotypes (p = 0.027). During the context test, DD displayed significantly fewer bouts of freezing relative to the WT (p = 0.05) (Fig 7B, left). In the CS test, DD mice displayed fewer freezing responses relative to WT and PD (p < 0.05) (Fig 7B, right). These findings suggest that DD, and to a lesser extent D/P mice, have impaired learning and/or memory performance.
DISCUSSION
WBS is a multi-system disorder involving abnormalities in growth, development, internal organs, connective tissue, bone formation as well as a characteristic cognitive-behavioural profile (Bellugi et al, 2001). Results of psychometric and behavioural studies have been linked with neuroanatomic, and structural and functional brain imaging studies offering insights into the dynamics of disordered brain development (Chiang et al, 2007; Hocking et al, 2008; Holinger et al, 2005; Meyer-Lindenberg et al, 2006; Mobbs et al, 2007). Although the cause of WBS, a recurrent microdeletion of ∼1.5 Mb on chromosome 7q11.23, was first recognized in 1993 (Ewart et al, 1993a), the contributions of individual genes within the deletion to the cognitive/behavioural phenotype have not been clearly established.
To dissect the molecular mechanisms underlying the unique features of WBS, we generated mouse models that replicate the human molecular deletion (D/P) and also divide it in half (DD and PD). The manipulations of ES cells required to make these two adjacent half-deletions are conceptually straightforward, but technically demanding. Cre-mediated loxP recombination over large genomic distances occurs infrequently and, therefore, the selection system for restored HPRT activity was essential to obtain 1loxP ES cells (Ramirez-Solis et al, 1995; Zheng et al, 1999). Mice with both loxP insertions, but still retaining the intervening sequence, appeared phenotypically normal. Such 2loxP mice, were used to obtain 1loxP DD mice by mating to transgenic mice expressing Cre-recombinase in oocytes. Our experimental system enabled us to compare DD, PD, D/P and WT littermates in phenotypical studies at the molecular, histological, imaging and neurobehavioural levels.
Effect of WBS region deletions on gene expression
To determine whether the genes that are heterozygously deleted are indeed dosage-sensitive at the transcript level, we focused on genes expressed in brain and carried out real-time quantitative RT-PCR analyses on brain tissue. Gene expression patterns follow the genotype expectation with only one exception: Baz1b expression is reduced by about 50% in both half-deletions, as well as in the double heterozygotes, D/P, when compared to WT. Although the absolute Bazb1 transcript levels are quite low in the brain, these results suggest a transcriptional regulatory circuit or trans-acting transcriptional control mechanism. Given the importance of the Bazb1 gene product for DNA replication and transcription, it is not surprising that a regulatory system has evolved. BAZ1B (bromodomain adjacent to zinc finger domain 1B), formerly WBSCR9 (Peoples et al, 1998), functions as a part of the ATP-dependent chromatin remodelling complexes that target heterochromatic replication foci (Bozhenok et al, 2002; Cavellan et al, 2006).
While gene expression data in human WBS brain tissue are not available, qRT-PCR studies of peripheral cells revealed that WBS deletion genes show dosage-dependent expression in fibroblasts and lymphoblasts, but the expression of neighbouring non-deleted genes may also be altered (Merla et al, 2006). To assess whether the PD, DD and D/P deletions affect the expression of genes located just outside of the deletion, we measured transcript levels of Hip1 (Huntingtin-interacting protein-1) in three brain regions. By qRT-PCR analysis, Hip1 expression was indistinguishable from WT in all three types of mutant mice (data not shown). Likewise, global gene expression studies of RNA from cortex, cerebellum and hippocampus showed normal transcript levels for flanking genes Hip1, Nsun5, and Pom121 (for location see Fig 1A) while confirming ∼50% reduction of 8–9 intra-deletion transcripts in all three brain regions (Illumina microarray studies done in collaboration with J. Li and R. M. Myers, Stanford Human Genome Center).
Phenotype studies—general considerations
The D/P double-heterozygotes model the complete WBS deletion in humans. Any phenotypes identified in D/P can potentially be assigned to the distal (DD) or the proximal (PD) half of the deletion. The initial phenotyping studies reported here focus on growth, skull morphology, brain structure and histology as well as cognitive and behavioural testing (Table I). In general, an abnormal phenotype observed in one of the half-deletions is also present in the D/P to about the same degree. In several parameters, however, D/P are more severely affected than either of the half-deletion mice, as in growth retardation and deficits in motor strength and coordination. These results indicate that loss of genes in both half-deletions may contribute to the phenotype. We cannot exclude, however, that homozygous deletion of Limk1 in the D/P mice also plays a role in some tests. Limk1−/− mice, however, were reported to have only few abnormal phenotypes such as dendritic spine defects and mild cognitive impairments (Meng et al, 2002). In addition, the previously postulated role of heterozygous LIMK1 deletion in the spatial cognition phenotype of WBS individuals was refuted in more recent studies (Gray et al, 2006).
Table 1.
Summary of WBS features and phenotypes in PD, DD and double heterozygous (D/P) mice
| WBS feature | Mouse findings | PD | DD | D/P | |
|---|---|---|---|---|---|
| General characterization | |||||
| Growth delay, short stature | Reduced body weight | *m | (+)m | ****m | |
| Short cranial base, midface hypoplasia | Foreshortened skull | − | + | ++ | |
| Vascular stenoses | Reduced aortic wall motion | − | *** | *** | |
| Hernia, prolapse, constipation | Hernia, rectal prolapse | − | + | ++ | |
| CNS abnormality | |||||
| Reduced brain weight | Reduced brain weight | **f | *** | **** | |
| Reduced volume of LVs | ** | − | * | ||
| Increased neuronal packing | Increased neuronal cell density | * | − | ** | |
| Behaviour | Test | ||||
| Social disinhibition | Social choice paradigm | SUMC | * | − | * |
| Partition test | BCM | * | * | * | |
| Direct social interaction | * | − | − | ||
| Tube test (social dominance) | BCM | * | − | * | |
| Anxiety | Open field exploration (centre ratio) | SUMC | * | * | * |
| BCM | * | * | * | ||
| Light–dark box | BCM | − | − | − | |
| Marble burying | BCM | − | − | − | |
| Learning/memory | Contextual fear | BCM | − | * | - |
| Motor deficit | Rotarod | SUMC | * | − | *** |
| BCM | − | − | * | ||
| Wire hanging | SUMC | − | − | ** | |
| Sensitivity to sound | Acoustic startle | BCM | * | − | − |
| Sensorimotor processing | Prepulse inhibition | BCM | * | − | − |
| Gap detection | BCM | − | * | − |
Abnormality observed; for quantitative data:
p < 0.05,
p < 0.01,
p < 0.001,
p < 0.0001.
female only; m, male only.
‘++’, defect relatively more severe; (+), non-significant trend; −, no difference between WT and deletion mice. Sites for behavioural tests, BMC, Baylor College of Medicine; SUMC, Stanford University Medical Center.
Distal deletion (DD) and D/P mice recapitulate the craniofacial dysmorphism in WBS individuals
The cranial base is shortened in WBS individuals, both anteriorly and posteriorly, and in females more than in males (Axelsson et al, 2005). The approach of measuring distances between defined points on three-dimensional CT image reconstructions of mouse skulls was successfully employed in studying mouse models for Down syndrome (Arron et al, 2006; Richtsmeier et al, 2002). When we compared measurements between landmarks in WBS subjects to point-to-point measurements in mice, we noted a strong agreement. While D/P females have shorter skulls, extending from the anterior to the occipital region along all axes, D/P males have significant shortening mostly at the posterior and occipital cranial bases. Although the overall skull shortening in D/P males is less severe than in females, their skull width is more significantly reduced. The cranial volumes, therefore, appear similarly reduced for both D/P males and females. As to the question which of the two half-deletions causes this phenotype, the data unequivocally support the DD region. DD mice of both genders have shortened skulls, whereas in PD, females appear normal and males exhibit an overall increase in length compared to WT. We, therefore, postulate that the craniofacial phenotype of WBS individuals is regulated mostly by genes in the DD region. Our conclusion contradicts the hypothesis that the GTF2IRD1 (Gtf2ird1 in mouse) gene is responsible for the craniofacial dysmorphism (Tassabehji et al, 2005). The reported data of a single human partial deletion case and a homozygous Gtf2ird1 deletion mouse are not comparable to our data.
Distinct CNS structural abnormalities associate with different genomic subregions
There are neuroanatomical similaritites and differences between WBS and Wbscr deletion brains. Adult brain weights of D/P and DD are reduced by ∼14% and 9.7%, respectively, similar to the 13% reduction in WBS brains (Chiang et al, 2007; Thompson et al, 2005). In contrast to humans, where the cerebellum is relatively spared, both cerebra and cerebella appear smaller in D/P and DD mice. Thus, deletion of DD region genes is responsible for reduced overall brain growth. Volume reduction of the LVs, observed in PD and D/P, has not been described in humans with WBS where detailed brain mapping revealed disproportionate reduction of white matter volumes and regional increases in cortical thickness (Thompson et al, 2005). Based on autopsy study of three WBS brains, Galaburda et al (1994) reported a significant increase in cell-packing density in layer IVCβ of the peripheral visual cortex of the left hemisphere. In addition, the left visual cortex had more small and fewer large neurons than controls in layers IVA, IVCa, IVCβ, V and VI. In the deletion mice, we did not detect a significant difference in cell density in the visual cortex or in layers II/III and IV of the somatosensory cortex. Instead, cell density was significantly increased within layer V of the somatosensory cortex in PD and D/P. Since there are no observable differences in cortical thickness between the different types of mice, we conclude that the increased cell density in layer V is due to increased cell number. This increase cannot be specifically attributed to any particular cell type as NeuN-staining does not distinguish between different neuronal subtypes, but preliminary data (not shown) also reveal increased numbers per cubic millimetre of GAD67 positive cells in PD compared to WT. While it is not known how increased cell density could specifically modulate behaviours relevant to WBS, cortical processing of sensory input altered by this mechanism, in principle, could subserve the enhanced sensory processing and even sensory-dependent social behaviour observed in the PD animals.
Behaviour phenotypes map to specific deletions
Social disinhibition represents the most unique and intriguing behavioural characteristic of individuals with the WBS deletion. An opposite, autism spectrum-type behaviour is associated with duplication of the WBS region (Berg et al, 2007). Therefore, the identification of the responsible dosage-sensitive loci is of great interest. Social interaction behaviour in the Wbscr deletion mice was evaluated with four different tests. In a direct social interaction test, PD displayed more bouts of active social interactions relative to the other genotypes. In the three-chamber sociability test, PD and D/P spent more time with the social partner relative to the non-social object. The difference between the D/P performance in the three-chamber test and the direct interaction test may indicate that D/P have an increased social interest when faced with a partner that cannot directly interact/respond, but may be less interactive in a setting with a partner that is free to move away or respond. This notion is supported by the partition test where the combined time near the partition during all three test phases was greater for D/P, DD and PD than for WT, suggesting an increase in social interest. Finally, PD and D/P respond differently in the tube test compared to WT and DD. In this test, mice are forced into a brief face-to-face confrontation with a strange mouse in a confined space. In this setting, both WT and DD are more likely to force the other mouse out of the tube. In contrast, PD and D/P are equally likely to ‘win’ this bout than to ‘lose’ it. The tube test is a very sensitive assay for social dominance, and our results reflect an abnormal social interaction. Considering all data, our Wbscr deletion models display a complex set of abnormal social interactions including increased social interest. Future studies will be necessary to more fully understand the nature of the social interactions in the Wbscr deletion mice and the situations and stimuli that elicit increased social interactions.
People with WBS have motor delays and difficulties with motor functions. In agreement with this observation, D/P mice also perform abnormally at motor tasks. They are significantly impaired on the rotarod from the start of testing, yet improve with training but do not reach WT level. This pattern of performance suggests that they have impaired motor skills, but that ‘skill learning’ is not significantly affected. Consistent with this interpretation, D/P alone did very poorly on the wire suspension tests. PD, and less so DD, are also impaired on the rotarod, but not to the level of D/P, suggesting that genes in both regions contribute to impaired motor skills and coordination. In the open field, DD and D/P are less active. PD and D/P spent less time in the centre of the field. While this may represent an increased anxiety-related response, no differences were observed in other tests for anxiety (light–dark test and marble-burying assay).
The PPI and gap inhibition tests both indicate abnormal processing of rapidly incoming sensory information in DD and PD, but in different ways, while D/P performed similar to WT. In the PPI test, the startle response of PD is significantly less inhibited by sound than that of WT, while DD showed a normal or increased level of inhibition. DD alone also displayed an increase in inhibition following a gap in background noise. The abnormal sensorimotor processing of auditory information in DD and PD is complemented in D/P that display normal inhibition in both the PPI and gap test. Interestingly, PD also showed an increased basal acoustic startle response, and an increased sensitivity to the painful stimulus associated with the hotplate test. Together these observations suggest that PD mice may have increased sensitivity to certain types of sensory stimuli, and that this effect transcends sensory modality. It is intriguing to speculate a relationship to the increased cell density observed in the PD somatosensory cortex. While 84–90% of children with WBS exhibit heightened sensitivity to sounds, they are not known to be overly sensitive to pain, but this has not been systematically evaluated. The basis of the hyperacusis in WBS is complex and appears to involve multiple systems (Gothelf et al, 2006).
The conditioned fear test, for a preliminary assessment of learning and memory responses, indicates that DD, and less so D/P, have a general fear conditioning impairment, suggesting that the DD region may contain gene(s) important for cognition. Additional tests of learning and memory including those that put a significant demand on visuo-spatial processes are necessary to understand better the role of the genes in the deletion region(s) in cognition.
Several of the behavioural tests described in this study were simultaneously performed at two sites on two different cohorts of mice with similar but not identical results. The reason for the differences could include differences in equipment, test protocols, genetic background, housing, feed and gender. Despite the many methodological differences, several behavioural observations were similar at BCM and SUMC suggesting that these particular traits are more robust and less likely to be influenced by procedural differences.
Linking phenotypes to deletions and genes
For phenotypes that clearly segregate with one or the other half-deletion (Table I), we consult the gene list (Fig 1A) for possible candidate loci. The DD region contains more genes but is associated with fewer phenotypes. The connective tissue phenotypes in DD, hernias, rectal prolapse and vascular abnormalities, are most likely attributable to loss of the Eln gene. A prime candidate for neuronal phenotypes is Stx1a. It encodes syntaxin1A, a synaptic vesicle membrane protein that is important for exocytosis, membrane trafficking and neurotransmitter release, and also regulates neurite sprouting and elongation. Homozygous Stx1a−/− mice, however, are remarkably unaffected (Fujiwara et al, 2006). Their normal growth, brain weight and brain structure exclude this locus as a major contributor to the short skull and small brain phenotypes of DD. Limited cognitive/behaviour testing of Stx1a−/− mice revealed normal performance in the open field and Morris water maze tests. Interestingly, contextual as well as cued fear memory were impaired, and the data for heterozygotes fell in between −/− and WT levels indicating a gene dosage effect. Therefore, loss of Stx1a may contribute to the impaired contextual fear phenotype of DD. Fzd9 could be a candidate for the neurobehavioural phenotype of DD. The Wnt receptor Fzd9 is highly expressed during development in parts of the nervous system (Wang et al, 1999), but homozygous Fzd9−/− mice created in our laboratory had no structural or neurobehavioural abnormalities (Ranheim et al, 2005). Another Fzd9−/− mouse showed abnormal spatial memory behaviours in the Morris water maze (Zhao et al, 2005). Although Fzd9+/− were only mildly impaired (data not significant), the authors proposed that Fzd9 contributes to the visuo-spatial defects in WBS. Relevant testing of DD mice will be necessary to address this hypothesis.
For PD region genes, phenotypes in heterozygous knock-out mice have been reported for Cyln2 and Gtf2ird. Cyln2 encodes the cytoplasmic linker protein CLIP-115 that regulates microtubule dynamics. Cyln2+/− mice have reduced body weight in females, reduced size of the corpus callosum, and a deficit in a 24 h contextual fear test, but no changes in cued fear conditioning, open field and wire hanging tests (Hoogenraad et al, 2002). GTF2I and GTF2IRD1 are closely related transcription factors of the TFII-I family. Phenotypes of an insertional mutant of Gtf2ird1 were only reported in homozygotes that are smaller and have craniofacial dysmorphology (Tassabehji et al, 2005) as well as enlarged lateral and 4th ventricles (van Hagen et al, 2007). Behavioural studies of heterozygotes for another Gtf2ird1 induced mutation, however, revealed a non-significant trend towards less anxiety on the elevated plus maze in heterozygotes, but a significant increase in centre/wall ratio in the open field test, in contradistinction to our PD data (Young et al, 2008). Likewise, reported deficits in auditory cued but not contextual fear conditioning are not replicated in our PD data. In a resident intruder test, Gtf2ird1+/− mutants showed a significant decrease in the number of aggressive interactions and a significant increase in the number and length of social interactions. It is, therefore, likely that Gtf2ird1 is a major contributing locus to the social interaction phenotype of PD mice. Caution must be exercised towards comparing between studies when the genetic backgrounds are not identical (Paylor et al, 2006a). Thus, future studies using congenic mice will be necessary to directly compare the WBS mouse models with single-gene and multi-gene deletions. In addition, the interpretation of observations in single-gene knockout models for explaining a deletion phenotype is limited by the possibility that a phenotype seen in the single-gene mutant could be modified or abolished in the deletion model due to complementary effects of the other genes in the deletion. Therefore, the single-gene knockout phenotype observed may not be relevant to the human deletion syndrome at all. The proper way to test the contribution of individual genes would be to make the multi-gene deletion model first, establish robust phenotypes and then try to correct the phenotypes by adding back, one gene at a time.
For Gtf2i, no phenotypes in knock-out mice have been reported, but the encoded TFII-I molecule has a multitude of interesting functions that make it a good candidate for the abnormal sound sensitivity and sensorimotor gating phenotype of PD. Originally found to activate gene transcription through binding to Inr and upstream elements of promoters, TFII-I family members also interact with HDACs and serve as negative regulators of transcription, specifically downregulating estrogen-responsive genes (Ogura et al, 2006; Tussie-Luna et al, 2002). A recently discovered role for TFII-I in the cytoplasm involves the regulation of TRPC3, a cell surface calcium channel (Caraveo et al, 2006).
We have documented solid phenotypes involving social and motor behaviour, skull and brain morphology, and brain histology, and have linked them to subsets of genes in the Wbscr. An abnormal social interaction phenotype, established in two independent cohorts of mice tested at different institutions, provides strong evidence that dosage-sensitive genes in the PD region are responsible. These regions can now be dissected further by genetic complementation studies. These mice will be useful for the study of sociability in mouse models of autism as well as in studies assessing the effect of drugs on social interaction behaviour. Lastly, other aspects of the human WBS phenotype such as hypertension, abnormal kidney function and the formation of vascular malformations can be examined in these mice in future studies.
MATERIALS AND METHODS
Generation of distal deletion (DD) mice
Mouse genomic DNA libraries with vector backbones containing all the essential elements for chromosome engineering were screened for suitable targeting constructs (Ramirez-Solis et al, 1995; Zheng et al, 1999). A 5′-Hprt clone was obtained by screening the mouse 5′-Hprt129/SvEvBrd-derived λ library using a Limk1 cDNA probe. The vector contains a 5.8 kb sequence from intron 2 to intron 4 of the Limk1 gene (Fig 1B,C). It was linearized with AatII and electroporated into AB2.2 ES cells (for cell culture methods see Supporting Information). Targeted ES cell clones, selected for neomycin resistance, were screened for the presence of the insert by Southern blot using a probe derived from Limk1 gene outside of the targeted region (Fig 1C, bottom left). The correct gene targeting event was confirmed by additional Southern blot using a probe upstream of the targeting region (data not shown). One of the correctly targeted clones, 1D4, was used in the secondary targeting with a 3′-Hprt vector construct.
To obtain the 3′-Hprt targeting construct, we inserted a 9.9 kb MluI fragment from the mouse bacterial artificial chromosome (BAC) RPCI 24-230L19 (http://bacpac.chori.org/) that contains a genomic sequence from the Fkbp6 to Trim50 region, into a 3′-Hprt vector (Ramirez-Solis et al, 1995). The construct was linearized by KpnI digestion and transformed into clone 1D4 (Fig 1C). Several puromycin-resistant clones were obtained and the correct insertion of the 3′-Hprt targeting vector was confirmed by Southern blot with probes flanking the targeted region on either side (Fig 1C, and data not shown).
To determine whether the 5′-Hprt and 3′-Hprt vectors are located on the same chromosome (in cis), all clones that contain both 5′-Hprt and 3′-Hprt inserts (2loxP) were transfected with plasmid pOG231 (O'Gorman et al, 1997), which expresses Cre recombinase. Upon transfection, ES cells that undergo loxP–loxP recombination have lost their neor and puror genes but express the reconstituted Hprt minigene, and thus will survive selection in medium supplemented with HAT (0.1 mM hypoxanthine, 0.4 µm aminopterin and 16 µm thymidine). HAT resistant clones were tested in a medium containing neomycin and puromycin. ES cells that contain the desired deletion do not survive this selection. Southern blot with a PCR fragment that includes exons 3–6 of the Hprt minigene cassette as probe confirmed the presence of the complete Hprt gene in 1loxP ES cells (Fig 1C, bottom).
Both 2loxP and 1loxP cells were injected into C57BL/6J blastocysts and only one chimera, derived from a 2loxP ES cell injection, was able to transmit the modified chromosome through the germline. To obtain 1loxP mice, N1 2loxP mice were crossed to C57BL/6J-Tg(Zp3-cre)93Knw/J transgenic mice that express Cre-recombinase in oocytes (The Jackson Laboratory Stock no. 003651).
Generation of proximal deletion (PD) mice
The 5′-Hprt129/SvEvBrd-derived λ library was screened using dCTP-P32 labelled probe generated by the forward 5′-tgcctgacggatctcagccg and reverse 5′-agcctcagagcctcgctccg Gtf2i genomic PCR primers. We obtained a 5′-Hprt targeting clone with a 5.0 kb Gtf2i genomic insert spanning exons 32 and 33 and the 3′-UTR (Fig 1D). The vector was linearized with NdeI and electroporated into AB2.2 ES cells that were then grown in a medium containing geneticin. Southern blots using flanking probes from outside the targeted regions confirmed the presence of the insert (Fig 1D).
To obtain the 3′-Hprt targeting construct for the PD, we removed the genomic insert from 5′-Hprt/Limk1 and placed it into the 3′-Hprt vector backbone. The vector was linearized with BglII and transformed into an ES cell clone previously targeted with 5′-Hprt/Gtf2i (Fig 1D). Several puromycin-resistant clones were selected and the insertion of the 3′-Hprt/Limk1 targeting vector was confirmed by Southern blot analyses with flanking probes (Fig 1D).
To obtain ES cell clones that carry the PD deletion, we introduced Cre-recombinase into 2loxP clones and selected for HATR, neoS and puroS ES cell colonies. Southern blot with a PCR amplified probe from intron 4 of the Limk1 gene confirmed the PD deletion (1loxP ES cells) (Fig 1D, lower right). Both 2loxP and 1loxP ES cells were injected into blastocysts from strain C57BL/6J and several chimeric mice were obtained. One male chimera each, derived from 1loxP and 2loxP ES cells, transmitted the modified allele through the germline.
Breeding of mice for studies
DD/+ and PD/+ mice of either sex were crossed to each other and sibship progenies were used in all studies. Strain background of mice used at SUMC was ∼85% C57BL/6J and ∼15% 129SvEv. Mice studied at BCM were of ∼92% C57BL/6J and 8% 129SvEv origin. Two to five mice were housed per cage in a room with a 12 h light–dark cycle (lights on at 6 AM, off at 6 PM) with access to food and water ad libitum. All housing and behavioural testing procedures were approved by the BCM and Stanford University Animal Care and Use Committees and NIH Guidelines were followed.
Skull morphometry
Littermates at 5–7 months of age were anaesthetized and scanned with an eXplore RS MicroCT System (GE Medical System) at 90 µm resolution. White voxels represent bone. The CT images were reconstructed and the distances between 27 landmark points were measured with the eXplore software.
Brain histology, volumetric and cell density measurements
Perfusion, fixation, sectioning and immunostaining procedures were done as described previously (Aravanis et al, 2007). Mouse anti-NeuN (1:100, Chemicon) was used as the primary antibody and biotinylated anti-mouse (1:500, Jackson) as the secondary antibody. Sections were mounted on slides in PVA-Dabco and imaged on a compound microscope. LV volumes were quantified with Neurolucida (MBF Bioscience Inc.). StereoInvestigator was used to quantify NeuN+ cells in the three major layers of somatosensory cortex and primary visual cortex (II/III, IV, V). A 63 × 63µm2 grid was superimposed onto the slide at 63× magnification and cells were counted within a single square using stereological sampling. Two slices (240 µM apart, centred at the anterior pole of the hippocampus) were counted for each brain. Cell density (cells/µm3) was estimated for each of the three cortical layers and normalized to the WT mean. Data are displayed graphically as a percentage of WT.
The paper explained
PROBLEM:
The human microdeletion Williams–Beuren syndrome (WBS) is associated with cardiovascular and neurobehavioural phenotypes. It is caused by de novo deletion events that are mediated by the same mechanism that is responsible for much of the copy number variation (CNV) that is currently being discovered in the human genome. How CNVs, in this case the simultaneous loss of one copy each of a set of 25 known adjacent genes, exert a phenotype is the challenging question asked here.
RESULTS:
Two strains of mice were generated with complementary half deletions of the conserved WBS orthologous region in mouse and crossed to generate mice carrying both deletions. The mouse models replicate several features of the human disorder and their analysis implies or excludes the contribution of individual genes or sets of genes for phenotypes such as increased sociability, growth retardation or reduced brain growth.
IMPACT:
The results provide a greater understanding of how these features come about in WBS individuals. To the best of our knowledge, this is the first study of mouse models for social disinhibition. The three new mouse strains will be useful for autism research as well as in studies of gene interactions that influence sociability, sensorimotor processing and auditory responses, and of drug effect on social interaction behaviour.
Behavioural testing
Independent behavioural assessments of different cohorts of Wbscr deletion mice were performed at BCM and SUMC.
At BCM, Wbscr deletion mice, received from SUMC, were rederived into the transgenic mouse facility. Mice were backcrossed to C57BL/6J mice for one additional generation. Male and female DD/+ mice were mated with female and male PD/+ mice, respectively. Offspring were genotyped following the SUMC protocol (Supporting Information). Male and female mice were subjected to a rapid version of a modified test battery, i.e. approximately 1–4 days between tests (Paylor et al, 2006b) as described previously (Spencer et al, 2005; Spencer et al, 2006; Walz et al, 2004) (For detailed methods see Supporting Information).
At SUMC, only male progeny of DD/+ and PD/+ crosses were tested when they were 12–20 weeks old. To measure sociability, we used a three-chambered apparatus (Nadler et al, 2004). During a 5 min acclimation period, the subject mouse was placed in the middle neutral chamber and allowed to explore all three chambers. Following acclimation, an unfamiliar juvenile mouse, which had no prior contact with the subject mouse, was placed in one of the side chambers enclosed in a clear plastic cylinder, with several holes. The other side contained an identical but empty plastic cylinder. The subject mouse was then allowed to explore the three-chambered apparatus for a period of 10 min. The location of the stranger in the left versus the right side chambers was alternated between trials. A video camera suspended from the ceiling recorded the sessions and Videotrak Automated Behavioural Analysis (Viewpoint Life Sciences Inc., Quebec, Canada) was used for data analysis.
For open field testing, the arena consisted of a square (40 × 40 × 38 cm3) box with black Plexiglas floor and walls. Mice were placed in one corner of the open field and allowed to explore freely for 10 min. A video camera suspended from the ceiling recorded the sessions and Videotrak Automated Behavioural Analysis (Viewpoint Life Sciences Inc., Quebec, Canada) was used for data analysis. Both central and peripheral activities were measured and centre time was defined as the time spent in the central 10 × 10 cm2 area of the open field. Rotarod and wire hanging tests were performed as described previously (Ding et al, 2008).
Statistical analyses
All data were analysed using a one-way ANOVA, followed with Least Significant Difference (LSD) post hoc where appropriate. For the behavioural studies at BCM, data were analysed by two-way (genotype × gender) or three-way (genotype × gender × repeated measure) ANOVA, followed by LSD post hoc where appropriate.
Acknowledgments
We are grateful to Alan Bradley for providing the libraries, to Monica Medina Sancho, Tereza Kolesnikov, Yelena Prints and Ellora Karmakar for technical assistance and to the Stanford Transgenic Mouse Facility for blastocyst injections. The work was supported by NIH R01 HD39927 (to U.F.) and by the MRDDRC neurobehavioural core at BCM. K.D. is supported by NIMH, NIDA, the NIH Director's Pioneer Award Program, and the Snyder and Coulter Foundations. L.A.M. was supported by a Stanford Bio-X predoctoral fellowship.
Supporting information is available at EMBO Molecular Medicine online (http://www.embomolmed.org/).
The authors declare that they have no conflict of interest.
For more information
OMIM, WBS:
http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=194050
Williams Syndrome Foundation:
Williams Syndrome Association:
http://www.williams-syndrome.org
Williams Syndrome Foundation:
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Aravanis AM, Wang LP, Zhang F, Meltzer LA, Mogri MZ, Schneider MB, Deisseroth K. An optical neural interface: in vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. J Neural Eng. 2007;4:S143–S1456. doi: 10.1088/1741-2560/4/3/S02. [DOI] [PubMed] [Google Scholar]
- Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, Yamasaki N, Miyakawa T, Francke U, Graef IA, Crabtree GR. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- Axelsson S, Kjaer I, Heiberg A, Bjornland T, Storhaug K. Neurocranial morphology and growth in Williams syndrome. Eur J Orthod. 2005;27:32–47. doi: 10.1093/ejo/cjh065. [DOI] [PubMed] [Google Scholar]
- Bayes M, Magano LF, Rivera N, Flores R, Perez Jurado LA. Mutational mechanisms of Williams-Beuren syndrome deletions. Am J Hum Genet. 2003;73:131–151. doi: 10.1086/376565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellugi U, Korenberg JR, Klima ES. Williams syndrome: an exploration of neurocognitive and genetic features. Clin Neurosci Res. 2001;1:217–229. [Google Scholar]
- Berg JS, Brunetti-Pierri N, Peters SU, Kang SH, Fong CT, Salamone J, Freedenberg D, Hannig VL, Prock LA, Miller DT, Raffalli P, Harris DJ, Erickson RP, Cunniff C, Clark GD, Blazo MA, Peiffer DA, Gunderson KL, Sahoo T, Patel A, Lupski JR, Beaudet AL, Cheung SW. Speech delay and autism spectrum behaviors are frequently associated with duplication of the 7q11.23 Williams-Beuren syndrome region. Genet Med. 2007;9:427–441. doi: 10.1097/gim.0b013e3180986192. [DOI] [PubMed] [Google Scholar]
- Bozhenok L, Wade PA, Varga-Weisz P. WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J. 2002;21:2231–2241. doi: 10.1093/emboj/21.9.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraveo G, van Rossum DB, Patterson RL, Snyder SH, Desiderio S. Action of TFII-I outside the nucleus as an inhibitor of agonist-induced calcium entry. Science. 2006;314:122–125. doi: 10.1126/science.1127815. [DOI] [PubMed] [Google Scholar]
- Cavellan E, Asp P, Percipalle P, Farrants AK. The WSTF-SNF2h chromatin remodeling complex interacts with several nuclear proteins in transcription. J Biol Chem. 2006;281:16264–16271. doi: 10.1074/jbc.M600233200. [DOI] [PubMed] [Google Scholar]
- Chiang MC, Reiss AL, Lee AD, Bellugi U, Galaburda AM, Korenberg JR, Mills DL, Toga AW, Thompson PM. 3D pattern of brain abnormalities in Williams syndrome visualized using tensor-based morphometry. Neuroimage. 2007;36:1096–1109. doi: 10.1016/j.neuroimage.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSilva U, Elnitski L, Idol JR, Doyle JL, Gan W, Thomas JW, Schwartz S, Dietrich NL, Beckstrom-Sternberg SM, McDowell JC, Blakesley RW, Bouffard GG, Thomas PJ, Touchman JW, Miller W, Green ED. Generation and comparative analysis of approximately 3.3 Mb of mouse genomic sequence orthologous to the region of human chromosome 7q11.23 implicated in Williams syndrome. Genome Res. 2002;12:3–15. doi: 10.1101/gr.214802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Li HH, Zhang S, Solomon NM, Camper SA, Cohen P, Francke U. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PloS ONE. 2008;3:e1709. doi: 10.1371/journal.pone.0001709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle TF, Bellugi U, Korenberg JR, Graham J. “Everybody in the world is my friend” hypersociability in young children with Williams syndrome. Am J Med Genet A. 2004;124:263–273. doi: 10.1002/ajmg.a.20416. [DOI] [PubMed] [Google Scholar]
- Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993a;5:11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- Ewart AK, Morris CA, Ensing GJ, Loker J, Moore C, Leppert M, Keating M. A human vascular disorder, supravalvular aortic stenosis, maps to chromosome 7. Proc Natl Acad Sci USA. 1993b;90:3226–3230. doi: 10.1073/pnas.90.8.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francke U. Williams-Beuren syndrome: genes and mechanisms. Hum Mol Genet. 1999;8:1947–1954. doi: 10.1093/hmg/8.10.1947. [DOI] [PubMed] [Google Scholar]
- Fujiwara T, Mishima T, Kofuji T, Chiba T, Tanaka K, Yamamoto A, Akagawa K. Analysis of knock-out mice to determine the role of HPC-1/syntaxin 1A in expressing synaptic plasticity. J Neurosci. 2006;26:5767–5776. doi: 10.1523/JNEUROSCI.0289-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaburda AM, Holinger DP, Bellugi U, Sherman GF. Williams syndrome: neuronal size and neuronal-packing density in primary visual cortex. Arch Neurol. 2002;59:1461–1467. doi: 10.1001/archneur.59.9.1461. [DOI] [PubMed] [Google Scholar]
- Galaburda AM, Wang PP, Bellugi U, Rossen M. Cytoarchitectonic anomalies in a genetically based disorder: Williams syndrome. Neuroreport. 1994;5:753–757. doi: 10.1097/00001756-199403000-00004. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Farber N, Raveh E, Apter A, Attias J. Hyperacusis in Williams syndrome: characteristics and associated neuroaudiologic abnormalities. Neurology. 2006;66:390–395. doi: 10.1212/01.wnl.0000196643.35395.5f. [DOI] [PubMed] [Google Scholar]
- Gray V, Karmiloff-Smith A, Funnell E, Tassabehji M. In-depth analysis of spatial cognition in Williams syndrome: a critical assessment of the role of the LIMK1 gene. Neuropsychologia. 2006;44:679–685. doi: 10.1016/j.neuropsychologia.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Hammond P, Hutton TJ, Allanson JE, Buxton B, Campbell LE, Clayton-Smith J, Donnai D, Karmiloff-Smith A, Metcalfe K, Murphy KC, Patton M, Pober B, Prescott K, Scambler P, Shaw A, Smith AC, Stevens AF, Temple IK, Hennekam R, Tassabehji M. Discriminating power of localized three-dimensional facial morphology. Am J Hum Genet. 2005;77:999–1010. doi: 10.1086/498396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking DR, Bradshaw JL, Rinehart NJ. Fronto-parietal and cerebellar contributions to motor dysfunction in Williams syndrome: a review and future directions. Neurosci Biobehav Rev. 2008;32:497–507. doi: 10.1016/j.neubiorev.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Holinger DP, Bellugi U, Mills DL, Korenberg JR, Reiss AL, Sherman GF, Galaburda AM. Relative sparing of primary auditory cortex in Williams syndrome. Brain Res. 2005;1037:35–42. doi: 10.1016/j.brainres.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Hoogenraad CC, Koekkoek B, Akhmanova A, Krugers H, Dortland B, Miedema M, van Alphen A, Kistler WM, Jaegle M, Koutsourakis M, Van Camp N, Verhoye M, van der Linden A, Kaverina I, Grosveld F, De Zeeuw CI, Galjart N. Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nat Genet. 2002;32:116–127. doi: 10.1038/ng954. [DOI] [PubMed] [Google Scholar]
- Jarvinen-Pasley A, Bellugi U, Reilly J, Mills DL, Galaburda A, Reiss AL, Korenberg JR. Defining the social phenotype in Williams syndrome: a model for linking gene, the brain, and behavior. Dev Psychopathol. 2008;20:1–35. doi: 10.1017/S0954579408000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan P, Wang PP, Francke U. Williams (Williams Beuren) syndrome: a distinct neurobehavioral disorder. J Child Neurol. 2001;16:177–190. doi: 10.1177/088307380101600305. [DOI] [PubMed] [Google Scholar]
- Kudryavtseva NN. Use of the “partition” test in behavioral and pharmacological experiments. Neurosci Behav Physiol. 2003;33:461–471. doi: 10.1023/a:1023411217051. [DOI] [PubMed] [Google Scholar]
- Lupski JR, Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1:e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- Merla G, Howald C, Henrichsen CN, Lyle R, Wyss C, Zabot MT, Antonarakis SE, Reymond A. Submicroscopic deletion in patients with Williams-Beuren syndrome influences expression levels of the nonhemizygous flanking genes. Am J Hum Genet. 2006;79:332–341. doi: 10.1086/506371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Mervis CB, Berman KF. Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition and behaviour. Nat Rev Neurosci. 2006;7:380–393. doi: 10.1038/nrn1906. [DOI] [PubMed] [Google Scholar]
- Mobbs D, Eckert MA, Menon V, Mills D, Korenberg J, Galaburda AM, Rose FE, Bellugi U, Reiss AL. Reduced parietal and visual cortical activation during global processing in Williams syndrome. Dev Med Child Neurol. 2007;49:433–438. doi: 10.1111/j.1469-8749.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- Nadler JJ, Moy SS, Dold G, Trang D, Simmons N, Perez A, Young NB, Barbaro RP, Piven J, Magnuson TR, Crawley JN. Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav. 2004;3:303–314. doi: 10.1111/j.1601-183X.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- O'Gorman S, Dagenais NA, Qian M, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci USA. 1997;94:14602–14607. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y, Azuma M, Tsuboi Y, Kabe Y, Yamaguchi Y, Wada T, Watanabe H, Handa H. TFII-I down-regulates a subset of estrogen-responsive genes through its interaction with an initiator element and estrogen receptor alpha. Genes Cells. 2006;11:373–381. doi: 10.1111/j.1365-2443.2006.00952.x. [DOI] [PubMed] [Google Scholar]
- Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, Sparks C, Choi CH, Oghalai J, Curran S, Murphy KC, Monks S, Williams N, O'Donovan MC, Owen MJ, Scambler PJ, Lindsay E. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci USA. 2006a;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paylor R, Spencer CM, Yuva-Paylor LA, Pieke-Dahl S. The use of behavioral test batteries, II: effect of test interval. Physiol Behav. 2006b;87:95–102. doi: 10.1016/j.physbeh.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Peoples R, Franke Y, Wang YK, Pérez-Jurado L, Paperna T, Cisco M, Francke U. A physical map, including a BAC/PAC clone contig, of theWilliams-Beuren syndrome deletion region at 7q11.23. Am J Hum Genet. 2000;66:47–68. doi: 10.1086/302722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peoples RJ, Cisco MJ, Kaplan P, Francke U. Identification of theWBSCR9 gene, encoding a novel transcriptional regulator, in theWilliams-Beuren syndrome deletion at 7q11.23. Cytogenet Cell Genet. 1998;82:238–246. doi: 10.1159/000015110. [DOI] [PubMed] [Google Scholar]
- Pober PR, Dykens EM. Williams syndrome: an overview of medical, cognitive, and behavioral features. Child Adolesc Psychiatr Clin North Am. 1996;5:929–943. [Google Scholar]
- Ramirez-Solis R, Liu P, Bradley A. Chromosome engineering in mice. Nature. 1995;378:720–724. doi: 10.1038/378720a0. [DOI] [PubMed] [Google Scholar]
- Ranheim EA, Kwan HC, Reya T, Wang YK, Weissman IL, Francke U. Frizzled 9 knock-out mice have abnormal B-cell development. Blood. 2005;105:2487–2494. doi: 10.1182/blood-2004-06-2334. [DOI] [PubMed] [Google Scholar]
- Richtsmeier JT, Zumwalt A, Carlson EJ, Epstein CJ, Reeves RH. Craniofacial phenotypes in segmentally trisomic mouse models for Down syndrome. Am J Med Genet. 2002;107:317–324. doi: 10.1002/ajmg.10175. [DOI] [PubMed] [Google Scholar]
- Spencer CM, Alekseyenko O, Serysheva E, Yuva-Paylor LA, Paylor R. Altered anxiety-related and social behaviors in the Fmr1 knockout mouse model of fragile X syndrome. Genes Brain Behav. 2005;4:420–430. doi: 10.1111/j.1601-183X.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- Spencer CM, Serysheva E, Yuva-Paylor LA, Oostra BA, Nelson DL, Paylor R. Exaggerated behavioral phenotypes in Fmr1/Fxr2 double knockout mice reveal a functional genetic interaction between Fragile X-related proteins. Hum Mol Genet. 2006;15:1984–1994. doi: 10.1093/hmg/ddl121. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Hammond P, Karmiloff-Smith A, Thompson P, Thorgeirsson SS, Durkin ME, Popescu NC, Hutton T, Metcalfe K, Rucka A, Stewart H, Read AP, Maconochie M, Donnai D. GTF2IRD1 in craniofacial development of humans and mice. Science. 2005;310:1184–1187. doi: 10.1126/science.1116142. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Lee AD, Dutton RA, Geaga JA, Hayashi KM, Eckert MA, Bellugi U, Galaburda AM, Korenberg JR, Mills DL, Toga AW, Reiss AL. Abnormal cortical complexity and thickness profiles mapped in Williams syndrome. J Neurosci. 2005;25:4146–4158. doi: 10.1523/JNEUROSCI.0165-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Blayney ML, Beck S, Hurles ME. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tussie-Luna MI, Bayarsaihan D, Seto E, Ruddle FH, Roy AL. Physical and functional interactions of histone deacetylase 3 with TFII-I family proteins and PIASxbeta. Proc Natl Acad Sci USA. 2002;99:12807–12812. doi: 10.1073/pnas.192464499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Borsel J, Curfs LM, Fryns JP. Hyperacusis in Williams syndrome: a sample survey study. Genet Couns. 1997;8:121–126. [PubMed] [Google Scholar]
- van Hagen JM, van der Geest JN, van der Giessen RS, Lagers-van Haselen GC, Eussen HJ, Gille JJ, Govaerts LC, Wouters CH, de Coo IF, Hoogenraad CC, Koekkoek SK, Frens MA, van Camp N, van der Linden A, Jansweijer MC, Thorgeirsson SS, De Zeeuw CI. Contribution of CYLN2 and GTF2IRD1 to neurological and cognitive symptoms in Williams syndrome. Neurobiol Dis. 2007;26:112–124. doi: 10.1016/j.nbd.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Walz K, Spencer C, Kaasik K, Lee CC, Lupski JR, Paylor R. Behavioral characterization of mouse models for Smith-Magenis syndrome and dup(17)(p11.2p11.2) Hum Mol Genet. 2004;13:367–378. doi: 10.1093/hmg/ddh044. [DOI] [PubMed] [Google Scholar]
- Wang YK, Sporle R, Paperna T, Schughart K, Francke U. Characterization and expression pattern of the frizzled gene Fzd9, the mouse homolog of FZD9 which is deleted in Williams-Beuren syndrome. Genomics. 1999;57:235–248. doi: 10.1006/geno.1999.5773. [DOI] [PubMed] [Google Scholar]
- Young EJ, Lipina T, Tam E, Mandel A, Clapcote SJ, Bechard AR, Chambers J, Mount HT, Fletcher PJ, Roder JC, Osborne LR. Reduced fear and aggression and altered serotonin metabolism in Gtf2ird1-targeted mice. Genes Brain Behav. 2008;7:224–234. doi: 10.1111/j.1601-183X.2007.00343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Aviles C, Abel RA, Almli CR, McQuillen P, Pleasure SJ. Hippocampal and visuospatial learning defects in mice with a deletion of frizzled 9, a gene in the Williams syndrome deletion interval. Development. 2005;132:2917–2927. doi: 10.1242/dev.01871. [DOI] [PubMed] [Google Scholar]
- Zheng B, Mills AA, Bradley A. A system for rapid generation of coat color-tagged knockouts and defined chromosomal rearrangements in mice. Nucleic Acids Res. 1999;27:2354–2360. doi: 10.1093/nar/27.11.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.